

Abstract
Prenylation of G protein gamma (γ) subunits is necessary for the membrane localization of heterotrimeric G proteins and for functional heterotrimeric G protein coupled receptor (GPCR) signaling. To evaluate GPCR signaling pathways during development, we injected zebrafish embryos with mRNAs encoding Gγ subunits mutated so that they can no longer be prenylated. Low-level expression of these prenylation-deficient Gγ subunits driven either ubiquitously or specifically in the primordial germ cells (PGCs) disrupts GPCR signaling and manifests as a PGC migration defect. This disruption results in a reduction of calcium accumulation in the protrusions of migrating PGCs and a failure of PGCs to directionally migrate. When co-expressed with a prenylation-deficient Gγ, 8 of the 17 wildtype Gγ isoforms individually confer the ability to restore calcium accumulation and directional migration. These results suggest that while the Gγ subunits possess the ability to interact with G Beta (β) proteins, only a subset of wildtype Gγ proteins are stable within PGCs and can interact with key signaling components necessary for PGC migration. This in vivo study highlights the functional redundancy of these signaling components and demonstrates that prenylation-deficient Gγ subunits are an effective tool to investigate the roles of GPCR signaling events during vertebrate development.
1. Introduction
Heterotrimeric G-Protein signaling is a versatile and broadly used mechanism in eukaryotes for the transduction of cellular signals such as those involved in the pathways controlling sight, neurotransmission and cell migration. Consisting of an alpha (α), beta (β) and gamma (γ) subunit, the heterotrimeric G-proteins are membrane-bound, cytosolically located signaling components that interact with and are activated by a diverse group of G protein coupled receptors (GPCRs). During heterotrimeric G-protein signal transduction, ligand binds a GPCR resulting in the activation of the Gα subunit to dissociate its bound GDP and bind the more highly concentrated, free cytosolic GTP (reviewed in [1]). The activated Gα subunit dissociates from the Gβγ dimer allowing both components to influence separate downstream effectors such as adenylyl cyclase [2-4], phospholipase Cβ [5, 6], phosphatidylinositol 3-kinase [7, 8], β adrenergic receptor kinase [9, 10], and ion channels [11, 12].
Heterotrimeric G proteins are capable of mediating a myriad of upstream and downstream signals, in part due to the large number of isoforms of the G protein α, β and γ subunits present in vertebrates. Humans and mice have 16 α, 5 β and 12 γ subunit isoforms [13] although not all combinations are formed. The constituents of the heterotrimeric G protein have been shown to differentially influence interactions between the heterotrimer and receptors [14, 15] as well as between the Gα subunit or Gβγ dimer and downstream effectors [16, 17]. The subcellular localization of the Gβγ dimer has also been hypothesized to increase the specificity of signal transduction [18]. Since prior work largely focused on cultured cells, it remains unclear as to what level of cross talk and redundancy exists among GPCRs, heterotrimers and their downstream effectors in vivo. The present study examines the role of Gγ subunit post-translational processing and GPCR mediated signaling in the complex environment of a live vertebrate.
Both Gα and Gγ subunits are post-translationally modified with lipids. The Gγ subunits are a family of proteins that have a carboxy terminal ‘CaaX’ motif (where C = cysteine, a = an aliphatic amino acid, and X = a variable amino acid) whose presence makes them substrates for the prenylation enzymes farnesyl transferase (FTase) or geranylgeranyl transferase I (GGTaseI). The amino acid makeup of the CaaX motif and, in particular, the identity of the ‘X’ largely determines the affinity of FTase versus GGTaseI for the CaaX motif-containing protein. Prenylation is the first step in a post-translation modification that involves adding either a 15-carbon farnesyl or 20-carbon geranylgeranyl group to the cysteine of the CaaX motif followed by proteolytic cleavage of the terminal ‘aaX’ amino acids and methylation of the carboxy-terminal prenyl cysteine. The process of prenylation results in the increased hydrophobicity necessary for these proteins to insert into membranes and interact with other hydrophobic proteins. Prenylation of Gγ subunits and palmitoylation of Gα subunits confer the heterotrimeric G protein with the ability to translocate to cell membranes [19, 20]. Prenylation of the Gγ subunit is also necessary for the Gβγ dimer to interact efficiently with a Gα subunit [21-23], receptors (Rhodopsin [24, 25]; A1 adenosine receptor [25]), and effectors [26-30] (Phosducin (pdc) [31]; PhospholipaseCβ (PLCβ and PLCβ2) [32]; adenylyl cyclase [33]).
While the functions of individual members of the large Gγ subunit family have been difficult to identify, some have been characterized through the creation of mouse gene knockouts (i.e. Gγ7 [34]; Gγ3 [35]). Quicker, alternative methods to determine the function of individual subunits have yielded inconsistent results. Although ribozymes targeted to individual Gγ subunits have shown the ability to assay in vitro function [36], their efficacy in vivo remains to be determined. Few zebrafish heterotrimeric G proteins have been identified [37-39]. The use of morpholinos (targeted antisense oligonucleotides) in zebrafish embryos to elucidate the biological function of specific G protein subunits is complicated by poor target efficacy, maternal protein contribution, off-target effects and/or functional redundancies between G protein subunits. In this study we show that knocking down many of the individual Gγ subunits is insufficient to disrupt primordial germ cell (PGC) migration, a developmental process known to rely on GPCR signaling in both insects and vertebrates [40-44]. Therefore, we evaluated an alternative method to determine the function of these important signaling proteins during zebrafish development. The tissue-specific dominant negative strategy reported here allows the study of G-protein signaling in the background of maternally provided proteins that would interfere with antisense-based methods.
In this study, we performed a comprehensive, comparative genomic analysis to identify the Gβ and Gγ subunit families in zebrafish. We investigated the capacity of 17 prenylation-deficient Gγ subunits to disrupt GPCR signaling by injecting embryos with mRNA and assaying PGC migration. PGCs, the cells that will later give rise to the gametes, arise at a site that is distinct from the future-developing gonad and therefore must migrate long distances during early embryogenesis. The pathways involved in this long-range migration exhibit parallels to several pathologies including chronic inflammation [45-47] and cancer metastasis [48-51]. PGCs are guided by a GPCR-mediated pathway that involves migration towards increasing concentrations of the attractant chemokine Sdf1a, discovered in the zebrafish to be dynamically expressed along the developing borders of the trunk mesoderm and the pronephros [42]. The GPCR that recognizes Sdf1a, Cxcr4b, is expressed within PGCs [41, 42]. Another pathway integral to the migration of PGCs involves the enzyme Geranylgeranyl transferase I [52, 53]. One way that these two pathways could work in concert is if the geranylgeranylation of Gγ subunits is necessary within the PGCs to localize and/or assemble the proper machinery for sdf1a signal transduction.
This study demonstrates that expressing prenylation-deficient Gγ subunits in zebrafish embryos is sufficient to interfere with GPCR signaling and disrupt PGC migration, resulting in a phenotype similar to that seen when Sdf1a signaling is disrupted. The finding that the majority of prenylation-deficient Gγ subunits disrupt PGC migration led us to test whether Gγ subunits mediate interactions with signaling components other than their obligate signaling partner, Gβ. Only a subset of co-administered wildtype Gγ subunits were able to restore proper migration in embryos expressing a prenylation-deficient Gγ subunit, suggesting that Gγ subunits can perform distinct and non-overlapping functions in vivo.
2. Materials and methods
2.1. Zebrafish strains and fish maintenance
Danio rerio of an AB background were outcrossed once to pet store zebrafish to restore hybrid vigor and inbred several generations for use as WT fish. Fish were raised and maintained as previously described [54, 55]. All zebrafish care and experimental procedures were carried out as specified in our Institutional Animal Care and Use Committee Protocol (#647A). Fish of the AB strain were used in the calcium labeling experiments.
2.2. RTPCR and cloning of the zebrafish G-protein β and γ subunits
Gβ and Gγ subunits were identified by performing tblastn searches of the human and mouse subunits against the current zebrafish assembly (Ensembl, Jun 2007, version 7). Reverse transcription polymerase chain reaction (RT-PCR) was performed on the Gβ and Gγ subunits using oligo-dT amplified, 4cell-6dpf cDNA and AMV Reverse transcriptase (NEB: M0277S) according to the manufacturer’s protocol. Primers for RT-PCR and for cloning of the zebrafish Gγ subunits into the pT3TS & nos1-3′UTR vectors are reported in supplementary information (Table S2). All RTPCR primers were designed to amplify a region that spanned an intron. The pT3TS vector contains the Xenopus ß-globin 5′ & 3′UTRs that result in a highly stable mRNA which is ubiquitously translated upon injection into zebrafish embryos [56]. The nos1-3′UTR vector contains the 3′UTR of the nanos1 gene which directs the transcribed mRNA to be translated in all cells at very early stages followed by mRNA degradation in somatic cells and stabilization in PGCs [57]. The Gβ subunits were cloned into the GFPnos1-3′UTR vector by disrupting the stop site of mGFP5 with the Gβ coding sequence.
2.3. Injection of Gγ-SaaX and Gγ-WT mRNA
Capped mRNA was in vitro transcribed from the pT3TS or nos1-3′UTR vectors using the T3 or SP6 mMessage mMachine kits, respectively (Ambion: AM1348, AM1340). Gγ-SaaX or Gγ-WT mRNAs were injected into embryos at the 1-2 cell stage. To test the ability of the wildtype mRNAs to reverse the phenotype induced by Gγ-SaaX, either gng2-SaaX mRNA was injected at 25pg/embryo or gng2-SaaX(nos) mRNA at 50pg/embryo, after being mixed with GFP-nos1-3′UTR mRNA [57] at a final concentration of 200pg/embryo. The level of fluorescence of the GFP-nos1-3′UTR mRNA serves as an internal control for the amount of the Gγ-SaaX mRNA administered. The injected embryos were split into two groups; one group received an injection of a Gγ-WT mRNA and the other group received no injection or an injection with an equal volume of control (phenol red; 0.2% final concentration). The ectopic PGC migration score for each larva was determined by counting the number of PGCs at 24-48hpf that were outside of the WT location at the anterior of the yolk extension (0 = 0-5% ectopic; 1 = 6-19% ectopic; 2 = 20-39% ectopic; 3 = 40-59% ectopic; 4 = 60-79% ectopic; 5 = 80-100% ectopic). Only larvae with a minimum of 25 fluorescent PGCs were scored.)
