

Abstract
The lethality of prostate cancer is due to the continuous growth of cancer initiating cells (CICs) which are often stimulated by Androgen Receptor (AR) signaling. However, the underlying molecular mechanism(s) for such AR-mediated growth stimulation are not fully understood. Such mechanisms may involve cancer cell-dependent induction of tumor stromal cells to produce paracrine growth factors or could involve cancer cell autonomous autocrine and/or intracellular AR signaling pathways. The present studies document that stromal AR expression is not required for prostate cancer growth, since tumor stroma surrounding AR-positive human prostate cancer metastases (N=127) are characteristically AR-negative. This lack of a requirement for AR expression in tumor stromal cells is also documented by the fact that prostate cancers derived from 5 independent patients grow equally well when xenografted in wild-type vs. AR-null nude mice. Using a series of AR-positive human prostate cancer cell lines, AR-dependent growth stimulation was documented to involve secretion, extracellular binding, and signaling by autocrine growth factors. Orthotopic xenograft animal studies documented that the cell autonomous autocrine growth factors which stimulate prostate CIC growth are not the andromedins secreted by normal prostate stromal cells. Such cell autonomous and extracellular autocrine signaling is necessary but not sufficient for the optimal growth of prostate CICs based upon the response to anti-androgen plus/or minus pre-conditioned media. Thus, AR-induced growth stimulation of human prostate CICs requires AR-dependent intracellular pathways. The identification of such AR-dependent intracellular pathways offers new leads for the development of effective therapies for prostate cancer.
Keywords: Androgen Receptor, Human Prostate Cancer, Oncogene, Cancer Initiating Cell
INTRODUCTION
The fact that systemic androgen is required for the maintenance of the cellular turnover in normal prostate has been known for more than 100 hundred years, and for more than 40 years it has been known that this involves androgen stimulation of proliferation and inhibition of cell death (1). Like normal prostate epithelial cells from which they arise, prostate cancer cells often retain responsiveness to androgen stimulation for their growth (1). In both normal prostate and prostate cancer, these androgenic responses are mediated by Androgen Receptor (AR) initiated signaling (2). Remarkably, while androgen ablation has been standard therapy for inhibiting AR signaling in metastatic prostate cancer patients for nearly 70 years (1), exactly how AR drives growth of prostate cancer cells is still not fully documented. In normal prostate, the epithelial compartment is organized in stem cell units composed of basally located adult stem cells which do not dependent on AR signaling to self renew or to give rise to either neuroendocrine or transit amplifying (TA) cell progeny (2). Cellular turnover in these progeny is driven by a transcriptionally-dependent cascade initiated by an indirect, extracellular-activated, reciprocal paracrine interaction between the stroma and epithelia (2,3). Factors secreted by prostate epithelium stimulate the supporting glandular stromal cells to express AR protein (2,3). Androgen supplied via circulation binds to the AR within prostate stromal cells and initiates AR-dependent transcription of specific target genes within prostate stromal cells resulting in their production and secretion of a series of peptide growth factors know as “andromedins” which include IGF-1, EGF, FGF7, FGF10 (2–6). These stromally derived paracrine andromedins diffuse across the basement membrane into the epithelial compartment where they bind to their respective cognate receptors and initiate cell signaling cascades. Under the appropriate context, these signaling cascades result in the AR negative prostate TA cells undergoing several rounds of proliferation before differentiating into cells termed intermediate cells (ICs) since they co-express both basal and luminal epithelial markers (7). These ICs differentiating into proliferatively quiescent, AR-positive secretory-luminal epithelial cells (2,8,9). Importantly, the proliferative pool of normal TA cells do not require AR expression, while in their AR-positive secretory-luminal progeny ligand induced AR signaling stimulates secretory functions but inhibits their entrance into the cell cycle (2,10).
In contrast to the growth suppressive effect of AR signaling in the subset of AR expressing normal prostate secretory-luminal epithelial cells, AR signaling stimulates the proliferation of prostate cancer cells (1). Indeed, this is the basis for androgen ablation therapy even though the mechanism of androgen stimulation growth of prostate cancer cells has not been fully resolved. Recent experimental studies document that the hierarchical expansion of cancer initiating cells (CICs) drives both the lethality and heterogeneity of prostate cancer (9,11–14). This is because CICs have unlimited self-renewal ability while also giving rise to a hierarchically expanding cascade of phenotypically diverse malignant progeny which have only a limited proliferative ability even though they share the malignant genotype inherited from their CICs parents (9,11–14). Prostate CICs exhibit characteristics of ICs in that they express AR, PSA, and PSCA while not expressing the basal cell marker ΔNp63 (7–9,15). This is consistent with prostate CICs being derived from a malignantly transformed intermediate cell, which has gained stem like self renewing ability (9,16,17). Theoretically, growth of these prostate CICs could involve multiple AR dependent mechanisms (Figure 1). One possibility could involve an indirect mechanism in which prostate cancer cells induce the expression of AR in the tumor stromal cells (Figure 1A). This AR induction, coupled with reprogramming of these stromal cells, could allow ligand-stimulated AR-dependent transcription of specific paracrine growth factor genes. The androgen driven secretion of these paracrine growth factors by tumor stromal cells would allow their extracellular binding to the cancer cell plasma membrane with resultant stimulation of cancer cell DNA replication and proliferation with no requirement of AR signaling within cancer cells themselves. A second indirect but cancer cell autonomous mechanism could involve AR-dependent transcription of specific target genes within the cancer cells themselves resulting in their secretion of autocrine growth factors which bind to plasma membrane receptors on cancer cell stimulating their proliferation with no additional requirement for AR signaling in the cancer cells (Figure 1B). A third possibility could involve a cell-autonomous mechanism whereby AR stimulates cancer cell proliferation not by the extracellular binding of autocrine growth factors but by direct intracellular AR signaling (Figure 1C). Finally, a fourth mechanism could require combination of both extracellular binding of autocrine growth factors plus additional intracellular AR signaling (Figure 1D). Resolving whether some or all of these mechanisms are involved in prostate CICs has significant implications for identifying novel targets for therapy. Therefore, the present studies were performed to resolve these issues.
