

Abstract
A method for the oxidative alkylation of ketones through intramolecular allyl-group transfer within preformed allyldimethylsilyl enol ethers is reported. A number of examples are detailed, including a study into the effects of resident sterocenters within cyclic enol ethers.
Ceric ammonium nitrate (CAN) is a powerful single electron oxidant that has significant demonstrated utility in promoting a wide range of useful synthetic transformations.1 Our interest in CAN as a reagent stemmed from an initial desire to elaborate the synthetic potential of silyl bis-enol ethers, an area we have been actively developing.2 While the 1,4-diketone adducts derived from silyl bis-enol ether oxidation are of significant value for synthesis, we wished to extend this general reactivity to other species and herein report the development of an intramolecular oxidative allylation from allylsilyl enol ethers.
We were drawn to the work published by the groups of Hwu3 and Flowers,4 who have each shown that 1,3-dicarbonyl species undergo effective oxidative allylation with allyltrimethylsilane in the presence of CAN.5,6 The scope of these transformations is limited to enolizable 1,3-dicarbonyls since the first step of the reaction pathway is most likely oxidation of the enol tautomer to an intermediate radical cation. We speculated that exposure of an allyldimethylsilyl enol ether (i.e., 1) to CAN might induce intramolecular bond formation via radical-cation 2, which would collapse to produce cyclic intermediate 3 (Figure 1). Si–C and Si–O bond cleavage, along with a second single electron oxidation would then produce the desired adduct 4.7 Related examples of intermolecular allylation of β-carbonyl imines,8 and one example of a vinylogous amide9 have been reported. Moeller and coworkers have shown that intramolecular allylation of enol ethers with allylsilanes is possible when the silane is part of the ketone (i.e., linked through the carbon backbone rather than the enol oxygen such as in our system).10 MacMillan and coworkers have elegantly demonstrated the power of their SOMO activation strategy by developing an enantioselective allylation of aldehydes using organocatalysis.11 Against this background, the method for intramolecular allylation of unactivated ketones (i.e., not 1,3 dicarbonyls) that we report herein expands the scope of compounds that may be accessed through oxidative coupling.
Figure 1.

Proposed Intramolecular Oxidative Allylation.
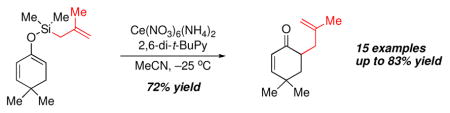
We began our study by preparing the allyldimethylsilyl ether derived from tetralone (i.e., 5a, R = H), since such ketones had proved exceptional in our prior work on silyl bis-enol ether oxidative coupling. Preparation of the substrate was trivial and involved the addition of commercially available chloro allyldimethylsilane to the lithium enolate of tetralone.12 Exposure of 5a (R = H) to the standard set of conditions known to promote oxidative coupling of enol silanes led to formation of the desired material in 51% yield (Table 1, entry 1). Conducting the reaction at a lower temperature (−30 °C) increased the yield to 57% (Table 1, entry 2), while use of 2,6-di-t-butylpyridine (DTBP) afforded a 46% yield of 6a (R = H). The addition of a base is required in these reactions in order to minimize decomposition of the acid-sensitive enol ether; exposure of 5a (R = H) to CAN in the absense of a base gave exclusively tetralone (Table 1, entry 4). We investigated the use of other oxidants known to promote enol silane coupling, such as Ag2O and Cu(OTf)2/Cu2O, but these provided none of the desired product. At this juncture, we prepared the allyldimethylsilyl enol ethers of several other ketones, but were disappointed to obtain generally low yields. We speculated that the general lack of scope might be due to the poor nucleophilicity of the allyl group. If intramolecular C–C bond formation from the initial radical-cation (akin to 2 → 3 in Figure 1) is slow, then unproductive Si–O bond cleavage may compete and lead to diminished yields. Allylsilanes possessing electron-releasing substituents at C2 are known to be more nucleophilic than unsubstituted systems,13 so we investigated the methallylsilyl enol ether 5b (R = Me).11 Exposure of 5b to CAN/NaHCO3 at −30 °C provided only 39% yield of 6b (Table 1, entry 5), but when conducted at −40 °C gave an enhanced 52% yield (Table 1, entry 6). Unlike for the simple allyl system (i.e., 5a), replacing NaHCO3 with 2,6-di-t-butylpyridine led to a further improvement in yield to 83% (Table 1, entry 7). Under these conditions the benzyloxy substituted silyl ether 5c (R = CH2OBn)9 gave 62% yield of the desired allylated ketone 6c (R = CH2OBn), indicating that functionalized allyl fragments may be employed with this chemistry.
Table 1.