2.4. In vitro translation
mRNAs were in vitro translated using the Retic Lysate IVT kit (Ambion: AM1200) in conjunction with 35S labeled methionine and visualized using SDS-PAGE.
2.5. Microscopy
GFP-expressing larvae were imaged under brightfield and a 488nm filter on a Nikon SMZ1500 or Zeiss StereoLUMAR microscope and captured by Zeiss AxioVision software.
2.6. Fluoresence colocalization
Colocalization studies were performed using a Leica TCSSP2-DMRXE confocal at 12-18hpf. Raw images of PGCs were processed using Metamorph software to determine the fluorescence density of GFP in regions corresponding to the plasma membrane and the cytosol. The ratio of cytosolic fluorescence density to membrane fluorescence density was calculated and the means were analyzed for statistical differences (ANOVA; Gabriel post hoc test).
2.7. Time-lapse movies
Embryos were injected with 180pg mRNA encoding GFP-nos3′UTR or GFP-CVLL-nos3′UTR, a modified version of mGFP5 that has a geranylgeranyl-specific CaaX box (-CVLL) added to its C-terminus, either alone or in conjunction with gng2-WT(nos) [100pg] or gng2-SaaX(nos) mRNA [50pg]. Embryos were mounted on coverslip-bottomed dishes in 0.7% agarose and imaged at 20X on a Leica DMIRE2 spinning disk confocal scope using Metamorph software. Images were montaged from 5-6 z-stacks taken every 8 minutes and 15 seconds from 10-30hpf. Migration of individual PGCs were tracked and measured for direction and velocity using Imaris software (Bitplane).
2.8. Measurement of calcium levels
Calcium levels in migrating PGCs were assayed in vivo by methods previously described [58].
2.9. In situ hybridization of zebrafish larvae
In situ hybridization was performed as described by Thisse et al., 2004 [59] with modifications for 2 colors according to Weidinger et al., 2002 [60].
2.10. Isolation of FLAG-tagged Gγ proteins
Embryos injected at the one-cell stage with 180pg of GFP-nos3′UTR mRNA and either 30pg of gng2-WT or gng2-SaaX mRNA were harvested at 24hpf, dechorionated, deyolked by shaking in deyolking buffer (55mM NaCl, 1.8mM KCl, 1.25mM NaHCO3) and washed in buffer (110mM NaCl, 3.5mM KCl, 2.7mM CaCl2, 10mM Tris/Cl pH 8.5). Following sonication in homogenization buffer (10mM Tris pH7.4, 250 mM Sucrose, 1 mM EDTA, 1% protease inhibitor), Cytosolic and membrane proteins were isolated as described in Mumby et al., 2002 [61] and subjected to immunoprecipitation using the FLAG-IP kit manufacturer’s protocol (Sigma-Aldrich: FLAGIPT1-1KT). Proteins were separated on a gradient 10-20% SDS-PAGE gel, transferred to a PVDF membrane and subjected to Western blotting using rabbit anti-FLAG Antibody (F7425, Sigma) and ECL-HRP-linked donkey anti-rabbit secondary (Amersham: NA934).
2.11. Morpholino injections
Morpholino antisense oligonucleotides designed to target the start sites of eight gamma subunits are as follows: Gγ2 (5′-CTGTGTTGTTGGTGGCCATGAGGCT-3′), Gγ3 (5′-GTTCACAGGGGTGTCTCCTTTCATC-3′), Gγ4 (5′-GTCCTTCATGATGAGTCCTGCACAC-3′), Gγ7 (5′-TGTTATTAGTCGTGGACATTCTCTG-3′), Gγ8 (5′-GACATCCTGTTCGTTTGTAGAGACC-3′), Gγ12a (5′-TTGAGCCCGACATTTTAGAGGACAT-3′), Gγ12b (5′-GAGCTTTGCATCTTGGACAACATTT-3′), and Gγ13 (5′-GGCAAGTCCATCTCGTCCATGACTG-3′). Each morpholino was injected with GFPnos1-3′UTR mRNA at the one-cell stage and titrated from 0.1 to 5ng per embryo.
3. Results
3.1. Zebrafish have at least 17 G Protein γ Subunits
We identified the zebrafish genes encoding G protein γ subunits. gngt1, gng3 and gng2 were previously characterized [37-39]. Identification and naming of the zebrafish Gγ subunits was based on homology to their human orthologs in conjunction with phylogenetic (Figure S1) and syntenic analysis (Figure S2). In contrast to S. cerevisiae that has only one Gγ subunit [62], or D. melanogaster that has two Gγ subunits [63, 64], the zebrafish has at least 17 Gγ subunits (Table 1 and Figure S1). These 17 Gγ subunits represent the vast majority of the known orthologs in the human and mouse [13, 65]. While we did not find a zebrafish gng11 ortholog, we identified two Gγ subunit paralogs (gngt2b, gng12b) that likely reflect gene duplications that occurred after the separation of the teleost and mammalian lineages. Furthermore, we identified 4 Gγ subunits with no clear mammalian ortholog (gng14, gng15, gng16, gng17), suggesting that these were either lost during mammalian evolution or evolved in fish after divergence from the common ancestor.
Table 1.
Zebrafish G protein γ subunit genes
| Subunit | Accession # | Chromosome | CaaX Motif |
Homology to Human Gamma Subunits |
|
|---|---|---|---|---|---|
| Nucleotide | Protein | % Identity / % Similarity | |||
| gngt1† | NM_199967 | NP_956261 | 19 | *CVIC | 66.7 / 81.3 |
| gngt2a | BC059612 | Q6PBR4 | 3 | *CIIT | 70.0 / 88.6 |
| gngt2b | none | none | 12 | *CIIT | 1 54.3 / 78.6 |
| gng2‡ | DQ109815 | Q15KE4 | 13 | CAIL | 94.4 / 98.6 |
| gng3§ | NM_131841 | NP_571916 | 21 | CALL | 93.4 / 98.7 |
| gng4 | XM_001335940 | XP_001335976 | 13 | CTIL | 88.2 / 100 |
| gng5 | - | NP_999886 | 2 | CSFL | 84.1 / 92.8 |
| gng7 | - | NP_001002397 | 2 | CIIL | 94.2 / 98.6 |
| gng8 | zgc:110316 | Q503Q6 | 21 | CTVL | 80.3 / 97.2 |
| gng10 | XM_001923181 | XP_001923216 | 5 | CTLL | 76.8 / 92.8 |
| gng12a | NM_213312 | NP_998477 | 2 | CTIL | 76.7 / 89.0 |
| gng12b | none | none | 6 | CTIL | 78.1 / 87.7 |
| gng13 | XM_001333427 | XP_001333463 | 3 | CVLL | 75.0 / 91.2 |
| gng14 | XM_001338349 | XP_001338385 | 1 | CVIL | 2 55.9 / 73.5 |
| gng15 | XM_681703 | XP_686795 | 6 | CVLL | 3 54.9 / 78.1 |
| gng16 | none | none | 1 | CGLL | 4 56.2 / 69.9 |
| gng17 | none | none | 3 | CVLL | 4 49.3 / 70.4 |
The identified zebrafish Gγ subunit genes, their associated sequences, chromosomal location and CaaX motif. The zebrafish Gγ subunits that lack an obvious human ortholog are compared to the human subunit that has the highest homology
gngt2b was compared to GNGT2
gng14 was compared to GNG13
gng15 was compared to GNG7
gng16 was compared to GNG5
gng17 was compared to GNG10.
= CaaX motifs that are preferred by farnesyl transferase over geranylgeranyl transferase.
Previously identified zebrafish subunits: Chen et al., 2007
3.2. Gγ-SaaX proteins are localized to the cytosol in vivo
Mutation of the cysteine of the prenylation-specific ‘CaaX’ motif into a serine (CaaX->SaaX) abolishes a protein’s ability to be prenylated and therefore disrupts membrane localization in vitro [19, 20, 31, 66-70]. To determine the effect of introducing prenylation-deficient Gγ subunits during zebrafish development, the 17 zebrafish Gγ subunits were separately cloned in the wildtype (WT) and mutant form (SaaX) into the pT3TS vector. mRNA in vitro transcribed from this construct is stable and ubiquitously translated when injected into zebrafish embryos [56]. Western blot analysis of proteins extracted from 24 hours post fertilization (hpf) larvae injected at the one cell stage with mRNA encoding N-terminally FLAG-tagged versions of Gγ2-WT or Gγ2-SaaX confirmed that the Gγ-SaaX proteins lose their membrane localization (Figure 1).
Fig. 1.

The membrane localization of Gβ and Gγ is disrupted when the CaaX motif of Gγ is mutated to SaaX. Embryos injected with mRNA encoding FLAG-tagged versions of the Gγ proteins were harvested at 24hpf, separated into cytosolic (C) and membrane (M) fractions and subjected to IP.
