

Abstract
Water-soluble polymers for the delivery of chemotherapeutic drugs passively target solid tumors as a consequence of reduced renal clearance and the enhanced permeation and retention (EPR) effect. Elimination of the polymers in the kidney occurs due to filtration through biological nanopores with a hydrodynamic diameter comparable to the polymer. Therefore we have investigated chemical features that may broadly be grouped as “molecular architecture” such as: molecular weight, chain flexibility, number of chain ends and branching, to learn how they impact polymer elimination. In this report we describe the synthesis of four pairs of similar molecular weight cyclic and linear polyacrylic acid polymers grafted with polyethylene glycol (23, 32, 65, 114 kDa) with low polydispersities using ATRP and “click” chemistry. The polymers were radiolabeled with 125I and their pharmacokinetics and tissue distribution after intravenous injection were determined in normal and C26 adenocarcinoma tumored BALB/c mice. Cyclic polymers above the renal threshold of 30kDa had a significantly longer elimination time (between 10 to 33 % longer) than did the comparable linear polymer (for the 66 kDa cyclic polymer, t1/2, β= 35 ± 2 h) and a greater area under the serum concentration time curve. This resulted in a greater tumor accumulation of the cyclic polymer than the linear polymer counterpart. Thus water-soluble cyclic comb polymers join a growing list of polymer topologies that show greatly extended circulation times compared to their linear counterparts and provide alternative polymer architecture for use as drug carriers.
Keywords: ATRP, click chemistry, mice, radiolabel, tumor accumulation
Introduction
In an attempt to improve polymeric drug delivery the role of polymer topology has been examined on how polymers are handled by the body after parenteral administration. [1] Knowledge concerning the circulation time and tumor accumulation of polymers with various architectures will inform us if a particular architecture has advantages for the delivery of drugs to solid tumors via the enhanced permeability and retention (EPR) effect. [2, 3] Previously we investigated star polymers and showed they have a longer circulation time and higher accumulation in tumors than linear polymers of the same molecular weight, [4] even though star polymers have a considerably smaller volume than the linear counterpart. [5] Linear polymers pass though nanopores in the kidneys and are eliminated more easily than star polymers. This is because linear chains reptate through nanopores whereas star polymers are excluded by the multiple arms when the nanopore diameter is smaller than the polymer diameter. [3, 5]
We extended this interest to the potential use of cyclic polymers in drug delivery. We expected that cyclic comb polymers would show a longer circulation time and higher accumulation in tumors than comparable linear comb polymers when their molecular weights were above the threshold (> 30 kDa)[6] for elimination in the kidneys. The reasoning is similar to that applied in the star polymer example; two chain segments each with an attached comb segment would need to traverse the pores for cyclic polymers to be eliminated (Scheme 1).
Scheme 1.
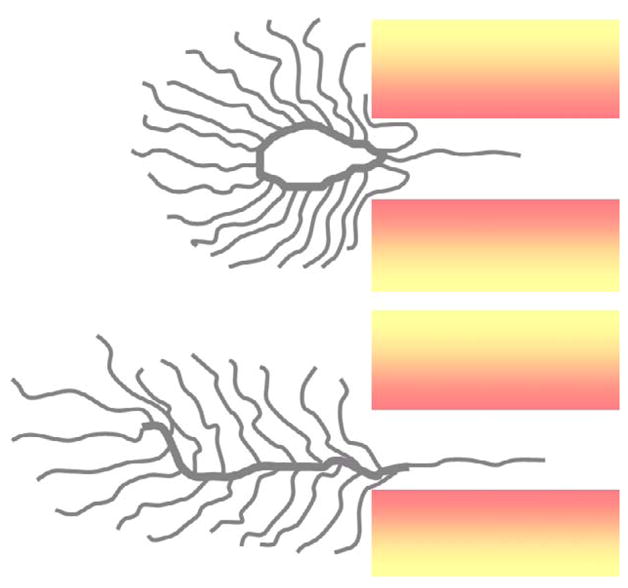
Schematic diagram depicting the PEGylated cyclic and linear polyacrylic acid comb structures with MWs larger than pores in the kidney. The hypothetical repation for the linear comb polymer through the pore is easier than for the cyclic comb polymer with same MWs.
Scientific curiosity concerning cyclic polymers began more than half a century ago with the discovery that double-stranded polyoma virus DNA exists as a closed circular macromolecule[7, 8] and the theoretical prediction that the cyclic polymers have different properties than linear polymers of the same molecular weight. [9] Synthetic routes to lower molecular weight cyclic molecules has allowed their properties to be explored by pharmacologists. [10–12] However, until recently, cyclic macromolecules have been difficult to synthesize. [13–17]
We reported that a biodegradable cyclic polymer had a longer elimination time and a greater area under the blood concentration versus time curve compared to a linear biodegradable polymer of the same molecular weight after intravenous injection. [6] We wanted to know if the observed pharmacokinetic difference was an intrinsic property of cyclic polymers or rather if it was due to the fact that we had studied biodegradable polymers; chain cleavage in a linear molecule results in two lower molecular weight polymers whereas a single chain cleavage in a cyclic polymer generates a linear chain with the same molecular weight as the initial cyclic polymer. The two lower molecular weight fragments would undergo renal elimination faster than would the intact linear polymer; hence hydrolysis and not the intrinsic physical properties related to the cyclic topology would account for the longer retention of the biodegradable cyclic polymer. [6]
In this study, we investigated the influence of the topology and MWs of more stable polymers on polymer circulation time and tumor uptake. It requires that cleavable sites on the cyclic and linear polymers be as few as possible so that the cyclic and linear polymers remain intact in the body. We synthesized cyclic poly(tert-butyl acrylate) (PtBA) using the method described by Grayson and coworkers (Figure 1). [18, 19] An advantage of the route is that each of the intermediates and the cyclic product can be isolated easily, yielding cyclic polymers with high conversion and narrow polydispersity. The MW and water solubility of polymers can be adjusted by conjugating polyethylene glycol amine to the carboxylic acid via an amide bond on the cyclic and linear poly(acrylic acid) (PAA) backbones after deprotection. A low number of tyramine (circa 1 per chain) was attached for labeling with radioactive iodine. We synthesized four pairs of cyclic and linear polymers with similar MWs and low polydispersities for each pair, radiolabeled them and determined the influence of polymer architecture and molecular weight on their pharmacokinetics and biodistribution. Using these more stable polymers we confirmed and extended the finding[6] that cyclic polymers with molecular weights above the renal elimination threshold have a longer circulation time in mice than do linear polymers of the same molecular weight.