Figure 1. Possible Mechanisms for AR-Mediated DNA Replication of Prostate Cancer Initiating Cells.
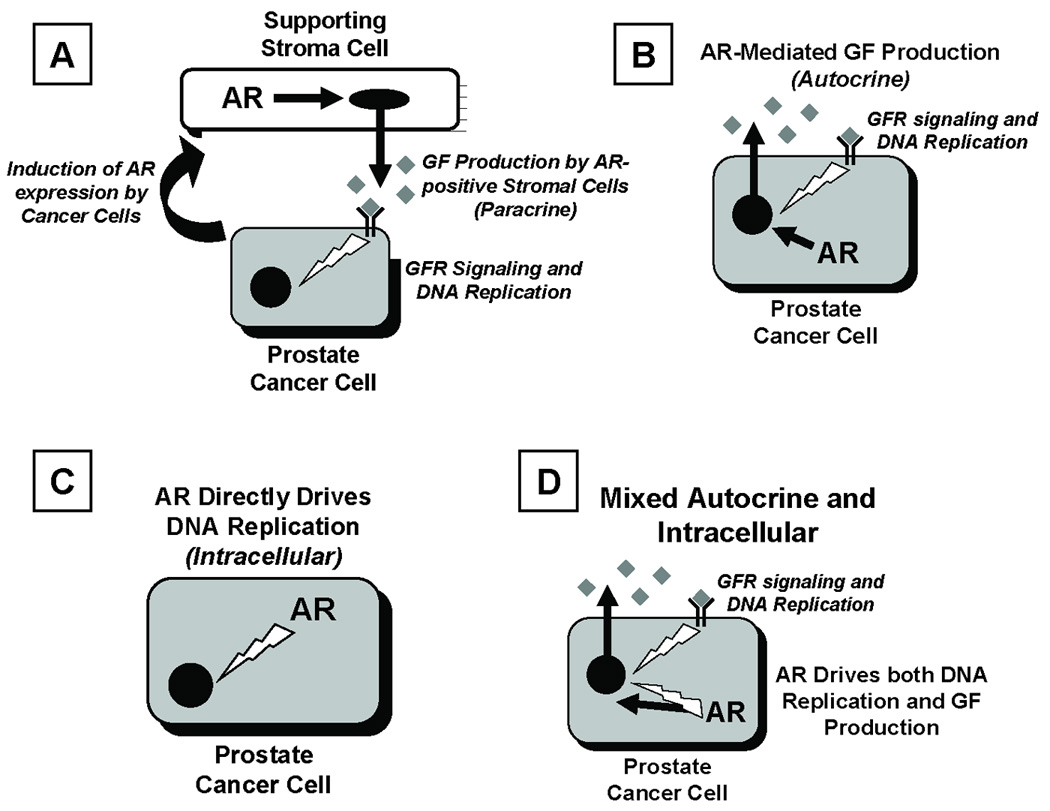
(A) Paracrine signaling whereby prostate CICs induce their surrounding stroma to express AR and consequently secrete AR-dependent growth factors (GFs, collectively termed “andromedins”). As a results, stromally-derived GFs binding to cognate GF receptors (GFRs) on prostate cancer cells, signaling for DNA replication and cell division.
(B) Autocrine signaling whereby AR drives the production of andromedin GFs, which bind to cognate GFRs on surrounding prostate CICs, thereby indirectly signaling DNA replication and cell division.
(C) Intracellular signaling whereby AR signaling directly mediates DNA replication and cell division in CICs.
(D) Combination of Autocrine signaling plus Intracellular signaling in prostate CICs.
MATERIALS AND METHODS
Materials and Cell Lines
The synthetic androgen R1881 was purchased from Perkin Elmer (Boston MA). The AR antagonist, Casodex (Bicalutamide) was purchased from LKT laboratories (St Paul, MN). All other chemicals were purchased from JT Baker (Phillipsburg NJ) or Sigma-Aldrich (St. Louis MO). LAPC-4, CWR22Rv1, and LNCaP human prostate lines were obtained and grown as previously described (8). LNCaP and CWR22Rv1cells were grown in RPMI-1640 media containing 10% fetal bovine serum (FBS). LAPC-4 were grown in Iscove’s media with 10 % FBS and 1nM R1881. All cells were routinely screened for the absence of mycoplasma contamination. Sealed silastic tubing packed with Testosterone was prepared and implanted subcutaneous in the flank of castrated male mice to restore and maintain serum testosterone as described previously (18).
In Vitro Growth Assays
Cell growth was measured by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay from Promega Corp. (Madison WI)) as previously described (10).
In Vivo Growth Assays
All animal studies were performed according to protocols approved by the Johns Hopkins Animal Care and Use Committee. In vivo growth assays were performed as previously described (18,19). For the in vivo studies the following five androgen sensitive human xenografts were used, each of which was derived from an independent patient: PC-82, CWR-22, LAPC-4, LNCaP, and MDA-PCA-2b. The history and characteristics of these xenografts have been described previously (8,18–21). Tfm-nude AR null mice were breed as described previously (22). Injection of CWR22rv1 cells into castrate nude mouse prostates was conducted by injecting one million cells in 20 µL HBSS into the prostates of male nude mice which had been castrated 11 days prior. Seven days later, the mouse prostates were removed, formalin fixed, and paraffin embedded for sectioning and staining.