3.3. Gγ-SaaX mRNAs disrupt PGC Migration
To visualize the effects that introducing prenylation-deficient Gγ mRNAs have on a known GPCR mediated event during development, PGC migration [71], embryos were co-injected with GFPnos1-3′UTR mRNA (Figures 2A and 2B). Appending the 3′UTR of the nanos1 (nos) gene to mRNA results in the inhibition of translation and degradation of the adjoined mRNA in somatic cells and its stabilization in PGCs [57]. Expression of GFPnos1-3′UTR allows visualization of the migrating PGCs from 8hpf until well after 24hpf, the stage by which PGCs reach their final target at the anterior of the yolk extension, where the gonad will later form. Embryos injected with in vitro transcribed, capped gng2-SaaX mRNA exhibited a profound disruption of PGC migration (Figures 2C and 2D). Morphological analysis and whole mount in-situ hybridization of sdf1a expression showed that while PGCs mislocalize after the injection of 30pg gng2-SaaX mRNA, somatic structures are not disrupted, suggesting that at the doses used, the effect is specific to PGCs (Figure 2 and S3). The lack of colocalization of ectopic PGCs with regions expressing the secreted chemoattractant sdf1a suggests that PGCs in gng2-SaaX mRNA injected larvae are insensitive to this guidance cue. The rare PGCs that are ectopically localized in wildtype embryos often end up in the hindbrain, another site of high sdf1a expression (personal observation, Movie S1, and [42]). Injection of the remaining Gγ subunit mRNAs in their SaaX form indicated that 13 of the 17 Gγ-SaaX subunits have the ability to disrupt PGC migration (Figures 2G and 2H; Table 2 and Figures 3A and S4). Injection of the wildtype versions of the Gγ subunit mRNAs resulted in no obvious disruption of PGC migration (Figures 2E and 2F) with the exception of the farnesylated Gγ subunits, gngt1 & gngt2a, which not only caused PGC migration defects at low doses but also somatic defects at higher concentrations (Figure 3B).
Fig. 2.

PGC migration in embryos overexpressing WT and SaaX mutated Gγ subunits. Bright-field image of an embryo injected with GFP-nos1-3′UTR mRNA [150pg] (A) results in GFP fluorescence restricted to the PGCs located at the anterior of the yolk extension (arrow in B). Bright-field image of an embryo injected with a mix of gng2-SaaX mRNA [20pg] driven ubiquitously (ubiq.) by the Xenopus β-globin 5′UTR & 3′UTR and the PGC driven GFP-nos1-3′ UTR mRNA (C). Fluorescent image of (C) reveals PGC migration defects (D) that are not observed when embryos are injected with gng2-WT mRNA [400pg] (E & F). Bright-field image of an embryo expressing gng8-SaaX [40pg] reveals no obvious alteration in somatic development (G) but fluorescent microscopy of the same embryo indicates PGC migration is perturbed (H). All embryos shown were injected with mRNAs at the 1 – 4 cell stage and subjected to microscopy at 24hpf.
Table 2.
Summary of the Gγ subunits’ ability to disrupt PGC migration in their WT and/or SaaX forms.
| Gamma Subunit |
PGC Disruption by γ-WT (T3TS) |
PGC Disruption by γ-SaaX (T3TS)? |
|---|---|---|
| gng1 | + * | + |
| gngt2a | + * | + |
| gngt2b | − | − |
| gng2 | − | + |
| gng3 | − | + |
| gng4 | − | + |
| gng5 | − | + |
| gng7 | − | + |
| gng8 | − | + |
| gng10 | − | # |
| gng12a | − * | + |
| gng12b | − | + |
| gng13 | − | + |
| gng14 | − | − |
| gng15 | − | ## |
| gng16 | − | ## |
| gng17 | − * | ## |
somatic disruption at high concentrations [x > 400pg/embryo]
PGC disruption only at relatively higher concentrations [x > 300pg/embryo]
PGC disruption at high concentrations potentially secondary to somatic disruption; See Figure S4 for detailed results
Fig. 3.

Ability of the Gγ subunits to disrupt PGC migration when injected as mRNA in their SaaX (A) or wildtype forms (B). Wildtype PGC migration is reflected by GFP-nos3′UTR injection alone (A). Injections of the remainder of the Gγ SaaX mRNAs are summarized in Figure S4. Each concentration is a summary of 2 – 5 experiments with a minimum of 20 embryos scored. (Ectopic PGC migration score is represented as Mean +/− S.D.)
In vivo time-lapse movies of PGCs migrating in zebrafish injected with prenylation-deficient Gγ mRNA indicated that despite the PGCs’ inability to reach the anterior of the yolk extension, PGCs maintain the ability to migrate actively and extend cellular protrusions. While the PGCs do not show a change in their migration speed (mean PGC speed: Gγ2-WT = 1.25μm/min (SD 0.37), n=266 cells tracked from 6 embryos; Gγ2-SaaX = 1.27μm/min (SD 0.20), n=114 cells, 3 embryos), they do show a loss of directional migration as is evident in their wildly uncoordinated migrations as well as the lack of congregation at the previously described sites of high sdf1a expression [60] (Movies S1-S3, Figure S3).
3.4. All G protein γ subunits are expressed in the first 24 hours of zebrafish development
To identify Gγ subunits important during development of the zebrafish and determine if any subunits are specifically expressed in PGCs during migration, we examined their expression by RT-PCR and whole mount in situ hybridization. RT-PCR analysis revealed that all of the Gγ subunits are expressed during the first 24hpf and, with the exception of gngt1, gng3, and gng14, are maternally deposited (Figure 4). In situ hybridization analysis revealed that none of the Gγ subunits have expression patterns that are restricted to the PGCs. Several of the Gγ subunits exhibit expression patterns that are either ubiquitous (e.g. gng5), in tissues where migrating PGCs are found (gng2[38], gng3[37]), or in other discrete tissues. gngt1, gngt2a, and gngt2b are restricted to the retina and epiphysis at later stages ([39, 72]; data not shown), suggesting roles in other developmental processes.
Fig. 4.

All Gγ subunits are expressed during the first 24 hours of development. With the exception of gngt1, gngt2a, gng3, and gng14, all the Gγ subunits are maternally deposited. The zebrafish gamma subunits are 204-228bp in length. All have a single intron interrupting their coding sequence after approximately 90 coding base pairs, as has been reported in humans and mice [101].
3.5. G protein β subunits are localized to the cytosol in the presence of Gγ-SaaX
One hypothesis as to how the majority of Gγ subunits disrupt PGC migration is that they interact with a common Gβ subunit that they sequester in the cytosol as a non-functional dimer. To test this in vivo, we co-injected mRNA encoding an N-terminal mGFP-fused Gβ subunit controlled by the nos1-3′UTR, membrane-driven mCherry-CVLL(nos) mRNA, and either gng2-WT or gng2-SaaX mRNA in order to visualize any changes in the subcellular localization of the Gβ subunit in the presence of a prenylation-deficient Gγ. When GFP-gnb1(nos) was injected alone, fluorescence was observed in the cytosol of migrating PGCs (presumably due to the lack of sufficient endogenous Gγ; Figure 5A). When co-injected with the wildtype form of either gng2 or gng5, GFP-Gβ1 localized to the plasma membrane (Figure 5B). However, when GFP-gnb1(nos) was coinjected with gng2-SaaX mRNA, Gβ1 was once again localized to the cytosol, supporting the sequestering hypothesis (Figure 5C). Quantification of the GFP fluorescence associated with the membrane and cytosolic compartments of PGCs revealed a significant shift in localization in the Gγ-WT injected groups when compared to the Gγ-SaaX and control injected groups (ANOVA, p < 0.0001; Gabriel test, p < 0.002; Figure 5D). To investigate whether the Gβ subunits have the ability to override the Gγ2-SaaX-induced phenotype and restore normal PGC migration, we cloned nine zebrafish Gβ subunits (gnb1, gnb1l, gnb2, gnb2l1, gnb3a, gnb3b, gnb4, gnb5a and gnb5b; Table S1). mRNAs encoding each Gβ were co-injected with gng2-SaaX mRNA, but none were able to reverse the SaaX-induced PGC migration defects. Injections of combinations of the Gβs were also incapable of overcoming the SaaX-induced PGC migration defects.
Fig. 5.

The membrane localization of Gβ1 is lost in the presence of Gγ-SaaX. Confocal images of PGCs migrating in vivo after embryos were injected with the plasma membrane-localized mCherry-CVLL(nos) mRNA (column 1) and GFP-gnb1 mRNA (column 2). Embryos were co-injected with either phenol red as a control (A), gng2-WT mRNA (B), or gng2-SaaX mRNA (C). Ratios of the GFP fluorescence density in the cytosol to the GFP fluorescence density in the membranes of embryos injected with or without Gγ subunits (D) (CFD = cytosol fluorescence density; mfd = membrane fluorescence density; * p < 0.002 when compared to phenol red and gng2-SaaX injected groups). (For interpretation of the references to color in this figure the reader is referred to the web version of this article.)
3.6. Gγ-SaaX induced migration defects can be reversed by a subset of Gγ-WT subunits
We postulate that Gγ-SaaX subunits perturb PGC migration through the binding and mislocalization of relevant Gβ subunits. The cytosolically located Gβγ-SaaX dimer would be incapable of regulating Gα and signaling downstream effectors. The addition of exogenous wildtype Gγ that binds the endogenously acting Gβ to form a dimer that can interact with the upstream and downstream proteins necessary for PGC migration should be capable of reversing the Gγ-SaaX phenotype. Only the Gβγ dimers that form a heterotrimer with the PGC migration-mediating Gα, interact with the relevant GPCR, and become activated to signal the appropriate downstream effectors would restore proper migration. To investigate the differences among Gγ subunits in their capacity to interact with signaling partners as a Gβγ dimer in vivo, we examined whether individual Gγ-WT subunits have the ability to reverse the Gγ-SaaX induced phenotype. To accomplish this we injected embryos with gng2-SaaX mRNA followed by a Gγ-WT mRNA and assayed PGC migration. Embryos were injected with a mixture of GFP-nos1-3′UTR and gng2-SaaX mRNA (Figures 6A and 6B). Half of the embryos injected with this cocktail were injected a second time with mRNA encoding one of the Gγ-WT subunits. Injection of gng2-WT mRNA resulted in a complete reversal of the Gγ2-SaaX induced PGC migration defects (Figures 6C and 6D). Furthermore, it was found that gng3, gng4, gng7, gng8, gng12a, gng12b, and gng13-WT mRNA also had the ability to reverse the Gγ2-SaaX-induced PGC migration defects. Phylogenetic analysis of these Gγ subunits elucidated that the ability to restore proper PGC migration is limited to members of 3 out of the 5 pre-defined groups in the Gγ family [73]. All of the members of groups 2 and 5 and a subset of group 3 were able to restore proper migration (Figure 6E). The failure of the remaining Gγ-WT subunits to reverse the SaaX-induced phenotype was not due to an inability to form protein, as determined by in vitro translation with 35S labeled Methionine and the immunoprecipitation of FLAG-tagged Gγ subunits and subsequent SDS-PAGE analysis performed on representative subunits (data not shown).