Initial Development of the Oxidative Allylationa
 | ||||
|---|---|---|---|---|
| entry | R | base | temp (°C) | yield (%)b |
| 1 | H (5a) | NaHCO3 | 0 | 51 |
| 2 | H (5a) | NaHCO3 | −30 | 57 |
| 3 | H (5a) | 2,6-di-t-BuPy | −30 | 46 |
| 4 | H (5a) | none | −30 | 0 |
| 5 | Me (5b) | NaHCO3 | −30 | 47 |
| 6 | Me (5b) | NaHCO3 | −40 | 52 |
| 7 | Me (5b) | 2,6-di-t-BuPy | −40 | 83 |
| 8 | CH2OBn (5c) | 2,6-di-t-BuPy | −40 | 62 |
1.0 mmol 5, 0.1 M MeCN.
Isolated yield after chromatography.
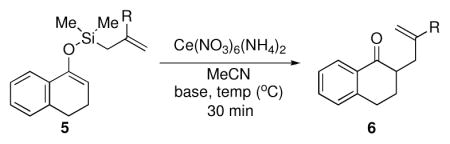
Given the limited results obtained with the simple allyldimethylsilyl enol ether when we attempted to use ketones other than tetralone, we wished to conduct an analysis of ketone scope for the 2-substituted allylsilanes (Table 2, prepared in 59–99% yield from the corresponding ketones, see Supporting Information). In general, the 2-substituted allylsilanes gave good yields of the allylated material on exposure to CAN/DTBP (Table 2, entries 1 and 2). When 2-methyltetralone was used as the ketone substrate, the oxidative allylation procedure led to smooth generation of a quaternary stereocenter in good yield (Table 2, entry 3). Acyclic α-aryl substituted ketones also proved to be suitable substrates for the reaction. For example, the allylsilanes (R = Me or CH2OBn) derived from propiophenone, gave 81% and 59% yields of the respective allylated adduct (Table 2, entry 4). The 2-acetylfuran derived system also performed well in the reaction (Table 2, entry 5).
Table 2.
Ketone Scope of the Oxidative Allylationa
| entry | ketone (7) | R | product (8) | R | yield[%]b(dr)c |
|---|---|---|---|---|---|
| 1 |  |
Me (7a) |  |
Me (8a) | 72 |
| CH2OBn (7b) | CH2OBn (8b) | 51 | |||
| 2 | 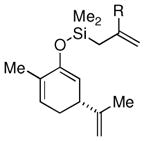 |
Me (7c) | 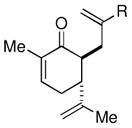 |
Me (8c) | 56 (1:1) |
| CH2OBn (7d) | CH2OBn (8d) | 67 (1:1) | |||
| 3 | 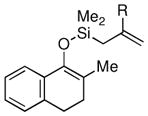 |
Me (7e) | 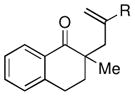 |
Me (8e) | 73 |
| CH2OBn (7f) | CH2OBn (8f) | 61 | |||
| 4 | 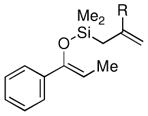 |
Me (7g) | 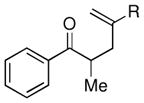 |
Me (8g) | 81 |
| CH2OBn (7h) | CH2OBn (8h) | 59 | |||
| 5 | 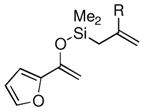 |
Me (7i) | 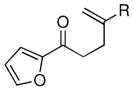 |
Me (8i) | 73 |
| 6 | 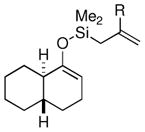 |
Me (7j) | 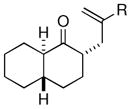 |
Me (8j) | 43d (3:1) |
| 7 | 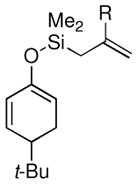 |
Me (7k) | 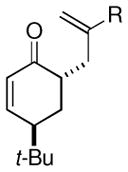 |
Me (8k) | 76 (20:1) |
| 8 | 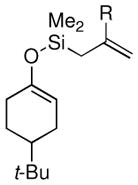 |
Me (7l) | 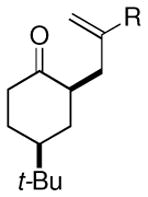 |
Me (8l) | 52 (5:1) |
Standard conditions for oxidative allylation: 7, CAN (2.0 equiv), 2,6-di-t-BuPy (4.0 equiv), 0.1 M MeCN, −25 °C, 0.5–24 h.
Isolated yield after chromatography.
Determined by 1H NMR spectroscopy.
Isolated yield of single isomer.