Western Blotting
Western blotting was performed as previously described (23). Whole-cell lysates collected from 100,000 cells were used per lane. Antibodies used were the anti-Beta Actin (Cell Signaling; Beverly, MA); anti EGF receptor (#2232, Cell Signaling); anti-IGF-type 1 receptor (Cell Signaling): anti-FGFR1 (Sigma); and anti-FGFR2IIIb (Sigma). All secondary horseradish peroxidase-conjugated antibodies and chemiluminescent detection reagents (ECL) were purchased from Amersham Biosciences (Piscataway NJ).
Immunocytochemical Detection
All prostate specimens used for immunohistochemical analysis were samples selected from the surgical pathology files at the Johns Hopkins Department of Pathology with Institutional Review Board approval and are part of The Johns Hopkins Brady Urological Research Institute Prostate Specimen Repository. Tissue microarrays containing normal prostate and pelvic lymph node metastases (n=47), and distant soft tissue (n=5), and bone metastases (n=2) from non-androgen deprived patients were created as described previously (24). These were stained for detection of AR protein expression as described previously (24). Staining for Ki67 and ΔNp63 were conducted as described previously (8,9,25).
Statistics
All of the values are presented as means ± SE. Statistical analysis was performed by a one-way ANOVA with the Newman-Keuls test for multiple comparisons.
RESULTS
Prostate CIC Growth in Vivo Does Not Require Stromal AR Expression or Stromal Andromedins but Does Require Cell-Autonomous AR Signaling
Factors secreted by normal prostate epithelium stimulate the supporting glandular stromal cells to express AR protein (2,3). This raises the question of whether prostate CICs induce their surrounding tumor stroma cells to express AR. Only if these tumor stromal cells express AR could binding of androgen to such AR within the tumor stromal cells induce their paracrine secretion of andromedins, thereby stimulating prostate cancer cell growth. Two approaches were taken to test this paracrine possibility. The first approach is to evaluate expression of AR protein expression in the tumor stromal cells using immunohistochemical staining within clinical prostate cancer specimens. To do this, tissue microarrays containing 127 prostate cancer metastases from 54 independent non-androgen deprived patients were immunohistochemically stained for AR (Figure 2). As a control for these studies, AR immunohistochemical staining was performed on normal human prostate. In areas of normal prostate from non-androgen deprived males, a subset of stromal cells (denoted by arrows in Figure 1A) and > 90% of the secretory-luminal epithelial cells express AR protein in their nuclei (Figure 1A). In contrast, analysis of metastatic sites of prostate cancer from non-androgen ablated patients demonstrated that <0.1 % of stromal cells express detectable levels of AR protein whereas >60% of prostate cancer cells express nuclear AR (Figure 1B–C, Table 1). These tissue data document that human prostate cancer cells do not induce their supporting tumor stroma to express AR protein and consequently androgen can not stimulate these AR negative tumor stromal cells to produce andromedins. These results are consistent with previous reports demonstrating a decrease in AR protein expression in the stroma surrounding primary prostate cancers correlates with poor patient survival (26–28).
Figure 2. Prostate Cancer Cells are surrounded by AR-Negative Stromal Cells.

(A) Upper Left Panel: Immunostaining showing nuclear AR protein expression in both the stroma and epithelium of normal prostate tissue. Arrows indicated AR-positive nuclei in the stroma. Upper Right and Lower Left Panels: No detectable AR staining in the stroma surrounding AR-positive metastases from soft tissue. Lower Right Panel: No detectable AR staining in the stroma surrounding AR-positive metastases from bone.
(B) Samples analyzed on the Tissue Microarray (TMA) for AR expression, documenting that the surrounding stroma of prostate cancer metastases do not express AR.
Table 1.
Growth of prostate cancer cell lines in Wild-Type or AR-Null male nude mice which were either hormonally intact or castrated.
| Human Cancer Line | AR Status of Cancer |
AR Status of Host Nude mouse |
Tumor Volume (mm3) 8-week Post-Inoculation in: | |
|---|---|---|---|---|
| Intact Male Hosta | Castrated Male Hosta | |||
| Pc-82 Primary CA | Wild Type AR | Wild Type | 388 ± 65 | <10 |
| AR Null | 315 ± 52 | <10 | ||
| CWR-22 Primary CA | Mutated (H874Y) | Wild Type | 213 ± 24 | <10 |
| AR | AR Null | 198 ± 30 | <10 | |
| LNCaP Lymph NOde | Mutated (H877A) | Wild Type | 524 ± 68 | <10 |
| Metastasis | AR | AR Null | 590 ± 73 | <10 |
| MDA-PCA 2B Bone | Double Mutated | Wild Type | 600 ± 97 | <10 |
| Metastasis | (L701H/T877A) AR | AR Null | 654 ± 83 | <10 |
| LAPC-4 Lymph Node | Wild Type A | Wild Type | 410 ± 38 | 100 ± 25 |
| Metastasis | AR Null | 450 ± 54 | 87 ± 12 | |
N=8 per grouping
In addition to this correlative approach, direct experimental validation was undertaken to confirm that AR signaling within tumor stromal cells is not required for optimal growth of androgen responsive human prostate cancer cells. To do this, a series of five androgen responsive human prostate cancer xenografts, each derived from a different patient expressing either wild type or mutated AR, were inoculated into intact mice of both the AR wild-type or AR-null genotype on the nude background. These AR-null male nude mice are produced by crossing nude mice to mice carrying the Tfm early stop codon AR mutation such that these Tfm-nude mice can not express any level of AR protein in any host tissue (22). All 5 of the androgen responsive prostate cancer cell lines tested grow equally well in both wild-type and AR-null intact male nude mice demonstrating that AR signaling by host stromal cells is not required for the optimal growth of human prostate cancer cells (Table 1).