Fig. 6.

Several wildtype G-protein γ subunits have the ability to reverse the Gγ2-SaaX induced disruption of PGC migration. Bright-field microscopy of a larvae ubiquitously expressing gng2-SaaX [20pg] reveals no somatic defects (A) while PGC migration in this same embryo is perturbed (B). Bright-field image of a larva ubiquitously expressing both gng2-WT mRNA [200pg] and gng2-SaaX mRNA [20pg] (C). The fluorescent image of (C) indicates a restoration of proper PGC migration (D). All embryos shown were injected at the 1 – 4 cell stage with mRNAs as described above, together with GFP-nos1-3′UTR mRNA and subjected to microscopy at 48hpf. Phylogenetic analysis of the zebrafish Gγ subunits and their ability to reverse the Gγ2-SaaX induced disruption of PGC migration (E). Method: Neighbor Joining; Best Tree; tie breaking = systematic. Distance: Poisson-correction; Gaps distributed proportionally. The Gγ subunits are divided into five groups according to their secondary structure as previously described [73]. Data was summarized from 2 – 5 experiments with a minimum of 20 embryos scored for the presence of ectopic PGCs at 24hpf. (For interpretation of the colors in this figure the reader is referred to the web version of this article.)
To address whether the failure of these wildtype Gγ subunits to restore proper migration in the presence of Gγ2-SaaX is due to their inability to compete with Gγ2 for PGC migration mediators (such as a common interacting Gβ subunit, upstream receptor or downstream effector) rather than their inability to interact with those mediators, wildtype mRNA of the Gγ subunits incapable of reversing the Gγ2-SaaX phenotype were injected in the presence of their own Gγ-SaaX mRNA. While mutation of the CaaX motif of Gγ subunits or truncation of the protein so that it is no longer prenylated does not affect the tight interaction of Gγ with its obligate partner, Gβ [74], we expect wildtype Gγ subunits would have a higher affinity for upstream or downstream effectors than their SaaX mutant counterparts due to the increased hydrophobicity imparted by the prenyl moiety. Wildtype Gγ subunits incapable of reversing the Gγ2-SaaX induced phenotype (Gγt1, t2a, 5, 10, 15, 16, 17) were also incapable of reversing the phenotype induced by their own Gγ-SaaX counterpart, suggesting that while they can interact with the endogenous Gβ subunit necessary for PGC migration (as assayed by their ability to disrupt PGC migration), they have an inability to interact with other essential PGC migration mediators. Alternatively these Gγ subunits could have reduced stability in the PGCs that would make them incapable of reversing the SaaX phenotype.
3.7. PGC-specific disruption of GPCR signaling results in ectopic PGCs
To address whether Gγ-SaaX proteins are acting within PGCs or within somatic cells to perturb PGC migration, we cloned wildtype and SaaX mutant Gγ subunits into the nanos1-3′UTR vector to drive their expression selectively in PGCs. While overexpression of gng3-WT mRNA in the PGCs has no effect on somatic development or PGC migration (Figures 7A and 7B), injection of gng3-SaaX(nos) resulted in PGC migration defects (Figures 7C and 7D). Similar results were found when these experiments were repeated with gng2-WT(nos) and gng2-SaaX(nos) (Figure 8). All of the nos1-3′UTR-directed wildtype and SaaX mRNAs tested (gngt1, gng2, gng3, gng4, gng5) behaved identically to their T3TS counterparts (except for the added ability of the nos1-3′UTR fused mRNAs to be injected at much higher concentrations before causing somatic disruption). This data is consistent with a cell-autonomous need for Gγ-mediated signaling within PGCs for proper migration. To assay for the disruption of somatic components of PGC migration, we tested the ability of a PGC-driven WT subunit to reverse the phenotype induced by ubiquitously driven gng2-SaaX. If Gγ2-SaaX is also disrupting GPCR signaling in somatic cells, a PGC-driven gng2-WT would be unable to reverse the SaaX-induced phenotype. At the low concentrations of ubiquitously translated gng2-SaaX necessary to specifically disrupt PGC migration, injection of the nos1-3′UTR controlled gng2-WT mRNA was able to restore proper PGC migration (Figures 7E, 7F, 8). However, injection of gng5-WT(nos) mRNA failed to reverse the SaaX-induced phenotype, providing additional evidence that Gγ subunits can have distinct signaling roles (Figures 7G and 7H).
Fig. 7.
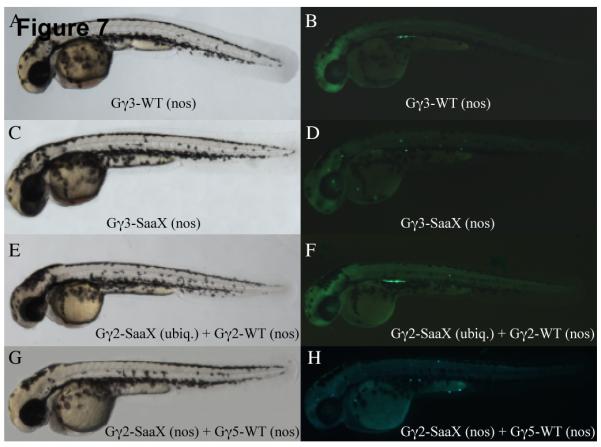
Gγ-SaaX expression specifically within the PGCs results in migration defects that can be reversed by injection of one of several Gγ-WT mRNAs. Overexpression of gng3-WT [300pg mRNA injection] specifically in the PGCs has no effect on either somatic development as assayed by bright-field microscopy (A) or PGC migration as revealed in the corresponding fluorescent image (B). gng3-SaaX expression [150pg] specifically in the PGCs also has no effect on somatic development (C) but profoundly disrupts PGC migration (D). The effect of gng2-SaaX on PGC migration is reversed by injection with PGC-driven gng2-WT mRNA [200pg] (E & F) but not PGC-driven gng5-WT mRNA [550pg] (G & H). All embryos shown were injected at the 1 – 4 cell stage with mRNAs as described above, together with GFP-nos1-3′UTR mRNA and subjected to microscopy at 48hpf.
Fig. 8.

Representative experiments exhibiting the reversal of the Gγ-SaaX phenotype by subsequent injection with Gγ-WT mRNA. When gng2-WT is driven either ubiquitously or in the PGCs (nos), it has the ability to reverse both the ubiquitously translated gng2-SaaX phenotype (A) and the phenotype resulting from gng2-SaaX being translated specifically in the PGCs (B). Each concentration is summarized from 2 – 5 experiments with a minimum of 20 embryos scored. (Ectopic PGC migration score is expressed as mean +/− S.D.)
3.8. Gγ-SaaX disrupts calcium accumulation in the protrusions of migrating PGCs
Disrupting Sdf1a signaling by targeting the chemokine sdf1a or its receptor cxcr4b with antisense morpholinos results in a decrease in calcium accumulation in the protrusions of migrating PGCs [58]. To investigate whether Gγ-SaaX expression disrupts this essential signaling pathway, we assayed calcium levels in the protrusions of PGCs migrating in larva injected with gng2-SaaX mRNA. Injection of gng2-SaaX mRNA resulted in a significant decrease of calcium accumulation in protrusions when compared to wildtype levels (Figure 9; Movie S4). This decrease is similar to that seen when embryos are injected with morpholinos targeted to cxcr4b and can be reversed by injection of gng2-WT mRNA (Movie S5). While injecting gng2-WT mRNA alone increases the amount of calcium present in these protrusions, it has no detectable effect on PGC migration (Movie S6).
Fig. 9.
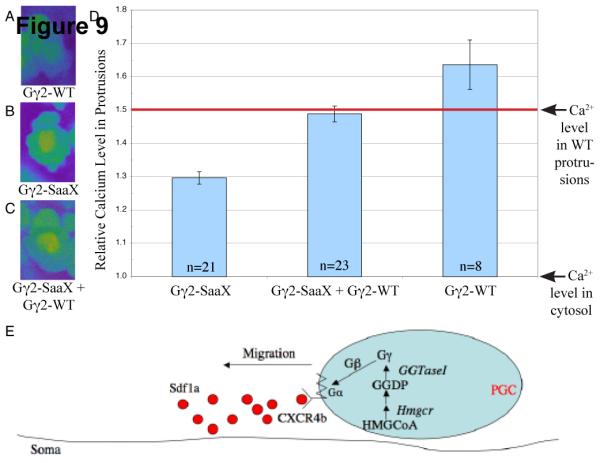
Overexpression of gng2-SaaX attenuates calcium accumulation within the protrusions of PGCs migrating in vivo. Injection of embryos at the 1-cell stage with the PGC-driven gng2-WT mRNA [75pg] results in an increase in the accumulation of calcium in the protrusions of PGCs migrating in vivo (A). PGC-driven gng2-SaaX mRNA [50pg] results in a suppression of the calcium accumulation in these protrusions (B), which is reversed by injection with gng2-WT mRNA [75pg] (C). Calcium levels were quantified with the calcium indicator Oregon green 488 BAPTA-1 [500 μM]. To identify the PGCs, embryos were co-injected with vasa-DsRedex1-nos1 RNA [180 pg] (A-C) and data is summarized in (D). (All PGCs described above were assayed 9-11hpf.) Model of a cell autonomous mechanism for HMGCR’s role in PGC migration (E). Internalization of the extracellular signal Sdf1a is transduced through a GPCR that utilizes heterotrimeric G-Proteins of which the γ subunit is a substrate for geranylgeranylation. (HMGCoA = 3-hydroxy-3-methylglutaryl-Coenzyme A; HMGCR = HMGCoA Reductase; GGDP = geranylgeranyl diphosphate synthase; GGTaseI = geranylgeranyl transferase I) (For interpretation of the colors in this figure the reader is referred to the web version of this article.)