Figure 1.

Synthesis of PEGylated cyclic and linear PAA comb polymers. (n ≈ 45, q ≈ 1, p+q+i=45) (i) CuBr, PMDETA, t-BA (ii)DMF, NaN3 (iii) CuBr, PMDETA, 120°C, 72h (iv) DCM, TFA (v) HATU, HOAT, mPEG amine, tyramine, DMF, 120°C, 48h
2. Materials and methods
2.1 Materials and instruments
Alumina, dichloro methylene (DCM) and dimethyl formide (DMF) with ACS grade were purchased from Fisher Scientific. Methoxypolyethylene glycol amine (mPEG amine) (5000) is bought from Laysan Bio Inc.. MPEG amine (2000) was synthesized as described previously. [20] All other chemicals of the highest grade available including solvents were purchased from Sigma-Aldrich and used without further purification. 1H NMR spectra were recorded on a Bruker Avance 300 MHz ultrashield NMR. NMR chemical shifts are reported in ppm and calibrated against solvent signals. MALDI-TOF MS data was collected on a PerSeptive Biosystems Voyager-DE instrument in positive ion mode using an iodoacetic acid (IAA) matrix and calibration against bovine insulin standards. Gel Permeation Chromatography (GPC) consisted of a Viscotek 1122 pump, a Viscotek 270 dual detector and a Viscotek 3500 differential refractive index (RI) detector. At 50°C GPC was performed at 1.0 mL/min in two Resipore (3 μm) columns (Polymer Laboratories, 300 × 7.5 mm) using THF as the mobile phase and poly(methyl methacrylate) (50kDa, Sigma-Aldrich) as the calibration standards.
2.2. Synthesis of APtBA-Br (2)
CuBr (334 mg, 2.34 mmol) and initiator 1 (Figure 1) (482 mg, 2.34 mmol) were added to a round bottom flask, which was subsequently evacuated for 15 min and back-filled with argon. Freshly distilled tert-butyl acrylate (15.0 g, 117 mmol) was added via a degassed syringe followed by degassed DMF (35 mL). The reaction flask was immediately submerged in liquid N2 for 5 minutes, then added PMDETA (391 mg) before evacuated for 15 min and backfilled with argon. When the mixture thawed completely, the flask was submerged in a 70ºC oil bath and stirred for 1 h. The reaction flask was frozen in liquid N2, then opened to air. The viscous, black mixture was diluted with DCM (120 mL). After thawing, the mixture was passed through a column of neutral alumina, concentrated on a rotary evaporator, precipitated in a 10:1 volume of 50-50 methanol-water three times, dissolved in diethyl ether, dried over MgSO4, filtered, concentrated on a rotary evaporator, and dried in vacuo for 1 d to yield APtBA-Br (4.30 g, Mn (NMR): 6,600 Da, PDI = 1.09 (GPC)) as a white solid. 1H NMR (300 MHz, CDCl3): δ 4.65 (s, 2 H), 4.11 (m, 1 H), 2.23 (br, 51 H), 1.81 (br, 27 H), 1.11-1.39 (b, 531 H)
2.3. Synthesis of APtBA-N3 (3)
Sodium azide (39 mg, 0.6 mmol) was added to a round-bottom flask containing compound PtBABr (0.2 g, 0.03 mmol) dissolved in DMF (20 ml). The reaction mixture was stirred at 50ºC for 1 d after which time it was allowed to cool to room temperature. The organics were concentrated on a rotary evaporator and precipitated into a 10:1 volume of 50-50 methanol-water. After decanting the methanol-water solution, the remaining solid was dissolved in diethyl ether, dried over MgSO4, filtered, concentrated on a rotary evaporator, and dried for 2 days in vacuo to yield APtBA-N3 (0.15 g) as a white solid. 1H NMR (300 MHz, CDCl3): δ 4.66 (s, 2 H), 3.65 (m, 1 H), 2.23 (br, 47 H), 1.83 (br, 25 H), 1.14–1.67 (br, 523 H).