The PC-82 and CWR22 xenografts were established from surgical material from localized prostate cancer in patients that were hormonally naïve (21,22). If the CICs in these cancer lines are derived from AR non-expressing transformed prostate adult stem cells then the growth of these CICs should occur even in a castrated host. In contrast, if the CICs are derived from AR-expressing ICs which gained stem like self-renewal ability, then cancer growth could be dependent upon AR signaling. To resolve between these two possibilities, PC-82 and CWR22 were inoculated into both wild-type and AR-null male nude mice that were castrated at the time of implantation. In contrast to the situation for intact male hosts, no tumors developed in castrated wild-type or AR null mice for either of lines by 8-weeks (Table 1). Even at one year post inoculation, no tumors developed. At one year post inoculation, these castrated animals were given androgen replacement via silastic implants containing testosterone which restored serum androgen from <0.1 ng/ml to >1–2 ng/ml (18,22). Even with such androgen replacement, no tumors developed over the next six months. These results document that growth of CICs from the PC-82 and CWR22 localized prostate cancers obtained from hormonally naive patients are dependent upon cell autonomous AR signaling consistent with their being derived from transformed ICs.
In contrast to PC-82 and CWR22, LNCaP, MDA-PCA 2B, and LAPC-4 were established from metastatic sites from patients who had failed androgen ablation. While these 3 metastatic lines are castration resistant, they are not independent of cell autonomous AR signaling. This is documented by fact that the growth of all three lines is the same in intact male wild type or AR null mice but much lower in castrated animals (Table 1). In fact only the LAPC-4 line produces detectably growing tumors in all castrated hosts by 8 weeks while such detectable tumors did eventually develop in ~90% of animals by 90–120 days post inoculation with the LNCaP and MDA-PCA 2B cells.
Androgen-Induced Growth Signaling in Prostate CICs Requires Both Autocrine and Intracellular Pathways and Synergizes With Endocrine Factors Supplied By Serum
These combined animal results document that in vivo growth of prostate CICs is characteristically stimulated by cell-autonomous androgen-induced AR-dependent signaling. A series of human prostate cancer cell lines which express either wild type AR (i.e., LAPC-4) or ligand binding domain mutated AR (i.e., LNCaP and CWR22Rv1) were used to clarify the nature of these cell-autonomous AR growth signaling. These lines were chosen because each responds to androgen in a cell autonomous manner in vivo (Table 1), their growth in vitro is dependent upon AR protein signaling (29–32), and they each contain CICs (9,33). The media for these cells are routinely supplemented with 10% Fetal Calf Serum (FCS) to provide additional nutrients like essential fatty acids and cholesterol and endocrine factors like androgen and tri-iodothyronine (T3) plus serum growth factors like insulin and EGF and cell attachment proteins like fetuin (34). To evaluate the growth response of CICs to androgen alone versus in combination with the serum growth factors, these cells were switched from media containing 10% FCS to serum-free media. When this switch to serum-free media is performed in combination with standard tissue culture flasks, prostate cancer cells become loosely attached to the flask, as documented by their acquiring a dendritic morphology, and they begin dying within ~ 48 hours. In contrast, if cells are grown on poly-D-lysine coated tissue culture flasks and then switched to serum-free media, the cells remain attached, do not undergo morphological changes, and are viable for more than two weeks. By one week in serum-free/poly-D-lysine coated culture, the viable cells are not growing as determined by MTT assays (Figure 3A), flow cytometry, or by c-Myc protein expression as determined by Western blots (data not shown). When an AR saturating dose (i.e., 1nM) of the synthetic androgen R1881 is added to the serum-free media, a growth response is induced, however it is only ~20% of the optimal growth response induced by adding media containing 10% FBS which simultaneously supplies both androgen and serum growth factors (Figure 3A). The limited growth induced by androgen in serum-free media is only ~ half that produced by adding serum containing media in combination with 10µM of the direct acting AR ligand antagonist casodex to neutralize the androgenic stimulation supplied by FCS (Figure 3A and B). Similar results were obtained with LAPC-4 and CWR22Rv1 cells documenting that growth stimulation of prostate CICs is characteristically synergized by the level of androgen and endocrine serum growth factors simultaneously supplied by 10% FBS. This serum dependent growth synergy is not unexpected since prostate cancers characteristically require insulin, T3, EGF, and fetuin and nutritional factors like essential fatty acids and cholesterol for their optimal growth which are supplied by the addition of 10% FCS to the media (34).
Figure 3. Androgen-Induced AR Signaling Involves Autocrine Growth Factors and their Receptors.