4. Discussion
We have described an in vivo method to disrupt G-protein coupled signaling in a tissue-specific manner and investigate the signaling components involved in a conserved cell migratory pathway. Here we show that introducing prenylation-deficient Gγ subunits results in the reduction of calcium within the protrusions of migrating PGCs and an inability of PGCs to migrate directionally.
4.1. Mechanism of Gγ-SaaX action
Gγ subunits have specific and overlapping affinities for the less-numerous Gβ subunits [68, 75-83]. The ability of the majority of Gγ-SaaX subunits to disrupt PGC migration is consistent with the hypothesis that Gγ-SaaX binds and sequesters the PGC migration-mediating Gβ subunits into nonfunctional, cytosolically located Gβγ-SaaX dimers. Gβγ dimer formation occurs prior to the prenylation and proteolytic cleavage of the carboxy terminal ‘aaX’ amino acids [23] and is thus unaffected by the loss of Gγ lipid modification. Sequestration of Gβ into Gβγ-SaaX dimers may result in the inactivation of not only Gβγ signaling but also downstream effectors of Gα subunit signaling as has been suggested in Saccharomyces [84] and Arabidopsis [85]. PGC defects may result from an inability of the Gβγ-SaaX dimer to form a heterotrimer with the Gα subunit or to interact with specific GPCRs and/or downstream effectors. Future studies will investigate whether the sequestration of Gβ alone or in concert with other signaling partners determines the ability of a Gγ-SaaX subunit to disrupt PGC migration. Swapping the central region of the Gγ subunits, the main site of interaction with the Gβ subunit [78, 86, 87], may confer the ability to restore or disrupt PGC migration. The ability of the transducin orthologs (gngt1 and gngt2) to disrupt PGC migration when expressed in their wildtype form suggests that they may direct the Gβγ dimer to an alternative subcellular localization in their wt form, similar to the ‘SaaX’ form, or they may interact with signaling molecules not involved in PGC migration.
The ability of wildtype Gγ subunits to reverse the Gγ-SaaX-induced phenotype suggests that Gγ-SaaX proteins are acting as competitive inhibitors of GPCR signaling that can be overcome by simultaneous overexpression of wildtype Gγ subunits. The finding that only a subset of the wildtype Gγ subunits reverse the Gγ-SaaX phenotype is consistent with the hypothesis that while many Gγ subunits can interact with the less-variable Gβ subunits, Gβγ dimers consisting of different Gβ or Gγ members do not interact with the same pool of GPCRs and effectors [15, 30, 32, 88-96]. This is evidenced by the ability of both Gγ2, which does reverse the Gγ-SaaX phenotype, and Gγ5, which does not reverse the Gγ-SaaX phenotype, to drive GFP-β1 to the plasma membrane.
Previous studies, primarily in cell culture, support the hypothesis that Gγ-SaaX could exert its potent effect on PGC migration by sequestering Gβ subunits, thereby, creating a pool of non-functional Gβγ-SaaX dimers incapable of mediating the interactions essential for directed migration. However, when we overexpressed the identified Gβ subunits either individually or together in embryos injected with Gγ-SaaX mRNA, the PGC migration defects remained. Although unlikely, it is possible that we were unable to express enough Gβ to reverse the effect of Gγ-SaaX mRNA (Gβ mRNA was injected in quantities 1-10 times more than the molar equivalent of Gγ2-SaaX). Alternatively, there may be an unidentified Gβ that is required for PGC migration. The failure of Gβ overexpression to reverse the Gγ-SaaX phenotype suggests Gβγ-SaaX dimers exert a dominant negative influence on GPCR signaling as opposed to being inactive in the cytosol. Gβγ-SaaX could exert a dominant negative effect by either sequestering the available Gα into a nonfunctional heterotrimer or sequestering other proteins such as the corresponding GPCR, and/or downstream effectors into larger nonfunctional units. Further studies will be necessary to elucidate the in vivo interactions between Gγ subunit family members and their signaling partners essential for PGC migration.
4.2. Morpholinos are insufficient to study Gγfunction
Historically, morpholinos have been a powerful antisense tool to study genetics in zebrafish. However, individual or combinatorial injection of morpholinos targeting the ATG sequence of the 8 Gγ-WT subunits that reverse the Gγ-SaaX phenotype had no effect on PGC migration. It is unclear whether this is due to an abundance of maternally deposited protein and mRNA or whether the morpholinos were unable to knock down their intended targets sufficiently. Until zebrafish lines containing mutations in each of the Gβ and Gγ subunits are established, viable alternative methods of disruption such as injecting Gγ-SaaX mRNA can be used to investigate the roles of heterotrimeric G proteins during development.
4.3. Gγ-SaaX mRNA injection as a general tool for GPCR disruption
Given the studies reporting differences in the specificities and affinities of Gγ subunits for Gβ subunits, we would expect that the Gγ subunits have variable efficiency in disrupting GPCR signaling pathways. Overexpression of a number of the Gγ-SaaX subunits results in failure of the dorsal tissues to converge at the midline, multiple tail tips, cyclopia, and gross edema. The amount of mRNA necessary to induce such phenotypes was variable between Gγ-SaaX subunits, which could either suggest a difference in translation efficiency in vivo or differences in their individual affinities for the Gβ and/or interacting partners.
4.4. Why are the PGCs so sensitive to Gγ-SaaX?
We expected that PGC-specific expression of Gγ-SaaX would be necessary to prevent disruptions in the somatic cells but found that the amount of ubiquitously translated mRNA injected could be titrated down to levels that exhibited a PGC migration-specific phenotype. Of all the signaling events that could be disrupted by Gγ-SaaX, such as those involving the mitogen-activated protein kinases ERK1 and ERK2 [97], Gα or sdf-mediated signaling necessary for convergence and extension [98] or endoderm migration [99], the signaling pathways operating in the PGCs seem to be especially sensitive to disruption by injection of Gγ-SaaX mRNA. One hypothesis why PGCs may be more susceptible to perturbation involves a limited level or delay of transcription in this cell type in comparison to the somatic cells such as has been described in Drosophila during early stages of development [100]. A smaller pool of endogenous wildtype Gγ could be more easily outcompeted by Gγ-SaaX in PGCs that are unable to synthesize new Gγ.
4.5. Signaling pathways that influence PGC migration
The chemokine SDF1 was recently shown to promote the accumulation of calcium in the protrusions of migrating PGCs [58]. The finding that Gγ-SaaX mRNAs also disrupt the accumulation of calcium in the protrusions of PGCs provides strong evidence that Sdf1a, Gγ and thus GGTaseI participate in the same pathway by functioning downstream of 3-Hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR). In this model, HMGCR acts within the PGCs to synthesize precursors of the isoprenoid geranylgeranyl diphosphate, which is post-translationally added by GGTaseI to the C-terminus of Gγ subunits necessary for the transduction of the Sdf1a signal (Figure 9E). Future experiments will explore whether abrogating prenylation through treatment with HMGCR inhibitors, such as atorvastatin, also disrupt calcium signaling.
If signaling through Sdf1a and its receptor CXCR4b is the same pathway disrupted in the presence of Geranylgeranyl transferase inhibitors (GGTIs), we might expect the migration defects caused by Gγ2-SaaX and GGTI-2166 to be identical, which is not the case. Gγ-SaaX mRNA injection results in directional migration defects and not in a decrease in PGC migration speed as was previously observed with GGTI-2166 treatment of zebrafish embryos [52]. These differences may result from maternally deposited, prenylated Gγ being present in the PGCs and unaffected by GGTI-2166. The lower concentrations of inhibitor required to maintain normal somatic development might be insufficient to completely block the geranylgeranylation of Gγ within PGCs. Alternatively, the geranylgeranylation of several protein targets may be necessary to orchestrate a cellular event as complex as the migration of PGCs in vivo.
5. Conclusion
Our findings provide evidence that prenylation of Gγ subunits is necessary within the PGCs for directed migration. These findings also suggest that multiple Gγ subunits present in the PGCs could act redundantly or as reserves to a primary Gγ subunit(s) during migration. It would be evolutionarily advantageous for an organism to have redundancies in the mechanisms involved in PGC migration given the limited ability of PGCs to synthesize new proteins coupled with the necessity of PGC migration for propagation. While there is currently no simple and efficient method for assaying the role of endogenous Gγ subunits in the zebrafish, the injection of Gγ-SaaX mRNAs provides robust data implicating the function of GPCR-mediated signaling during development.
Supplementary Material
Fig. S1. Phylogenetic tree of the Danio rerio (Dr) and Homo sapien (Hs) Heterotrimeric Gγ subunit genes. Method: Neighbor Joining; Best Tree; tie breaking = systematic. Distance: Poisson-correction; Gaps distributed proportionally.
Fig. S2. Syntenic maps of the zebrafish G protein γ subunits. Each panel shows the syntenic relationship of the human (Hs) and zebrafish (Dr) chromosomes. Green lines represent genes likely duplicated in the zebrafish genome. Homology of genes lacking annotation is based on the best hit from TBLASTN searches of the Homo sapien reference release and Danio rerio assembly v7.
Fig. S3. While the wildtype sdf1a expression pattern is maintained, PGCs are ectopic in embryos injected with gng2-SaaX mRNA. Whole mount in situ hybridization at the 13 somite stage shows the expression pattern of sdf1a (red - labels several somatic structures including the pronephros, somites, branchial arches and rhombomeres 4, 6 and 7) and nanos (blue - labels the PGCs) in embryos that were uninjected (A) and injected with 30pg gng2-SaaX mRNA (B). Arrows show wildtype locations for the PGCs in uninjected embryos (overlapping the sdf1a signal lateral to somites 4-7 and along the pronephros). Arrowheads show the ectopic PGCs in embryos injected with gng2-SaaX. gng2-SaaX mRNA injected zebrafish exhibit wildtype sdf1a expression which serves a dual role of evidencing the proper patterning of somatic structures and the expression of an essential PGC attractant.