2.4. Synthesis of cyclic PtBA (5)
To a 500 ml round bottomed flask was added CuBr 0.477 g (2.2 mmol), 1.03g 4, 4’-bipyridine (bpy) and 300 ml of DMF degassed by Ar. A separate flask containing 0.1 g (0.015 mmol) APtBA-N3 dissolved in 5 ml DMF was degassed through argon bubbling. This solution was then added to the CuBr/bpy reaction solution at 120°C via a syringe pump at a rate of 0.5 μl/min. Once the addition of polymer to the catalyst solution was complete, the reaction was allowed to proceed at 120°C for an additional 5 h. The reactions were concentrated on a rotary evaporator and precipitated into a 10:1 volume of 50-50 methanol-water. After decanting the methanol-water solution, the remaining solid was dissolved in diethyl ether, dried over MgSO4, filtered, concentrated on a rotary evaporator, and dried for 2 days in vacuo to give a white solid.1H NMR (300 MHz, CDCl3): δ 7.74 (d, 1 H), 5.19 (s, 2 H), 2.24 (br, 52 H), 1.84 (br, 26 H), 1.14–1.67 (br, 535 H)
1HNMR and GPC results of cyclic and linear PtBA are listed in Table 1.
Table 1.
1H NMR and GPC results of cyclic and linear PtBA.
| polymer | Mn (kDa) | Mn (kDa) | Mw (kDa) | PDI |
|---|---|---|---|---|
| (NMR) | (GPC) | (GPC) | (Mw/Mn) | |
| linear PtBA | 6.6 | 9.9 | 10.8 | 1.09 |
| cyclic PtBA | 6.8 | 7.2 | 8.1 | 1.13 |
2.5. Synthesis of polyacrylic acid
Into 1 mL of dichloromethane solution of PtBA (100 mg), trifluoroacetic acid (0.5 ml) was slowly added at 0°C with vigorous stirring. The reaction was kept at 0°C for 3 h and then overnight at room temperature. The reaction mixture was carefully evaporated and washed with ether. The product was dried in vacuo for overnight.
2.6. Synthesis of PEGylated polyacrylic acid
2-(1H-7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium (HATU, 0.4 g) and 1-Hydroxy-7-Azabenzotriazole (HOAT, 0.124 g) were added to a flask containing cyclic or linear polyacrylic acid (16 mg, 0.224 mmol of acid groups) in 1.6 ml of anhydrous DMF under argon. The mixture was stirred for 2 h, followed by the addition of N, N-diisopropylethylamine (DiPEA, 0.2 ml), tyramine (0.63 mg) and methoxypolyethylene glycol amine (mPEGA). The PEGylated linear polymers were dialyzed against water to remove the free PEG chains while the PEGylated cyclic polymers were dialyzed against methanol to prevent hydrolysis of the ester bond on the cyclic backbones. [6] A Spectra/Por Cellulose Ester Membrane with a MW cutoff of 100 kDa from Spectrum Laboratories was used for all samples. Solvents were changed every 12 h and dialyses were stopped after 72 h. The suitable amount of mPEGA added in the reaction and the results of polymer MWs and polydispersity characterized by 1HNMR and GPC are listed in Table 2.
Table 2.
Synthesis conditions and 1H NMR and GPC results of PEGylated polyacrylic acid.
| samples | PAA | PEG | molar ratio | Mn (kDa) | Mn (kDa) | Mw (kDa) | PDI |
|---|---|---|---|---|---|---|---|
| (kDa) | (acid/PEG) | by NMR | by GPC | GPC | |||
| L22k | linear | 2 | 5 | 16.2 | 22.1 | 26.1 | 1.18 |
| C23k | cyclic | 2 | 5 | 18.1 | 23.4 | 27.4 | 1.17 |
| L32k | linear | 2 | 3 | 26.2 | 34.2 | 37.4 | 1.09 |
| C32k | cyclic | 2 | 3 | 27.4 | 32.1 | 34.5 | 1.08 |
| L65k | linear | 2 | 0.91 | 43.4 | 65.2 | 67.2 | 1.03 |
| L66k | cyclic | 2 | 0.91 | 44.5 | 65.7 | 67.7 | 1.03 |
| L114k | linear | 5 | 2 | 76.4 | 114.2 | 117.6 | 1.03 |
| L115k | cyclic | 5 | 2 | 77.0 | 115.2 | 118.4 | 1.03 |
2.7. Animal experiments
All animal experiments were performed in compliance with the National Institutes of Health guidelines for animal research under a protocol approved by the Committee on Animal Research at University of California (San Francisco, CA) (UCSF).
2.8. Biodistribution studies
To track the fate of the polymers in table 2 in vivo, the phenolic groups were radiolabeled with 125I using the chloramine T method as previously described. [21] Polymer solutions (200 μL) at a dose of approximately 50 mg/kg were administered intravenously via the tail vein to 6–8 week old BALB/c female mice. The mice were sacrificed at seven different time points (three mice per group) ranging from 5 min to 48 h postinjection for biodistribution analyses. The blood (collected by heart puncture), heart, lungs, liver, stomach, spleen, intestines, kidney, body, tail and head were weighed, and the amount of radioactivity present in each organ was quantified. Periodically, blood was also collected from the retroorbital sinus at intermediate time points to determine the dose of polymer in the blood. For the 24 h time points, mice were housed in metabolic cages to allow for the collection of urine and feces. The % injected dose per gram (% ID/g) of blood versus time curve was analyzed using a two-compartment model. [22]
2.9. Biodistribution studies in tumored mice
Female BALB/c mice were injected with C26 colon carcinoma cells on the right rear flank by subcutaneous administration. Twelve days after tumor inoculation, the mice (three per group) were injected intravenously via the tail vein with 200 μL of radiolabeled polymer solution prepared as described above. The mice were sacrificed at 48 h postinjection injected with ~115 and 32 kDa polymers and at 24, 48, 72 and 168 h with ~66 kDa polymers. Their blood, liver, muscle and tumor were collected and weighed, and the radioactivity in each was quantified. Distribution of radioactivity in organ was not corrected for the blood remaining in the organ.