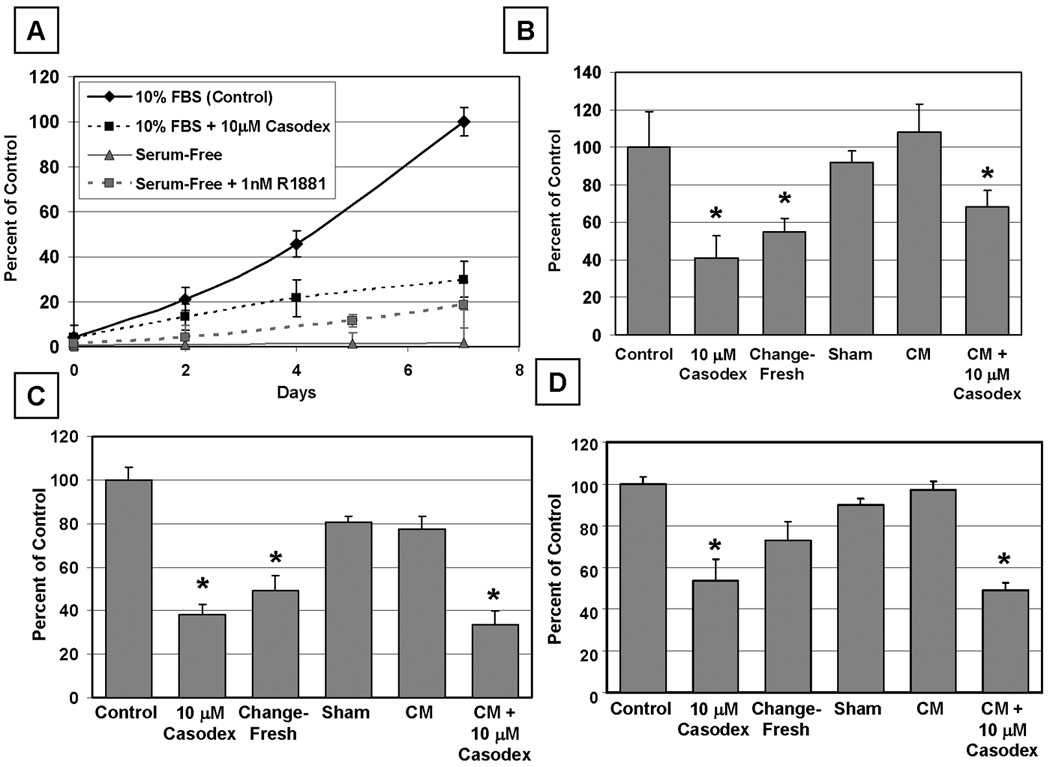
(A) Both serum and AR activity are required for prostate cancer cell growth. Growth of LNCaP cells in vitro in the presence of 10 µM of the anti-androgen casodex or in the absence of serum growth factors. 1.0 nM of the synthetic androgen R1881 is insufficient to restore the growth of serum-deprived LNCaP cells.
(B–D) Conditioned media (CM) from exponentially growing cells was tested for its ability to stimulate growth of CICs in casodex-inhibited cultures. LNCaP (B), LAPC-4 (C), or CWR22Rv1 (D) cells were left untreated (Control), growth-inhibited with 10 µM of the anti-androgen casodex (10 µM Casodex), changed with fresh media (Change-Fresh), the media was re-administered (Sham), or changed with Conditioned Media (CM) for seven days. Finally, cells were growth inhibited with casodex and the ability of conditioned media from exponentially growing cultures to stimulate growth was tested (CM + 10 µM Casodex). Such conditioned media only partially stimulated growth of the cells inhibited by casodex.
Such synergistic growth also involves androgen stimulation of secretion of autocrine growth factors which need to be simultaneous present with the endocrine growth factors provided by FCS for optimal growth of prostate cancer cells. Such a requirement for autocrine factors is documented by the following results. Fresh serum containing media was replaced daily to limit the concentration of secreted autocrine growth factors in the media and the growth was compared to that in cultures without daily media change to maintain the highest concentration of autocrine factors. The growth of LNCaP (Figure 3A and B), LAPC-4 (Figure 3C), and CWR22Rv1 (Figure 3D) CICs is inhibited (p<0.05) when the media is change daily. This growth inhibition is due to the lack of growth factor conditioning and not mechanical stress since daily removal and immediate replacement of this conditioned media does not inhibit the growth (Figure 3B–D). This requirement for a critical level of autocrine growth factors as well as serum growth factors is also documented by the observation that daily removal of serum-containing media and replacement with serum-containing media pre-conditioned for four days by exponentially growing cells fully maintains optimal growth (Figure 3B–D). The fact that addition of 10 µM casodex produces a greater degree of growth inhibition than daily media replacement (Figure 3B–D), however, suggests that androgen stimulation of prostate CICs includes additional intracellular signaling besides those induced by autocrine and serum derived endocrine growth factors.
If autocrine plus serum-derived endocrine growth factor signaling are not the only requirements for optimal prostate CIC growth, then pre-conditioned media containing autocrine growth factors secreted by optimally growing prostate cancer cells should not maximally stimulate growth of these cells if casodex is subsequently added to conditioned media. To evaluate this possibility, LNCaP and LAPC-4 cells were exposed to 10 µM casodex in the simultaneous presence of conditioned media containing both serum-derived endocrine growth factors and autocrine growth factors secreted over a four day period by exponentially growing cells not exposed to casodex. The results of these studies demonstrated that serum-containing CM from growing cells in the co-presence of casodex only partially rescues the growth inhibition of either LNCaP (Figure 3B) or LAPC-4 cells (Figure 3C), and has little effect on CWR22Rv1 cells (Figure 3D). These data document that androgen-stimulated AR signaling within prostate cancer cells activates additional intracellular functions needed for full growth stimulation in addition to the production of autocrine growth factors.
An additional intracellular function needed for full androgen-induced AR growth signaling could involve stimulating the expression of cell surface receptors needed for response to the autocrine and/or serum derived endocrine growth factors on prostate CICs. To test this, the levels of EGFR, IGF-1R, FGFR1 and FGFR2IIIb receptor proteins were determined in prostate cancer cells grown for 2 days in 10% FBS containing media vs. 10% FBS containing media with 10 µM casodex vs. serum containing CM from growing cells to which 10µM casodex is subsequently added. These results documented that casodex-induced inhibition of AR signaling decreases EGFR protein level by >3 fold and IGF-1R protein levels by >20 fold in LNCaP cells with no effect on the expression of either FGFR1 or FGR2IIIb (Figure 4). Similar results were obtained with LAPC-4 and CWR22Rv1 cells demonstrating that enhanced expression of plasma membrane growth factor receptors is a characteristic response in AR expressing human prostate CICs. The addition of serum-containing CM to which casodex is subsequently added resulted in the nearly full AR sensitive expression of the EGF receptor while not fully maintaining the expression of the IGF receptor (Figure 4). This partial response is consistent with the inability of the combination of CM + 10 µM casodex to induce optimal growth of any of the prostate CICs (Figure 3B–D). These results implicate IGF signaling as an important component of AR signaling cascade.