Fig. S4. Summary of the ability of the Gγ subunits to disrupt PGC migration when injected as mRNA in their SaaX form. Each concentration is a summary of 2 – 5 experiments with a minimum of 20 embryos scored. (Ectopic PGC migration score is represented as Mean +/− S.D.)
Table S1. Syntenic maps of the duplicated zebrafish G protein β subunits (A). Each panel shows the syntenic relationship of the human (Hs) and zebrafish (Dr) chromosomes. Green lines represent genes that are likely duplicated in the zebrafish genome. Homology of genes lacking annotation is based on the best hit from TBLASTN searches of the Homo sapien current reference release and Danio rerio assembly v7. Identified zebrafish G protein β subunits are summarized in (B). Except where noted, the zebrafish Gβ subunits exhibited the highest homology to their human orthologs (1 gnb1l was compared to GNB1; 2 gnb5a was compared to GNB5 isoform a; 3 gnb5b was compared to GNB5 isoform b.)
Table S2. Primers used for cloning and RTPCR of the zebrafish Gβ and Gγ subunit genes. Gγ PCR products were cut with BglII and SpeI for cloning into pT3TS except for gng2 and gng10 which were cut with EcoRV and SpeI. All Gγ PCR products were cut with BglII and XhoI for cloning into the nos1-3′UTR vector except for gng2 which was cut with HindIII and XhoI. gnb1, gnb1l, gnb3a, gnb3b, gnb4, and gnb5b PCR products were cut with KpnI and XhoI for cloning into the GFPnos3′UTR vector. gnb3 was cut with KpnI and StuI and gnb2l1 and gnb5a were cut with XhoI. Primers used for cloning were also used for RTPCR except for gngt1, gngt2b, gng8, gng14, gng15, bactin1 and ef1a, whose RTPCR primers are listed separately.
Movie Stills. Still images from movies S1-S3. The paths of migrating PGCs are designated by the colored lines. Anterior is at the top and posterior at the bottom. The orientation of the future midline is represented by the dotted white line. The target of wildtype PGCs, the anterior of the yolk extension where the gonad will later form, is just lateral to the midline and posterior to the frame shown in the movie.
Movie S1. Dorsal view of the PGCs migrating in vivo from 10-30 hours post fertilization. The PGCs are visable due to the injection of GFP-CVLL-(nos) mRNA (pictures taken every 8 minutes and 15 seconds; 10 frames per second)
Movie S2. Dorsal view of the PGCs migrating in vivo from 10-30hpf after zebrafish were injected with GFP-CVLL-(nos) and gng2-wt(nos) mRNA at the one cell stage (pictures taken every 8 minutes and 15 seconds; 10 frames per second)
Movie S3. Dorsal view of the PGCs migrating in vivo from 10-30hpf after injection of GFP-CVLL-nos3′UTR and Gγ2-SaaX(nos3′UTR) mRNA at the one cell stage (pictures taken every 8 minutes and 15 seconds; 10 frames per second)
Movie S4. Zebrafish injected with 50pg gng2-SaaX(nos) mRNA. PGCs migrating in vivo labeled with the free calcium indicator Oregon green 488 BAPTA-1. Images were captured at 1.5 second intervals for 27 seconds and played at 9 frames per second. (PGCs in Movies S4-S6 all assayed between 9 and 11hpf)
Movie S5. Zebrafish injected with 50pg gng2-SaaX(nos) and 75 pg gng2-WT(nos) mRNA. PGCs migrating in vivo labeled with the free calcium indicator Oregon green 488 BAPTA-1. Images were captured at 1.5 second intervals for 27 seconds and played at 9 frames per second.
Movie S6. Zebrafish injected with 75 pg gng2-WT(nos) mRNA. PGCs migrating in vivo labeled with the free calcium indicator Oregon green 488 BAPTA-1. Images were captured at 1.5 second intervals for 27 seconds and played at 9 frames per second.
Acknowledgments
The authors would like to thank Amy Kowalski and Mohan Bolisetty for technical assistance, Jennifer Anderson for editorial assistance, and Mahmud Siddiqi for microscopy guidance. We would also like to thank Marnie Halpern, Alex Bortvin, Mark Van Doren, Haiqing Zhao, and Fang Lin for critical reading and advice.
This work was supported in part by NIH grant GM63904 (SAF). HB was supported by funds from the Deutsche Forschungsgemeinschaft (DFG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Oldham WM, Hamm HE. Nat Rev Mol Cell Biol. 2008;9(1):60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- [2].Federman AD, Conklin BR, Schrader KA, Reed RR, Bourne HR. Nature. 1992;356(6365):159–161. doi: 10.1038/356159a0. [DOI] [PubMed] [Google Scholar]
- [3].Tang WJ, Gilman AG. Science. 1991;254(5037):1500–1503. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- [4].Tang WJ, Iniguez-Lluhi JA, Mumby S, Gilman AG. Cold Spring Harb Symp Quant Biol. 1992;57:135–144. doi: 10.1101/sqb.1992.057.01.017. [DOI] [PubMed] [Google Scholar]
- [5].Boyer JL, Graber SG, Waldo GL, Harden TK, Garrison JC. J Biol Chem. 1994;269(4):2814–2819. [PubMed] [Google Scholar]
- [6].Lee SB, Shin SH, Hepler JR, Gilman AG, Rhee SG. J Biol Chem. 1993;268(34):25952–25957. [PubMed] [Google Scholar]
- [7].Thomason PA, James SR, Casey PJ, Downes CP. J Biol Chem. 1994;269(24):16525–16528. [PubMed] [Google Scholar]
- [8].Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. Cell. 1994;77(1):83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- [9].Kameyama K, Haga K, Haga T, Kontani K, Katada T, Fukada Y. J Biol Chem. 1993;268(11):7753–7758. [PubMed] [Google Scholar]
- [10].Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Science. 1992;257(5074):1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- [11].Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Nature. 1996;380(6571):258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- [12].Ikeda SR. Nature. 1996;380(6571):255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- [13].Hurowitz EH, Melnyk JM, Chen YJ, Kouros-Mehr H, Simon MI, Shizuya H. DNA Res. 2000;7(2):111–120. doi: 10.1093/dnares/7.2.111. [DOI] [PubMed] [Google Scholar]
- [14].McIntire WE, MacCleery G, Murphree LJ, Kerchner KR, Linden J, Garrison JC. Biochemistry. 2006;45(38):11616–11631. doi: 10.1021/bi0604882. [DOI] [PubMed] [Google Scholar]
- [15].Richardson M, Robishaw JD. J Biol Chem. 1999;274(19):13525–13533. doi: 10.1074/jbc.274.19.13525. [DOI] [PubMed] [Google Scholar]
- [16].Bommakanti RK, Vinayak S, Simonds WF. J Biol Chem. 2000;275(49):38870–38876. doi: 10.1074/jbc.M007403200. [DOI] [PubMed] [Google Scholar]
- [17].Hildebrandt JD. Biochem Pharmacol. 1997;54(3):325–339. doi: 10.1016/s0006-2952(97)00269-4. [DOI] [PubMed] [Google Scholar]
- [18].Hansen CA, Schroering AG, Carey DJ, Robishaw JD. J Cell Biol. 1994;126(3):811–819. doi: 10.1083/jcb.126.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Takida S, Wedegaertner PB. J Biol Chem. 2003;278(19):17284–17290. doi: 10.1074/jbc.M213239200. [DOI] [PubMed] [Google Scholar]
- [20].Simonds WF, Butrynski JE, Gautam N, Unson CG, Spiegel AM. J Biol Chem. 1991;266(9):5363–5366. [PubMed] [Google Scholar]
- [21].Matsuda T, Takao T, Shimonishi Y, Murata M, Asano T, Yoshizawa T, Fukada Y. J Biol Chem. 1994;269(48):30358–30363. [PubMed] [Google Scholar]
- [22].Dietrich A, Scheer A, Illenberger D, Kloog Y, Henis YI, Gierschik P. Biochem J. 2003;376(Pt 2):449–456. doi: 10.1042/BJ20030578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Higgins JB, Casey PJ. J Biol Chem. 1994;269(12):9067–9073. [PubMed] [Google Scholar]
- [24].Ohguro H, Fukada Y, Takao T, Shimonishi Y, Yoshizawa T, Akino T. EMBO J. 1991;10(12):3669–3674. doi: 10.1002/j.1460-2075.1991.tb04934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yasuda H, Lindorfer MA, Woodfork KA, Fletcher JE, Garrison JC. J Biol Chem. 1996;271(31):18588–18595. doi: 10.1074/jbc.271.31.18588. [DOI] [PubMed] [Google Scholar]
- [26].Dietrich A, Brazil D, Jensen ON, Meister M, Schrader M, Moomaw JF, Mann M, Illenberger D, Gierschik P. Biochemistry. 1996;35(48):15174–15182. doi: 10.1021/bi960305j. [DOI] [PubMed] [Google Scholar]
- [27].Nakajima Y, Nakajima S, Kozasa T. FEBS Lett. 1996;390(2):217–220. doi: 10.1016/0014-5793(96)00661-8. [DOI] [PubMed] [Google Scholar]
- [28].Dietrich A, Meister M, Brazil D, Camps M, Gierschik P. Eur J Biochem. 1994;219(12):171–178. doi: 10.1111/j.1432-1033.1994.tb19927.x. [DOI] [PubMed] [Google Scholar]