3. Results and discussion
3.1. Cyclic polymer synthesis
To obtain meaningful and reproducible pharmacokinetic data of polymers, it is critical to produce the water-soluble cyclic polymers with narrow polydispersity (<1.2). [4] Only a few methods can prepare cyclic polymers with this criterion though various ways have explored the synthesis of cyclic polymers. [17, 23–29] Most of the cyclic polymers with narrow polydispersities were prepared by controlled living polymerizations. For example, cyclic [styrene (S)-b-isoprene (I)-b-methyl methacrylate (MMA)] with narrow polydispersity was synthesized under high-dilution conditions by the end-to-end intramolecular amidation reaction of the corresponding linear-α, ω-amino acid precursor [S-b-I-b-MMA] prepared by anionic polymerization. In this case, tedious purifications are necessary to isolate cyclic polymers from acyclic impurities due to the low efficiency of the DCC coupling. [30] Grayson and coworkers have synthesized well-defined cyclic homopolymers and block copolymers by combining atomic transfer radical polymerization (ATRP) with “click chemistry” to cyclize α, ω-difunctional linear precursors with a high diluted concentration. [18, 19] The conversion of cyclization by “click chemistry” is near-quantitative so time-consuming purification was circumvented. This approach can be used to prepare a broad diversity of cyclic polymers with a range of monomer functionalities. Recently, Waymouth and colleagues have successfully developed a fast and highly efficient zwitterionic strategy for the controlled synthesis of high-molecular-weight cyclic polylactide[31] and poly(β-butylactone)[32] with narrow polydispersity even at relatively high monomer concentrations. However, further modifications are needed to make these cyclic polymers water-soluble to be used for drug delivery. The approach taken in this work is one way to prepare water-soluble cyclic polymers.
In this study, poly(tert-Butyl acylate) (PtBA) was employed as cyclic and linear backbone because carboxylic acid can be provided for further PEGylation for MW tuning by removing the t-butyloxy protection and the bromide at the end of linear polyacrylate chain is readily to be substituted by azide compared to polymethacrylate (Figure 1). [33] PtBA was prepared by atomic transfer radical polymerization (ATRP) using a previously reported alkyne initiator, [34] 1, catalyzed by Cu(I)Br with PMDETA ligand at 60°C under the protection of argon after three freeze-pump-thaw cycles to remove oxygen.(Figure 1) The resulting alkyne terminated PtBA (APtBA-Br) homopolymer has a narrow distribution (PDI < 1.09) with molecular weight of ~6.6 kDa (Table 1). The hydrogen a close to the bromide with chemical shift of 4.11 ppm was completely replaced by hydrogen c with chemical shift of 3.65 ppm (Figure 2A) due to the substitution of bromide by azide as reported by Johnson et al. [35] Thus, as we expected, the acrylate bromide end group of APtBA-Br was stoichiometrically transformed to the azide of A-PtBA-N3 using sodium azide as a nucleophile.
Figure 2.

Characterization of cyclic and linear polymers. (A) 1HNMR spectra of linear APtBA-Br (2), linear APtBA-N3 (3) and cyclic PtBA (5). The hydrogens a – e and numbers 2, 3 and 5 are corresponding to the hydrogens and polymers in scheme 1, respectively. (B) The retention volume of linear (3, black) and cyclic (5, green) PtBA measured by GPC in THF. Numbers 2, 3 and 5 are corresponding to the polymers in Figure 1. (C) Linear polyacrylic acid (APAA-N3) and cyclic PAA characterized by matrix assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF MS)
The desired cyclic polymers were efficiently produced by the Huisgen cycloaddition known as “click chemistry” using a syringe pump for continuous addition of α, ω-functionalized linear polymer precursor, APtBA-N3 [19] From 1H NMR spectroscopy (Figure 2A), the new hydrogens d and e (Figure 1) with chemical shifts of 7.7 and 5.19 ppm replaced the hydrogens b and c with chemical shifts of 3.65 and 4.66 ppm, respectively. This means that cycloaddition is complete. The MW of the cyclized polymer is almost identical to APtBA-N3 measured 1HNMR (Table 1). The longer elution time of cyclic polymer and narrow polydispersity (PDI < 1.13) observed by GPC (Figure 2B) indicates intramolecular cyclization has occurred. This is because the hydrodynamic volume of cyclic polymers is smaller compared to linear polymers, consequently, [19, 36, 37] the MWs of the cyclic polymers measured by GPC are smaller than the linear counterparts.(Table 1).
The linear polyacrylic acid (PAA), 4 and cyclic PAA, 6 formed after deprotection of APtBA-N3, 2 and cyclic PtBA, 3, respectively, have the MWs of 3640 and 3590 Da measured by matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF MS). We are unable to obtain mass spectroscopy of PtBA 2 and 3. The MWs of 4 and 6 are consistent with the MWs of the cyclic and linear PtBA by NMR (Figure 2). This strongly supports the assignment of an intramolecular cyclization instead of an intermolecular condensation in the reaction.