Figure 4. Level of Various Cell Surface Receptors on LNCaP Cells Exposed to Casodex Alone or in Combination with Conditioned Media.

Exposure of LNCaP cells for 4 days to 10 µM casodex decreases the level of EGFR and IGFR, but not FGFRI or FGFR2IIIb. The addition of CM from untreated cells to casodex treated cells inhibits such decreases in receptor expression.
Autocrine Growth Factors Produced by Prostate CICs are Different from Andromedins Produced by Normal Prostate Stromal Cells
The in vitro results demonstrate that AR stimulated autocrine growth factor signaling pathways are necessary but not sufficient for the optimal growth of prostate cancer cells. This raises the issue of whether the autocrine growth factors secreted by prostate CICs are identical to the andromedins secreted by normal prostate stromal cells which stimulate the proliferation of normal prostatic epithelial cells. If they are the same then prostate cancer cells inoculated orthotopically into the regressed prostate of previously castrated nude mice should stimulate proliferation of the mouse prostatic epithelial cells. To test this possibility, the CWR22Rv1 human prostate cancer line was used because its growth requires expression of AR protein (29) and while its growth is enhanced in intact male nude mice, it does grow in a castrated male mouse, albeit at a slower rate (19). Therefore, nude male mice were castrated, two weeks allowed for the prostate to regression, and then CWR22Rv1 cells were inoculated orthotopically into the regressed ventral prostate of male nude mice. Inoculation of CWR22Rv1 cells into the castrated mouse prostate produced growing cancers which within 5 days invade and surround regressed mouse prostate glandular acini (Figure 5, upper left panel). AR is expressed in the nuclei of the majority of the human CWR22Rv1 cancer cells even though these cancer cells are growing in a castrated male host (Figure 5, lower right panel). These CWR22Rv1 prostate CICs are producing biologically relevant levels of autocrine growth factor as documented by the fact that they are proliferating as detected by their nuclear expression of the cell proliferation marker, Ki67 (Figure 5, upper and lower left panels). These growing prostate cancer cells surround normal prostatic acini (Figure 5) which are composed of a well-developed stroma of smooth muscle cells, fibroblasts and endothelial cells, and epithelium composed of basal layer of ΔNp63 positive epithelial cells upon which are scattered ΔNp63-negative cuboidal cells which produce a small but patent lumen (Figure 5, upper right panel). These cuboidal mouse prostate epithelial cells, but not the ΔNp63 positive basal prostate cells, express AR protein in their cytoplasm but not their cell nuclei (Figure 5, lower right panel). Importantly, there is no proliferation in the regressed normal mouse acini as documented by a complete lack of Ki67 nuclear expression in these cells (Figure 5, upper and lower left panels) even though they are surrounded by CWR22Rv1 prostate cancer cells which are secreting autocrine growth factors at levels sufficient for stimulation of the proliferation of the cancer cells. This lack of a proliferative response occurs even though these normal mouse epithelial cells express the receptors for stromally derived andromedins. This is documented by the fact, as we have previously reported (35), that at little as 2–3 day of exogenous androgen replacement induces proliferation of these regressed normal prostate epithelial cells in response to the andromedins produced by the androgen stimulated normal prostate stromal cell. These results demonstrate that the autocrine growth factors produced by and stimulating prostate CICs are not identical to the andromedins produced by normal prostate stromal cells.
Figure 5. CWR22Rv1 Prostate Cancer Cells Do Not Secrete Factors Which Stimulate the Proliferation of Normal Mouse Prostate Cells.

Male nude mice which had been castrated 11 days previously were injected with CWR22Rv1 cells into the atrophied ventral prostate. The growing prostate tumor did not produce secreted factors to induce the regeneration of quiescent mouse glands. Upper Left Panel: castrate mouse prostate containing growing CWR22Rv1 tumors (10x). Arrows denote mouse prostatic ducts, and the box encompasses the region of interest (20x). Lower Left Panel: no Ki67 expression is observed in the mouse gland while the surrounding prostate tumor is proliferating. Upper Right Panel: expression of the basal cell marker ΔNp63 shows mouse basal cells within the gland. Lower Right Panel: diffuse cytoplasmic AR staining the mouse gland with strong nuclear staining in the growing CWR22Rv1 tumor. Such data document that CWR22Rv1 cells do not secrete any growth factors capable of supporting the growth of normal mouse prostate acini.
DISCUSSION
The present study documents that AR promotes malignant growth of prostate CICs via cell autonomous signaling pathways. This conclusion is based upon tissue microarray analysis and human prostate cancer xenograft studies in AR null nude mice which document that this cancer cell autonomous signaling is not associated with AR expression by the cancer associated stromal cells and does not require these stromal cells to secrete AR dependent growth factors. This is in contrast to the situation in the normal prostate where epithelial turnover is regulated via reciprocal paracrine interactions between prostate stromal and epithelial cells. Under normal conditions, AR signaling within stromal cells induces their secretion of andromedin growth factors while within normal prostate epithelial cells, AR functions as a growth suppressor (2,10). These results emphasize that during prostate carcinogenesis, AR acquires gain of function oncogenic ability to stimulate malignant growth. Using a series of human prostate cancer cell lines, this cell autonomous ability of prostate cancer cells to stimulate their own malignant growth was demonstrated to require novel AR signaling which enhances both the secretion of autocrine growth factors and the expression of their cognate receptors. In additional orthotopic xenograft studies, the autocrine growth factors secreted by prostate CICs were found not to be identical to the andromedins secreted by the normal prostate stromal cells. This is consistent with that fact that during prostate carcinogenesis, there is a switch in the splice variant of the Fibroblast Growth Factor receptors (FGFR) expressed by malignant prostate cells (36). Normal prostate epithelial cells express the FGFR2IIIb isoform which is the primary receptor for andromedins FGF7 (i.e., KGF) and FGF10 (36). In contrast, prostate cancer cells express the FGFR1 and FGFR2IIIc isoforms which are primary receptors for FGF1 and 2 which are not andromedins (36).