- [29].Matsuda T, Hashimoto Y, Ueda H, Asano T, Matsuura Y, Doi T, Takao T, Shimonishi Y, Fukada Y. Biochemistry. 1998;37(27):9843–9850. doi: 10.1021/bi973194c. [DOI] [PubMed] [Google Scholar]
- [30].Myung CS, Yasuda H, Liu WW, Harden TK, Garrison JC. J Biol Chem. 1999;274(23):16595–16603. doi: 10.1074/jbc.274.23.16595. [DOI] [PubMed] [Google Scholar]
- [31].Lukov GL, Myung CS, McIntire WE, Shao J, Zimmerman SS, Garrison JC, Willardson BM. Biochemistry. 2004;43(19):5651–5660. doi: 10.1021/bi035903u. [DOI] [PubMed] [Google Scholar]
- [32].Fogg VC, Azpiazu I, Linder ME, Smrcka A, Scarlata S, Gautam N. J Biol Chem. 2001;276(45):41797–41802. doi: 10.1074/jbc.M107661200. [DOI] [PubMed] [Google Scholar]
- [33].Iniguez-Lluhi JA, Simon MI, Robishaw JD, Gilman AG. J Biol Chem. 1992;267(32):23409–23417. [PubMed] [Google Scholar]
- [34].Schwindinger WF, Betz KS, Giger KE, Sabol A, Bronson SK, Robishaw JD. J Biol Chem. 2003;278(8):6575–6579. doi: 10.1074/jbc.M211132200. [DOI] [PubMed] [Google Scholar]
- [35].Schwindinger WF, Giger KE, Betz KS, Stauffer AM, Sunderlin EM, Sim-Selley LJ, Selley DE, Bronson SK, Robishaw JD. Mol Cell Biol. 2004;24(17):7758–7768. doi: 10.1128/MCB.24.17.7758-7768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Robishaw JD, Guo ZP, Wang Q. Methods Mol Biol. 2004;237:169–180. doi: 10.1385/1-59259-430-1:169. [DOI] [PubMed] [Google Scholar]
- [37].Kelly GM, Vanderbeld B, Krawetz R, Mangos S. Int J Dev Neurosci. 2001;19(4):455–467. doi: 10.1016/s0736-5748(01)00002-8. [DOI] [PubMed] [Google Scholar]
- [38].Leung T, Chen H, Stauffer AM, Giger KE, Sinha S, Horstick EJ, Humbert JE, Hansen CA, Robishaw JD. Blood. 2006;108(1):160–166. doi: 10.1182/blood-2005-09-3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen H, Leung T, Giger KE, Stauffer AM, Humbert JE, Sinha S, Horstick EJ, Hansen CA, Robishaw JD. Gene Expr Patterns. 2007;7(5):574–583. doi: 10.1016/j.modgep.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kunwar PS, Starz-Gaiano M, Bainton RJ, Heberlein U, Lehmann R. PLoS Biol. 2003;1(3):E80. doi: 10.1371/journal.pbio.0000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Knaut H, Werz C, Geisler R, Nusslein-Volhard C. Nature. 2003;421(6920):279–282. doi: 10.1038/nature01338. [DOI] [PubMed] [Google Scholar]
- [42].Doitsidou M, Reichman-Fried M, Stebler J, Koprunner M, Dorries J, Meyer D, Esguerra CV, Leung T, Raz E. Cell. 2002;111(5):647–659. doi: 10.1016/s0092-8674(02)01135-2. [DOI] [PubMed] [Google Scholar]
- [43].Molyneaux KA, Zinszner H, Kunwar PS, Schaible K, Stebler J, Sunshine MJ, O’Brien W, Raz E, Littman D, Wylie C, Lehmann R. Development. 2003;130(18):4279–4286. doi: 10.1242/dev.00640. [DOI] [PubMed] [Google Scholar]
- [44].Ara T, Nakamura Y, Egawa T, Sugiyama T, Abe K, Kishimoto T, Matsui Y, Nagasawa T. Proc Natl Acad Sci U S A. 2003;100(9):5319–5323. doi: 10.1073/pnas.0730719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. J Exp Med. 1996;184(3):1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Murdoch C. Immunol Rev. 2000;177:175–184. doi: 10.1034/j.1600-065x.2000.17715.x. [DOI] [PubMed] [Google Scholar]
- [47].Gonzalo JA, Lloyd CM, Peled A, Delaney T, Coyle AJ, Gutierrez-Ramos JC. J Immunol. 2000;165(1):499–508. doi: 10.4049/jimmunol.165.1.499. [DOI] [PubMed] [Google Scholar]
- [48].Lee BC, Lee TH, Avraham S, Avraham HK. Mol Cancer Res. 2004;2(6):327–338. [PubMed] [Google Scholar]
- [49].Kang H, Mansel RE, Jiang WG. Int J Oncol. 2005;26(5):1429–1434. [PubMed] [Google Scholar]
- [50].Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- [51].Gilbert DC, Chandler I, McIntyre A, Goddard NC, Gabe R, Huddart RA, Shipley J. J Pathol. 2009;217(1):94–102. doi: 10.1002/path.2436. [DOI] [PubMed] [Google Scholar]
- [52].Thorpe JL, Doitsidou M, Ho SY, Raz E, Farber SA. Dev Cell. 2004;6(2):295–302. doi: 10.1016/s1534-5807(04)00032-2. [DOI] [PubMed] [Google Scholar]
- [53].Santos AC, Lehmann R. Dev Cell. 2004;6(2):283–293. doi: 10.1016/s1534-5807(04)00023-1. [DOI] [PubMed] [Google Scholar]
- [54].Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Dev Dyn. 1995;203(3):253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- [55].Westerfield M. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) University of Oregon Press; Eugene: 2000. [Google Scholar]
- [56].Hyatt TM, Ekker SC. Methods Cell Biol. 1999;59:117–126. doi: 10.1016/s0091-679x(08)61823-3. [DOI] [PubMed] [Google Scholar]
- [57].Koprunner M, Thisse C, Thisse B, Raz E. Genes Dev. 2001;15(21):2877–2885. doi: 10.1101/gad.212401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Blaser H, Reichman-Fried M, Castanon I, Dumstrei K, Marlow FL, Kawakami K, Solnica-Krezel L, Heisenberg CP, Raz E. Dev Cell. 2006;11(5):613–627. doi: 10.1016/j.devcel.2006.09.023. [DOI] [PubMed] [Google Scholar]
- [59].Thisse C, Thisse B. Nat Protoc. 2008;3(1):59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- [60].Weidinger G, Wolke U, Koprunner M, Thisse C, Thisse B, Raz E. Development. 2002;129(1):25–36. doi: 10.1242/dev.129.1.25. [DOI] [PubMed] [Google Scholar]
- [61].Mumby SM. Methods Enzymol. 2002;344:383–403. doi: 10.1016/s0076-6879(02)44729-5. [DOI] [PubMed] [Google Scholar]
- [62].Dohlman HG. Annu Rev Physiol. 2002;64:129–152. doi: 10.1146/annurev.physiol.64.081701.133448. [DOI] [PubMed] [Google Scholar]
- [63].Ray K, Ganguly R. J Biol Chem. 1992;267(9):6086–6092. [PubMed] [Google Scholar]
- [64].Schulz S, Huber A, Schwab K, Paulsen R. J Biol Chem. 1999;274(53):37605–37610. doi: 10.1074/jbc.274.53.37605. [DOI] [PubMed] [Google Scholar]
- [65].Downes GB, Gautam N. Genomics. 1999;62(3):544–552. doi: 10.1006/geno.1999.5992. [DOI] [PubMed] [Google Scholar]
- [66].Hochholdinger F, Baier G, Nogalo A, Bauer B, Grunicke HH, Uberall F. Mol Cell Biol. 1999;19(12):8052–8065. doi: 10.1128/mcb.19.12.8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hirschman JE, Jenness DD. Mol Cell Biol. 1999;19(11):7705–7711. doi: 10.1128/mcb.19.11.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pronin AN, Gautam N. Proc Natl Acad Sci U S A. 1992;89(13):6220–6224. doi: 10.1073/pnas.89.13.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Muntz KH, Sternweis PC, Gilman AG, Mumby SM. Mol Biol Cell. 1992;3(1):49–61. doi: 10.1091/mbc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dietrich A, Meister M, Spicher K, Schultz G, Camps M, Gierschik P. FEBS Lett. 1992;313(3):220–224. doi: 10.1016/0014-5793(92)81195-r. [DOI] [PubMed] [Google Scholar]
- [71].Dumstrei K, Mennecke R, Raz E. J Cell Sci. 2004;117(Pt 20):4787–4795. doi: 10.1242/jcs.01362. [DOI] [PubMed] [Google Scholar]
- [72].Thisse B, Heyer V, Lux A, Alunni V, Degrave A, Seiliez I, Kirchner J, Parkhill JP, Thisse C. Methods Cell Biol. 2004;77:505–519. doi: 10.1016/s0091-679x(04)77027-2. [DOI] [PubMed] [Google Scholar]
- [73].Ray K, Kunsch C, Bonner LM, Robishaw JD. J Biol Chem. 1995;270(37):21765–21771. doi: 10.1074/jbc.270.37.21765. [DOI] [PubMed] [Google Scholar]
- [74].Schillo S, Belusic G, Hartmann K, Franz C, Kuhl B, Brenner-Weiss G, Paulsen R, Huber A. J Biol Chem. 2004;279(35):36309–36316. doi: 10.1074/jbc.M404611200. [DOI] [PubMed] [Google Scholar]
- [75].Rosskopf D, Koch K, Habich C, Geerdes J, Ludwig A, Wilhelms S, Jakobs KH, Siffert W. Cell Signal. 2003;15(5):479–488. doi: 10.1016/s0898-6568(02)00140-7. [DOI] [PubMed] [Google Scholar]
- [76].Schmidt CJ, Thomas TC, Levine MA, Neer EJ. J Biol Chem. 1992;267(20):13807–13810. [PubMed] [Google Scholar]
- [77].Garritsen A, Simonds WF. J Biol Chem. 1994;269(39):24418–24423. [PubMed] [Google Scholar]
- [78].Lee C, Murakami T, Simonds WF. J Biol Chem. 1995;270(15):8779–8784. doi: 10.1074/jbc.270.15.8779. [DOI] [PubMed] [Google Scholar]
- [79].Yan K, Kalyanaraman V, Gautam N. J Biol Chem. 1996;271(12):7141–7146. doi: 10.1074/jbc.271.12.7141. [DOI] [PubMed] [Google Scholar]
- [80].Asano T, Morishita R, Ueda H, Kato K. J Biol Chem. 1999;274(30):21425–21429. doi: 10.1074/jbc.274.30.21425. [DOI] [PubMed] [Google Scholar]
- [81].Dingus J, Wells CA, Campbell L, Cleator JH, Robinson K, Hildebrandt JD. Biochemistry. 2005;44(35):11882–11890. doi: 10.1021/bi0504254. [DOI] [PubMed] [Google Scholar]
- [82].Mervine SM, Yost EA, Sabo JL, Hynes TR, Berlot CH. Mol Pharmacol. 2006;70(1):194–205. doi: 10.1124/mol.106.022616. [DOI] [PubMed] [Google Scholar]