3.2. PEGylation of cyclic and linear polymers
The polyacrylic acid (PAA) backbone deprotected from PtBA was further modified by conjugation of mPEG amine (Figure 1) to increase water solubility and increase the MW. The grafting density of PEG chains with MW of 2 and 5 kDa on the linear and cyclic PAA backbones can be tuned by changing the molar ratio between PEG chains and backbones, so that the MWs of polymers can be adjusted above and below the elimination threshold (~30 – 40 kDa) of the kidney. HATU was employed as coupling agent since DCC and EDCI were not efficient enough to achieve high MWs. Approximately one tyramine per polymer molecule were also attached before polymer PEGylation for iodination.
Four pairs of PEGylated cyclic and linear polymers were prepared with MWs from ~20 to 115 kDa.(Table 2) Each pair have very similar MWs and narrow polydispersities (PDI < 1.2), which are essential to obtain reproducible pharmacokinetics and ensure that any difference of pharmacokinetics and biodistribution between cyclic and linear polymers is caused by the difference in polymer architecture.
3.3. Biodistribution studies of radiolabeled polymer
The 22 kDa linear and 23 kDa cyclic polymers showed very rapid elimination because the MWs of both polymers are below the nominal MW cutoff (30–40 kD) of the renal filtration of linear PEG (Table 3, Figure 3). [38] The t1/2,β and AUC0-∞ are almost identical for the cyclic and linear polymer but it is interesting that the cyclic 23kDa polymer is eliminated slightly faster than the linear 22kDa. This is consistent with our previous findings on the cyclic polycaprolactone where, at the lowest MW, the cyclic polymer is eliminated faster than the linear polymer [6].
Table 3.
Biodistribution study of PEGylated polyacrylic acid.
| polymers | t1/2, α (h) | t1/2, β (h) | AUC0→∞ (%ID.h/g) | Vd (g blood) |
|---|---|---|---|---|
| C115k | 0.84±0.23 | 43.9±2.2 | 3210±310 | 1.61±0.17 |
| L114k | 0.75±0.11 | 38.9±1.8 | 2920±220 | 1.62±0.09 |
| C66k | 0.71±0.13 | 35.4±1.9 | 2710±140 | 1.68±0.15 |
| L65k | 0.55±0.20 | 25.0±1.4 | 1520±120 | 1.87±0.13 |
| C32k | 0.47±0.04 | 29.6±0.7 | 1660±90 | 1.73±0.08 |
| L32k | 0.29±0.17 | 22.3±2.3 | 1260±170 | 1.80±0.13 |
| C23k | 0.15±0.06 | 16.6±1.1 | 690±130 | 1.79±0.07 |
| L22k | 0.30±0.08 | 17.5±1.4 | 700±110 | 1.88±0.11 |
Figure 3.

Blood circulation profile of radiolabeled cyclic and linear polymers given as the average percent injected dose of polymer in the blood over time (n = 3 mice per time point).
All of the polymers ranging in MW from 32 to 115 kDa had long plasma circulation time (> 20 h) in blood (Table 3). As expected, the greater the molecular weight of the cyclic and linear polymers, the longer the blood retention time. As the number of the PEG chains (2 kDa) was increased from around 10, 15 to 31, the MWs of the linear polymers increased from 22.1 (L22k), 32.3 (L32k) to 65.2 kDa (L65k) and the elimination half lives (t 1/2,β) increases from 17.5, 22.3 to 25 h and the areas under the % dose/g versus time curve (AUC0→ ∞) increases from 700, 1260 to 1520 %ID.h/g, respectively. Similar to the linear polymers, as the number of the PEG chains (2 kDa) increased from 10, 15 to 31, the MWs increase from 23.4 (C23k), 32.1 (C32k) to 65.7 kDa (C66k), the t 1/2,β of cyclic polymers increases from 16.6, 29.6 to 35.4 h and AUC0→ ∞ increases from 690, 1660 to 2710 %ID.h/g, respectively.(Table 3) The cyclic polymer C115k and linear polymer L114k with ~22 PEG chains (5 kDa) have the longest t 1/2,β and the largest values of AUC0→ ∞ in the cyclic polymers and linear polymers, respectively. The results confirm the important influence of PEG number, MW and architecture on polymer circulation time in the body.
Similar to the results of cyclic and linear PEGylated poly(ε-caprolactone)[6], cyclic polymers with molecular size greater than the renal filtration threshold (>30 kDa) have longer plasma circulation times and larger values of AUC0→ ∞ than linear polymers of similar mass (Figure 4). Linear polymers can reptate through the nanopores in the glomeruli more easily than cyclic polymers (Scheme 1) and we always found more radioactivity from linear polymers in the urine (Figure 5B). The largest difference of t1/2,β and AUC0→ ∞ occurs in the 66 kDa polymers (Figure 4). For the 114 kDa polymer pair, the difference in elimination time is less than the 66 kDa polymer pair. The long circulation time (38.9±1.8 h) of 114 kDa linear polymer shows that it is difficult for linear polymers to pass through the nanopores in the kidney when the polymer molecular weight is high enough. High molecular weight diminishes the significance of polymer architecture in determining the elimination time. Also as the cyclic polymer circulates, the ester linkage hydrolyzes. Over time the cyclic polymer is converted into a linear polymer. Thus the half-lives for high MW cyclic and linear polymers will start to converge.
Figure 4.

T 1/2,β (A) and AUC0→ ∞ (B) of cyclic and linear polymers as function of molecular weight. (n = 3 mice per time point)
Figure 5.