Such gain of autocrine signaling and FGFR isotype switching are not the only characteristic changes in cell autonomous AR function by prostate CICs. Using an anti-androgen direct binding antagonist plus conditioned media approach, the present studies document that AR characteristically acquires oncogenic functions in prostate CICs involving intracellular pathway(s) in addition to autocrine signaling. Recently, we have identified that one of these additional intracellular functions in prostate cancer cells involves direct AR participation in “licensing” of DNA for replication (25,37,38). These studies have identified that a major point of androgen regulation in prostate CIC proliferation occurs in early G1 involving licensing of DNA for replication (37). Such intracellular AR signaling provides a novel therapeutic target and is the focus of our future studies.
Acknowledgements
We wish to acknowledge the expert assistance of Jessica Hicks for the AR and Ki67 immunostaining and the Brady Urological Institute Prostate Specimen Repository
Funding: NIH Grant R01DK52645 and the Maryland Stem Cell Research Fund MSCRFII-0428-00 has generously supported this research. Donald Vander Griend was supported by a Urology Training Grant, NIH T32DK07552 and is now supported by a DOD Post-Doctoral Training Award, PC060843. Jason D’Antonio is supported by a Urology Training Grant (NIH T32DK07552-21A1).
Abbreviations
- AR
Androgen Receptor
- CM
Conditioned media
- FBS
Fetal bovine serum
- Tfm
Testicular feminizing mutation
- TMA
Tissue Microarray
REFERENCES
- 1.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2(5):389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles' heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88(7):2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 3.Cunha GR, Cooke PS, Kurita T. Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol. 2004;67(5):417–434. doi: 10.1679/aohc.67.417. [DOI] [PubMed] [Google Scholar]
- 4.Le H, Arnold JT, McFann KK, Blackman MR. DHT and testosterone, but not DHEA or E2, differentially modulate IGF-I, IGFBP-2, and IGFBP-3 in human prostatic stromal cells. Am J Physiol Endocrinol Metab. 2006;290(5):E952–E960. doi: 10.1152/ajpendo.00451.2005. [DOI] [PubMed] [Google Scholar]
- 5.Lu W, Luo Y, Kan M, McKeehan WL. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J Biol Chem. 1999;274(18):12827–12834. doi: 10.1074/jbc.274.18.12827. [DOI] [PubMed] [Google Scholar]
- 6.Nakano K, Fukabori Y, Itoh N, Lu W, Kan M, McKeehan WL, Yamanaka H. Androgen-stimulated human prostate epithelial growth mediated by stromal-derived fibroblast growth factor-10. Endocr J. 1999;46(3):405–413. doi: 10.1507/endocrj.46.405. [DOI] [PubMed] [Google Scholar]
- 7.van Leenders G, Dijkman H, Hulsbergen-van de Kaa C, Ruiter D, Schalken J. Demonstration of intermediate cells during human prostate epithelial differentiation in situ and in vitro using triple-staining confocal scanning microscopy. Lab Invest. 2000;80(8):1251–1258. doi: 10.1038/labinvest.3780133. [DOI] [PubMed] [Google Scholar]
- 8.Litvinov IV, Vander Griend DJ, Xu Y, Antony L, Dalrymple SL, Isaacs JT. Low-Calcium Serum-Free Defined Medium Selects for Growth of Normal Prostatic Epithelial Stem Cells. Cancer Res. 2006;66(17):8598–8607. doi: 10.1158/0008-5472.CAN-06-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Griend DJ, Karthaus WK, Dalrymple S, Meeker AK, De Marzo AM, Isaacs JT. The Role of CD133 in Normal Human Prostate Stem Cells and Malignant Cancer Initiating Cells. Cancer Res. 2008;68(23):9703–9711. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litvinov IV, Antony L, Dalrymple SL, Becker R, Cheng L, Isaacs JT. PC3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006 doi: 10.1002/pros.20483. [DOI] [PubMed] [Google Scholar]
- 11.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 12.Gu G, Yuan J, Wills M, Kasper S. Prostate Cancer Cells with Stem Cell Characteristics Reconstitute the Original Human Tumor In vivo. Cancer Res. 2007;67(10):4807–4815. doi: 10.1158/0008-5472.CAN-06-4608. [DOI] [PubMed] [Google Scholar]
- 13.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical Organization of Prostate Cancer Cells in Xenograft Tumors: The CD44+{alpha}2{beta}1+ Cell Population Is Enriched in Tumor-Initiating Cells. Cancer Res. 2007;67(14):6796–6805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- 14.Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98(4):756–765. doi: 10.1038/sj.bjc.6604242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharifi N, Hurt EM, Farrar WL. Androgen receptor expression in prostate cancer stem cells: is there a conundrum? Cancer Chemother Pharmacol. 2008;62(5):921–923. doi: 10.1007/s00280-007-0659-5. [DOI] [PubMed] [Google Scholar]
- 16.Schalken JA, van Leenders G. Cellular and molecular biology of the prostate: stem cell biology. Urology. 2003;62(5 Suppl 1):11–20. doi: 10.1016/s0090-4295(03)00758-1. [DOI] [PubMed] [Google Scholar]