- [83].Rosskopf D, Nikula C, Manthey I, Joisten M, Frey U, Kohnen S, Siffert W. FEBS Lett. 2003;544(13):27–32. doi: 10.1016/s0014-5793(03)00441-1. [DOI] [PubMed] [Google Scholar]
- [84].Grishin AV, Weiner JL, Blumer KJ. Mol Cell Biol. 1994;14(7):4571–4578. doi: 10.1128/mcb.14.7.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chakravorty D, Botella JR. Gene. 2007;393(12):163–170. doi: 10.1016/j.gene.2007.02.008. [DOI] [PubMed] [Google Scholar]
- [86].Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Nature. 1996;379(6563):369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- [87].Mende U, Schmidt CJ, Yi F, Spring DJ, Neer EJ. J Biol Chem. 1995;270(26):15892–15898. doi: 10.1074/jbc.270.26.15892. [DOI] [PubMed] [Google Scholar]
- [88].Kisselev O, Pronin A, Ermolaeva M, Gautam N. Proc Natl Acad Sci U S A. 1995;92(20):9102–9106. doi: 10.1073/pnas.92.20.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lim WK, Myung CS, Garrison JC, Neubig RR. Biochemistry. 2001;40(35):10532–10541. doi: 10.1021/bi010950c. [DOI] [PubMed] [Google Scholar]
- [90].Hou Y, Azpiazu I, Smrcka A, Gautam N. J Biol Chem. 2000;275(50):38961–38964. doi: 10.1074/jbc.C000604200. [DOI] [PubMed] [Google Scholar]
- [91].Yasuda H, Lindorfer MA, Myung CS, Garrison JC. J Biol Chem. 1998;273(34):21958–21965. doi: 10.1074/jbc.273.34.21958. [DOI] [PubMed] [Google Scholar]
- [92].Akgoz M, Azpiazu I, Kalyanaraman V, Gautam N. J Biol Chem. 2002;277(22):19573–19578. doi: 10.1074/jbc.M201546200. [DOI] [PubMed] [Google Scholar]
- [93].Jian X, Clark WA, Kowalak J, Markey SP, Simonds WF, Northup JK. J Biol Chem. 2001;276(51):48518–48525. doi: 10.1074/jbc.M107129200. [DOI] [PubMed] [Google Scholar]
- [94].Kerchner KR, Clay RL, McCleery G, Watson N, McIntire WE, Myung CS, Garrison JC. J Biol Chem. 2004;279(43):44554–44562. doi: 10.1074/jbc.M406071200. [DOI] [PubMed] [Google Scholar]
- [95].Myung CS, Lim WK, DeFilippo JM, Yasuda H, Neubig RR, Garrison JC. Mol Pharmacol. 2006;69(3):877–887. doi: 10.1124/mol.105.018994. [DOI] [PubMed] [Google Scholar]
- [96].Jian X, Sainz E, Clark WA, Jensen RT, Battey JF, Northup JK. J Biol Chem. 1999;274(17):11573–11581. doi: 10.1074/jbc.274.17.11573. [DOI] [PubMed] [Google Scholar]
- [97].Krens SF, He S, Lamers GE, Meijer AH, Bakkers J, Schmidt T, Spaink HP, Snaar-Jagalska BE. Dev Biol. 2008;319(2):370–383. doi: 10.1016/j.ydbio.2008.04.032. [DOI] [PubMed] [Google Scholar]
- [98].Lin F, Sepich DS, Chen S, Topczewski J, Yin C, Solnica-Krezel L, Hamm H. J Cell Biol. 2005;169(5):777–787. doi: 10.1083/jcb.200501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Mizoguchi T, Verkade H, Heath JK, Kuroiwa A, Kikuchi Y. Development. 2008;135(15):2521–2529. doi: 10.1242/dev.020107. [DOI] [PubMed] [Google Scholar]
- [100].Van Doren M, Williamson AL, Lehmann R. Curr Biol. 1998;8(4):243–246. doi: 10.1016/s0960-9822(98)70091-0. [DOI] [PubMed] [Google Scholar]
- [101].Yang W, Hildebrandt JD. Cell Signal. 2006;18(2):194–201. doi: 10.1016/j.cellsig.2005.04.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phylogenetic tree of the Danio rerio (Dr) and Homo sapien (Hs) Heterotrimeric Gγ subunit genes. Method: Neighbor Joining; Best Tree; tie breaking = systematic. Distance: Poisson-correction; Gaps distributed proportionally.
Fig. S2. Syntenic maps of the zebrafish G protein γ subunits. Each panel shows the syntenic relationship of the human (Hs) and zebrafish (Dr) chromosomes. Green lines represent genes likely duplicated in the zebrafish genome. Homology of genes lacking annotation is based on the best hit from TBLASTN searches of the Homo sapien reference release and Danio rerio assembly v7.
Fig. S3. While the wildtype sdf1a expression pattern is maintained, PGCs are ectopic in embryos injected with gng2-SaaX mRNA. Whole mount in situ hybridization at the 13 somite stage shows the expression pattern of sdf1a (red - labels several somatic structures including the pronephros, somites, branchial arches and rhombomeres 4, 6 and 7) and nanos (blue - labels the PGCs) in embryos that were uninjected (A) and injected with 30pg gng2-SaaX mRNA (B). Arrows show wildtype locations for the PGCs in uninjected embryos (overlapping the sdf1a signal lateral to somites 4-7 and along the pronephros). Arrowheads show the ectopic PGCs in embryos injected with gng2-SaaX. gng2-SaaX mRNA injected zebrafish exhibit wildtype sdf1a expression which serves a dual role of evidencing the proper patterning of somatic structures and the expression of an essential PGC attractant.
Fig. S4. Summary of the ability of the Gγ subunits to disrupt PGC migration when injected as mRNA in their SaaX form. Each concentration is a summary of 2 – 5 experiments with a minimum of 20 embryos scored. (Ectopic PGC migration score is represented as Mean +/− S.D.)
Table S1. Syntenic maps of the duplicated zebrafish G protein β subunits (A). Each panel shows the syntenic relationship of the human (Hs) and zebrafish (Dr) chromosomes. Green lines represent genes that are likely duplicated in the zebrafish genome. Homology of genes lacking annotation is based on the best hit from TBLASTN searches of the Homo sapien current reference release and Danio rerio assembly v7. Identified zebrafish G protein β subunits are summarized in (B). Except where noted, the zebrafish Gβ subunits exhibited the highest homology to their human orthologs (1 gnb1l was compared to GNB1; 2 gnb5a was compared to GNB5 isoform a; 3 gnb5b was compared to GNB5 isoform b.)
Table S2. Primers used for cloning and RTPCR of the zebrafish Gβ and Gγ subunit genes. Gγ PCR products were cut with BglII and SpeI for cloning into pT3TS except for gng2 and gng10 which were cut with EcoRV and SpeI. All Gγ PCR products were cut with BglII and XhoI for cloning into the nos1-3′UTR vector except for gng2 which was cut with HindIII and XhoI. gnb1, gnb1l, gnb3a, gnb3b, gnb4, and gnb5b PCR products were cut with KpnI and XhoI for cloning into the GFPnos3′UTR vector. gnb3 was cut with KpnI and StuI and gnb2l1 and gnb5a were cut with XhoI. Primers used for cloning were also used for RTPCR except for gngt1, gngt2b, gng8, gng14, gng15, bactin1 and ef1a, whose RTPCR primers are listed separately.
Movie Stills. Still images from movies S1-S3. The paths of migrating PGCs are designated by the colored lines. Anterior is at the top and posterior at the bottom. The orientation of the future midline is represented by the dotted white line. The target of wildtype PGCs, the anterior of the yolk extension where the gonad will later form, is just lateral to the midline and posterior to the frame shown in the movie.
Movie S1. Dorsal view of the PGCs migrating in vivo from 10-30 hours post fertilization. The PGCs are visable due to the injection of GFP-CVLL-(nos) mRNA (pictures taken every 8 minutes and 15 seconds; 10 frames per second)
Movie S2. Dorsal view of the PGCs migrating in vivo from 10-30hpf after zebrafish were injected with GFP-CVLL-(nos) and gng2-wt(nos) mRNA at the one cell stage (pictures taken every 8 minutes and 15 seconds; 10 frames per second)
Movie S3. Dorsal view of the PGCs migrating in vivo from 10-30hpf after injection of GFP-CVLL-nos3′UTR and Gγ2-SaaX(nos3′UTR) mRNA at the one cell stage (pictures taken every 8 minutes and 15 seconds; 10 frames per second)
Movie S4. Zebrafish injected with 50pg gng2-SaaX(nos) mRNA. PGCs migrating in vivo labeled with the free calcium indicator Oregon green 488 BAPTA-1. Images were captured at 1.5 second intervals for 27 seconds and played at 9 frames per second. (PGCs in Movies S4-S6 all assayed between 9 and 11hpf)
Movie S5. Zebrafish injected with 50pg gng2-SaaX(nos) and 75 pg gng2-WT(nos) mRNA. PGCs migrating in vivo labeled with the free calcium indicator Oregon green 488 BAPTA-1. Images were captured at 1.5 second intervals for 27 seconds and played at 9 frames per second.
Movie S6. Zebrafish injected with 75 pg gng2-WT(nos) mRNA. PGCs migrating in vivo labeled with the free calcium indicator Oregon green 488 BAPTA-1. Images were captured at 1.5 second intervals for 27 seconds and played at 9 frames per second.