Polymer amount in organs at 24 h postinjection (A) as the percent of injected dose per gram tissue (B) as percent of the injected dose per organ. (n = 3 mice per time point)
No specific tissue accumulation was observed for all the polymers with respect to other organs (Figure 5). The cyclic and linear PEGylated PAA comb polymers with around 15 PEG (2000 Da) chains and 32 kDa MW have larger values of t1/2,β and AUC0→ ∞ than the 50 kDa cyclic and linear poly(ε-caprolactone) with ~20 PEG chains (2000 Da) probably because PEG chains are packed more tightly on the non-hydrolysable PAA backbones. [6] The 32 kDa PEGylated PAA comb polymers have similar t1/2,β to 34.1 kDa PEGylated poly-L-lysine dendrimers with 16 PEG (2000 Da) chains[39] but smaller t 1/2,β than PEGylated triazine dendrimer (30 kDa) with around 13 PEG (2000) chains. [40] The difference in t1/2,β between the PEGylated PAA polymers and PEGylated triazine dendrimer could arise if PEGylated triazine dendrimers are self-associated so have a greater effective hydrodynamic diameter.
Polymers biodistributions were also evaluated in a C26 tumor bearing animal model in which the tumor was implanted on the rear flank. Cyclic polymers with the MWs of 115, 67 and 32 kDa and linear polymers with the MWs of 114, 66 and 32 kDa were selected to span a range of circulation life-times. Compared to the results in healthy BALB/c mice, the concentrations of polymers in blood of tumor bearing mice for the three pairs of polymers were lower after 48h (Figure 6). The concentrations of these polymers in the liver were maintained at the same level, circa 10 %ID/g, after 48h and are not related to the polymer concentrations in the blood. The lower blood levels of the polymers in the tumored animals compared to the normal mice are probably caused by the retention of polymers in the tumors due to the EPR effect.
Figure 6.

Polymer concentration as the percent injected dose per gram of tissue in tumor bearing mice at 48 h. (n = 3 mice per time point)
3.4. Biodistribution studies in tumor-bearing mice
High concentrations of the polymers accumulated in tumors and the amount increased as the polymer MWs increased (Figure 6). For cyclic polymers, the polymer concentrations in the tumors increased to 45.9 %ID/g as MWs increased to 115.2 kDa. The values for the linear polymers also increased, however, at 48 h, the tumor accumulation levels of cyclic polymers are higher than that of linear polymers with the same MWs. The greatest difference occurs between the 66 kDa cyclic and 65 kDa linear polymer which corresponds to the polymer pair that had the greatest difference in circulation time (Figure 4).
The tumor uptake of cyclic 66 kDa and linear 65 kDa polymers over a seven day period was quantified in the C26 tumor bearing animals. The mice were sacrificed at 24, 48, 72 and 168 h and the polymer concentrations were measured in blood, liver, muscle and tumor (Figure 7). Both polymers are gradually eliminated from the blood and the polymer blood concentration decreased from 24 to 168 h. The polymer concentrations in liver are maintained at the same level over this period. This suggests that either there is an initial uptake of polymer in the early times that is not metabolized or polymer that is eliminated form the liver is replenished by polymer eliminated from the blood into the liver. This confirms that the polymers and the associated label are stable in the biophase for an extended time and provides an assurance that the difference in the elimination rate between the cyclic and linear polymers of similar MW is due to topology and not to properties of the label.
Figure 7.

Polymer concentration variations for 66 kDa cyclic and 65 kDa linear polymers as the average percent injected dose in per gram of tumor. (n = 3 mice per time point)
From 24 to 48 h, the tumor accumulation of both polymers exhibits an increase, then a continuous decrease from 48 to 168h to a similar value 18.5 %ID/g (Figure 7). The level of tumor uptake for cyclic 66 kDa polymer is higher than the uptake of linear 65 kDa during over the time (Figure 7), which corresponds to the longer circulation time and higher AUC of cyclic polymers compared to their linear counterparts in non-tumored mice (Figure 3, Table 3) The highest uptake difference, ~ 9.9 %ID/g occurs at 72 h. The high tumor uptake (~20 %ID/g) for both polymers is maintained for a week, which suggests a strong potential to deliver a drug to the tumor sites.
The finding that stable cyclic water-soluble polymers above the renal threshold are eliminated less rapidly than are their linear counterparts confirms our previous report using biodegradable cyclic polymers. [6] Although we now know that cyclic polymer architecture significantly influences polymer circulation time, we have yet to examine if polymer architecture influences tumor cell uptake of the polymer. Such studies will help to identify those aspects of polymer structure that can be modified to further improve polymer drug delivery to tumors.
Conclusion
We have synthesized four pairs of cyclic and linear PEGylated polyacrylic acid comb polymers. Each pair of polymers has the same MWs with narrow polydispersity. Cyclic polymers showed longer circulation times and higher tumor uptake than do their linear polymer counterparts. The polymer pair with MWs of ~66 kDa showed the largest difference in circulation time and tumor uptake between the cyclic and linear versions of the four pairs of polymers at 48 h. The study confirms[6] that cyclic polymer topology significantly increases circulation time and tumor uptake among polymers of similar MW.