- 17.van Leenders G, van Balken B, Aalders T, Hulsbergen-van de Kaa C, Ruiter D, Schalken J. Intermediate cells in normal and malignant prostate epithelium express c-MET: implications for prostate cancer invasion. Prostate. 2002;51(2):98–107. doi: 10.1002/pros.10073. [DOI] [PubMed] [Google Scholar]
- 18.Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001;61(13):5038–5044. [PubMed] [Google Scholar]
- 19.Denmeade SR, Sokoll LJ, Dalrymple S, Rosen DM, Gady AM, Bruzek D, Ricklis RM, Isaacs JT. Dissociation between androgen responsiveness for malignant growth vs. expression of prostate specific differentiation markers PSA, hK2, and PSMA in human prostate cancer models. Prostate. 2003;54(4):249–257. doi: 10.1002/pros.10199. [DOI] [PubMed] [Google Scholar]
- 20.Dalrymple S, Antony L, Xu Y, Uzgare AR, Arnold JT, Savaugeot J, Sokoll LJ, De Marzo AM, Isaacs JT. Role of notch-1 and E-cadherin in the differential response to calcium in culturing normal versus malignant prostate cells. Cancer Res. 2005;65(20):9269–9279. doi: 10.1158/0008-5472.CAN-04-3989. [DOI] [PubMed] [Google Scholar]
- 21.Nagabhushan M, Miller CM, Pretlow TP, Giaconia JM, Edgehouse NL, Schwartz S, Kung HJ, de Vere White RW, Gumerlock PH, Resnick MI, Amini SB, Pretlow TG. CWR22: the first human prostate cancer xenograft with strongly androgen-dependent and relapsed strains both in vivo and in soft agar. Cancer Res. 1996;56(13):3042–3046. [PubMed] [Google Scholar]
- 22.Gao J, Isaacs JT. Development of an androgen receptor-null model for identifying the initiation site for androgen stimulation of proliferation and suppression of programmed (apoptotic) death of PC-82 human prostate cancer cells. Cancer Res. 1998;58(15):3299–3306. [PubMed] [Google Scholar]
- 23.Uzgare AR, Xu Y, Isaacs JT. In vitro culturing and characteristics of transit amplifying epithelial cells from human prostate tissue. J Cell Biochem. 2004;91(1):196–205. doi: 10.1002/jcb.10764. [DOI] [PubMed] [Google Scholar]
- 24.Faith DA, Isaacs WB, Morgan JD, Fedor HL, Hicks JL, Mangold LA, Walsh PC, Partin AW, Platz EA, Luo J, De Marzo AM. Trefoil factor 3 overexpression in prostatic carcinoma: prognostic importance using tissue microarrays. Prostate. 2004;61(3):215–227. doi: 10.1002/pros.20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litvinov IV, Vander Griend DJ, Antony L, Dalrymple S, De Marzo AM, Drake CG, Isaacs JT. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci U S A. 2006;103(41):15085–15090. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olapade-Olaopa EO, MacKay EH, Taub NA, Sandhu DP, Terry TR, Habib FK. Malignant transformation of human prostatic epithelium is associated with the loss of androgen receptor immunoreactivity in the surrounding stroma. Clin Cancer Res. 1999;5(3):569–576. [PubMed] [Google Scholar]
- 27.Wikstrom P, Ohlson N, Stattin P, Bergh A. Nuclear androgen receptors recur in the epithelial and stromal compartments of malignant and non-malignant human prostate tissue several months after castration therapy. Prostate. 2007;67(12):1277–1284. doi: 10.1002/pros.20569. [DOI] [PubMed] [Google Scholar]
- 28.Ricciardelli C, Choong CS, Buchanan G, Vivekanandan S, Neufing P, Stahl J, Marshall VR, Horsfall DJ, Tilley WD. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate. 2005;63(1):19–28. doi: 10.1002/pros.20154. [DOI] [PubMed] [Google Scholar]
- 29.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68(13):5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62(4):1008–1013. [PubMed] [Google Scholar]
- 31.Li TH, Zhao H, Peng Y, Beliakoff J, Brooks JD, Sun Z. A promoting role of androgen receptor in androgen-sensitive and -insensitive prostate cancer cells. Nucleic Acids Res. 2007;35(8):2767–2776. doi: 10.1093/nar/gkm198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, Pollak M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR) Cancer Res. 2001;61(16):6276–6280. [PubMed] [Google Scholar]
- 33.Pfeiffer MJ, Schalken JA. Stem Cell Characteristics in Prostate Cancer Cell Lines. Eur Urol. 2009 doi: 10.1016/j.eururo.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 34.Hedlund TE, Miller GJ. A serum-free defined medium capable of supporting growth of four established human prostatic carcinoma cell lines. Prostate. 1994;24(5):221–228. doi: 10.1002/pros.2990240502. [DOI] [PubMed] [Google Scholar]
- 35.Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O'Malley BW, Isaacs JT, Dahiya R, Cunha GR. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ. 2001;8(2):192–200. doi: 10.1038/sj.cdd.4400797. [DOI] [PubMed] [Google Scholar]
- 36.Acevedo VD, Ittmann M, Spencer DM. Paths of FGFR-driven tumorigenesis. Cell Cycle. 2009;8(4):580–588. doi: 10.4161/cc.8.4.7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Antonio J, Vander Griend D, Isaacs J. DNA licensing as a novel androgen receptor mediated therapeutic target for prostate cancer. Endocr Relat Cancer. 2009 doi: 10.1677/ERC-08-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle. 2007;6(6):647–651. doi: 10.4161/cc.6.6.4028. [DOI] [PubMed] [Google Scholar]