Acknowledgments
This work was supported by NIH grant R01-EB 002047. We thank Ms. Nicole Macaraeg for excellent technical assistance in the animal experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Uzgiris E. The role of molecular conformation on tumor uptake of polymeric contrast agents. Investigative radiology. 2004;39(3):131–137. doi: 10.1097/01.rli.0000107495.48025.97. [DOI] [PubMed] [Google Scholar]
- 2.Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clinical pharmacokinetics. 2003;42(13):1089–1105. doi: 10.2165/00003088-200342130-00002. [DOI] [PubMed] [Google Scholar]
- 3.Lee CC, MacKay JA, Frechet JM, Szoka FC. Designing dendrimers for biological applications. Nature biotechnology. 2005;23(12):1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 4.Gillies ER, Dy E, Frechet JM, Szoka FC. Biological evaluation of polyester dendrimer: poly(ethylene oxide) "bow-tie" hybrids with tunable molecular weight and architecture. Molecular pharmaceutics. 2005;2(2):129–138. doi: 10.1021/mp049886u. [DOI] [PubMed] [Google Scholar]
- 5.de Gennes PG. Polymers in Confined Environments. Vol. 138. 1999. pp. 91–105. [Google Scholar]
- 6.Nasongkla N, Chen B, Macaraeg N, Fox ME, Frechet JM, Szoka FC. Dependence of Pharmacokinetics and Biodistribution on Polymer Architecture: Effect of Cyclic versus Linear Polymers. Journal of the American Chemical Society. 2009;131(11):3842–3843. doi: 10.1021/ja900062u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weil R, Vinograd J. The Cyclic Helix and Cyclic Coil Forms of Polyoma Viral DNA. Proceedings of the National Academy of Sciences of the United States of America. 50(1963):730–738. doi: 10.1073/pnas.50.4.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dulbecco R, Vogt M. Evidence for a Ring Structure of Polyoma Virus DNA. Proceedings of the National Academy of Sciences of the United States of America. 50(1963):236–243. doi: 10.1073/pnas.50.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloomfield V, Zimm BH. Viscosity Sedimentation, et cetera, of Ring- and Straight-Chain Polymers in Dilute Solution. Journal of Chemical Physics. 1966;44(1):315. [Google Scholar]
- 10.Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nature reviews. 2004;3(12):1023–1035. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- 11.Cody WL, Mahoney M, Knittel JJ, Hruby VJ, Castrucci AMD, Hadley ME. Cyclic Melanotropins.9. 7-D-Phenylalanine Analogs of the Active-Site Sequence. J Med Chem. 1985;28(5):583–588. doi: 10.1021/jm50001a008. [DOI] [PubMed] [Google Scholar]
- 12.Craik DJ. Chemistry. Seamless proteins tie up their loose ends. Science. 2006;311(5767):1563–1564. doi: 10.1126/science.1125248. [DOI] [PubMed] [Google Scholar]
- 13.Oike H. Supramolecular approach for synthesis and functionalization of cyclic macromolecules. Reactive & functional polymers. 67(2007):1157–1167. [Google Scholar]
- 14.Schappacher M, Deffieux A. Synthesis of macrocyclic copolymer brushes and their self-assembly into supramolecular tubes. Science. 2008;319(5869):1512–1515. doi: 10.1126/science.1153848. [DOI] [PubMed] [Google Scholar]
- 15.Tam JP, Lu YA, Yu QT. Thia zip reaction for synthesis of large cyclic peptides: Mechanisms and applications. Journal of the American Chemical Society. 1999;121(18):4316–4324. [Google Scholar]
- 16.Bielawski CW, Benitez D, Grubbs RH. An "endless" route to cyclic polymers. Science. 2002;297(5589):2041–2044. doi: 10.1126/science.1075401. [DOI] [PubMed] [Google Scholar]
- 17.Li HY, Debuigne A, Jerome R, Lecomte P. Synthesis of macrocyclic poly(epsilon-caprolactone) by intramolecular cross-linking of unsaturated end groups of chains precyclic by the initiation. Angewandte Chemie-International Edition. 2006;45(14):2264–2267. doi: 10.1002/anie.200503961. [DOI] [PubMed] [Google Scholar]
- 18.Eugene DM, Grayson SM. Efficient preparation of cyclic poly(methyl acrylate)-block-poly(styrene) by combination of atom transfer radical polymerization and click cyclization. Macromolecules. 2008;41(14):5082–5084. [Google Scholar]
- 19.Laurent BA, Grayson SM. An efficient route to well-defined macrocyclic polymers via "Click" cyclization. Journal of the American Chemical Society. 2006;128(13):4238–4239. doi: 10.1021/ja0585836. [DOI] [PubMed] [Google Scholar]
- 20.Harris J Milton, Struck Evelyn C, Case Martha G, Paley M Steven, Yalpani Manssur, Van Alstine James M, Brooks DE. Synthesis and characterization of poly(ethylene glycol) derivatives. Journal of Polymer Science: Polymer Chemistry Edition. 1983;22(2):12. [Google Scholar]
- 21.Larwood DJ, Szoka FC. Synthesis, Characterization, and In vivo Disposition of Iodinatable Polyethylene-Glycol Derivatives - Differences Invivo as a Function of Chain-Length. Journal of Labelled Compounds & Radiopharmaceuticals. 1984;21(7):603–614. [Google Scholar]
- 22.Welling PG. Pharmacokinetics. Americal Chemical Society; Washington: 1986. [Google Scholar]
- 23.Hadjichristidis N, Pitsikalis M, Pispas S, Iatrou H. Polymers with complex architecture by living anionic polymerization. Chemical reviews. 2001;101(12):3747–3792. doi: 10.1021/cr9901337. [DOI] [PubMed] [Google Scholar]
- 24.Pang XC, Wang GW, Jia ZF, Liu C, Huang JL. Preparation of the amphiphilic macro-rings of poly(ethylene oxide) with multi-polystyrene lateral chains and their extraction for dyes. Journal of Polymer Science Part a-Polymer Chemistry. 2007;45(24):5824–5837. [Google Scholar]
- 25.Takano A, Nonaka A, Kadoi O, Hirahara K, Kawahara S, Isono Y, Torikai N, Matsushita Y. Preparation and characterization of cyclic polystyrene with short poly(2-tert-blutylbutadiene) sequences. Journal of Polymer Science Part B-Polymer Physics. 2002;40(15):1582–1589. [Google Scholar]
- 26.Ben-Haida A, Colquhoun HM, Hodge P, Williams DJ. Cyclic oligomers of poly(ether ketone) (PEK): synthesis, extraction from polymer, fractionation, and characterisation of the cyclic trimer, tetramer and pentamer. Journal of Materials Chemistry. 2000;10(9):2011–2016. [Google Scholar]
- 27.Boydston AJ, Xia Y, Kornfield JA, Gorodetskaya IA, Grubbs RH. Cyclic ruthenium-alkylidene catalysts for ring-expansion metathesis polymerization. Journal of the American Chemical Society. 2008;130(38):12775–12782. doi: 10.1021/ja8037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou HY, Leung KCF, Lanari D, Nelson A, Stoddart JF, Grubbs RH. Template-directed onestep synthesis of cyclic trimers by ADMET. Journal of the American Chemical Society. 2006;128(48):15358–15359. doi: 10.1021/ja065572j. [DOI] [PubMed] [Google Scholar]
- 29.Blackwell HE, Sadowsky JD, Howard RJ, Sampson JN, Chao JA, Steinmetz WE, O'Leary DJ, Grubbs RH. Ring-closing metathesis of olefinic peptides: Design, synthesis, and structural characterization of macrocyclic helical peptides. Journal of Organic Chemistry. 2001;66(16):5291–5302. doi: 10.1021/jo015533k. [DOI] [PubMed] [Google Scholar]
- 30.Pantazis D, Schulz DN, Hadjichristidis N. Synthesis of a model cyclic triblock terpolymer of styrene, isoprene, and methyl methacrylate. Journal of Polymer Science Part a-Polymer Chemistry. 2002;40(10):1476–1483. [Google Scholar]
- 31.Culkin DA, Jeong W, Csihony S, Gomez ED, Balsara NP, Hedrick JL, Waymouth RM. Zwitterionic polymerization of lactide to cyclic poly(lactide) by using N-heterocyclic carbene organocatalysts. Angewandte Chemie (International ed. 2007;46(15):2627–2630. doi: 10.1002/anie.200604740. [DOI] [PubMed] [Google Scholar]
- 32.Jeong W, Hedrick JL, Waymouth RM. Organic spirocyclic initiators for the ring-expansion polymerization of beta-lactones. Journal of the American Chemical Society. 2007;129(27):8414–8415. doi: 10.1021/ja072037q. [DOI] [PubMed] [Google Scholar]
- 33.Coessens V, Pintauer T, Matyjaszewski K. Functional polymers by atom transfer radical polymerization. Progress in Polymer Science. 2001;26(3):337–377. [Google Scholar]
- 34.Tsarevsky NV, Sumerlin BS, Matyjaszewski K. Step-growth "click" coupling of telechelic polymers prepared by atom transfer radical polymerization. Macromolecules. 2005;38(9):3558–3561. [Google Scholar]
- 35.Johnson JA, Lewis DR, Az D, Finn MG, Koberstein JT, Turro NJ. Synthesis of degradable model networks via ATRP and click chemistry. Journal of the American Chemical Society. 2006;128(20):6564–6565. doi: 10.1021/ja0612910. [DOI] [PubMed] [Google Scholar]
- 36.Grayson SM, Godbey WT. The role of macromolecular architecture in passively targeted polymeric carriers for drug and gene delivery. Journal of drug targeting. 2008;16(5):329–356. doi: 10.1080/10611860801969616. [DOI] [PubMed] [Google Scholar]
- 37.Ohta Y, Masuoka K, Takano A, Matsushita Y. Chain dimension of cyclic polymers in solutions. 2006. pp. 532–534. [Google Scholar]
- 38.Yamaoka T, Tabata Y, Ikada Y. Distribution and Tissue Uptake of Poly(Ethylene Glycol) with Different Molecular-Weights after Intravenous Administration to Mice. Journal of Pharmaceutical Sciences. 1994;83(4):601–606. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- 39.Kaminskas LM, Boyd BJ, Karellas P, Krippner GY, Lessene R, Kelly B, Porter CJ. The impact of molecular weight and PEG chain length on the systemic pharmacokinetics of PEGylated poly l-lysine dendrimers. Molecular pharmaceutics. 2008;5(3):449–463. doi: 10.1021/mp7001208. [DOI] [PubMed] [Google Scholar]
- 40.Lim J, Guo Y, Rostollan CL, Stanfield J, Hsieh JT, Sun X, Simanek EE. The role of the size and number of polyethylene glycol chains in the biodistribution and tumor localization of triazine dendrimers. Molecular pharmaceutics. 2008;5(4):540–547. doi: 10.1021/mp8000292. [DOI] [PubMed] [Google Scholar]