Surprisingly, the oxidative allylation of carvone afforded low levels of diastereoselectivity, which stood in contrast to what we have observed in our ketone–ketone cross-coupling.2b The locked bicyclic enol silane derived from trans-decalone, however, produced a 3:1 mixture favoring of the axial product 8j (43% isolated yield of single diastereomer, Table 2, entry 6). We also wished to determine the effect a more remote stereocenter at C4 would have on the facial selectivity. To probe this issue, the enol allylsilanes of 4-t-Bu-cyclohexenone and of 4-t-Bu-cyclohexanone were prepared and exposed to conditions for effecting oxidative bond formation. Allylation of the cyclohexenone derivative proceeded in 76% yield as a 20:1 mixture of diastereomers, favoring the 1,3-anti isomer 8k (Table 2, entry 6). The corresponding saturated derivative gave a slightly diminished yield (52%), but favored the syn isomer 8l (5:1 syn to anti).
At first glance the preferential formation of the 1,3-syn isomer 8l from t-Bu-cyclohexanone derivative 7l (Table 2, entry 7) was surprising, given the results for 7k (Table 2, entry 6) and the well documented preference for axial alkylation in similar systems.14 We suspected that under the mildly basic reaction conditions, the less stable 1,3-anti isomer would undergo epimerization to the thermodynamically favored 1,3-syn isomer. Independent synthesis of the 1,3-anti isomer,15 and its subsequent exposure to the reaction conditions confirmed that significant isomerization was occuring (9:1 anti:syn → 1:5 anti:syn after 24 h at −25 °C). In light of this result, we assessed the diastereoselectivity as a function of reaction time (Figure 3). Typical reaction times for the oxidative allylation of 7l were about 24 hours and afforded a 5:1 ratio of syn to anti isomers. Quenching the reaction after just 3 hours, however, revealed a modest preference for the anti-diastereomer indicating that kinetic axial alkylation was occuring to a significant extent, but the extended reaction times required for all the starting material to be consumed allowed for significant product isomerization. In light of these details, the poor diastereoselectivity of the carvone-derived allyl silanes (7c and 7d) may be attributed to an internal competition between axial addition leading to the syn isomer and allylation anti to the isopropenyl substituent. In this instance there appears to be no bias for one stereochemical course over the other.16
Figure 3.

Product stereochemistry with respect to reaction time.
The diminished yields for saturated systems (i.e., entries 6 and 8, Table 2) relative to aryl and unsaturated derive systems, is most likely a consequence of slower oxidation of the less electron-rich nonconjugated π-system and subsequent generation of a less stable initially formed radical-cation intermediate. We probed this issue by conducting an experiment where an equimolar ratio of 7k and 7l we exposed to only enough CAN to oxidize one substrate completely. Our hypothesis regarding the different reactivity proved correct: only the unsaturated derivative 7k underwent oxidative allylation to form 8k, and the corresponding adduct of the saturated derivative 7l was not observed (eq. 1, Figure 4).
Figure 4.

Experiments to gain insight into the reaction process.
Cross-over experiments were conducted to determine whether the reaction was proceeding solely through intramolecular delivery of the allyl fragment (Figure 4). Accordingly, a 1:1 mixture of the allylsilanes 5a and 7a, each of which performed well on their own, was exposed to the oxidation conditions and the product mixture analyzed (eq. 2). Only the products of intramolecular delivery were observed. We also investigated whether intermolecular oxidative allylation could be achieved when using an external nucleophile. In the event, exposure of the TMS-enol ether of 4-t-Bu-cyclohexanone (i.e., 9) to CAN/DTBP in the presence of one equivalent of allylsilane 10 gave little of the desired adduct 8l. By increasing the amount of allylsilane used to ten equivalents, a chemically efficent allylation could be achieved (57% yield), albeit as a 1.5:1 mixture of diastereomers. The necessary use of excess silane negates this alternative as a possible fragment coupling for advanced precursors, and serves to highlight the utility of the intramolecular allylation in the context of complex molecule synthesis.
To summarize, we have successfully expanded the scope of silicon-tethered oxidative carbon–carbon bond formation to allow for allylation of ketones. The reaction was shown to be compatible with aryl ketones, enones, and saturated systems. This new method for allylation offers an additional synthetic tool for chemists for cases where direct enolate alkylation, Claisen rearrangement or palladium-catalyzed methods are insufficient to achieve construction of the desired molecule.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH/NIGMS 1R01GM085322), Northwestern University (NU), and the NU Intergrated Molecular Structure and Education Research Center (DMR0114235 and CHE9871268).
Footnotes
Supporting Information Available Experimental procedures and spectral data (PDF). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For a comprehensive recent review, see: Nair V, Deepthi A. Chem Rev. 2007;107:1862–1891. doi: 10.1021/cr068408n.
- 2.Clift MD, Taylor CN, Thomson RJ. Org Lett. 2007;9:4667–4669. doi: 10.1021/ol702306z.Avetta CT, Konkol LC, Taylor CN, Dugan KC, Stern CL, Thomson RJ. Org Lett. 2008;10:5621–5624. doi: 10.1021/ol802516z.Clift MD, Thomson RJ. J Am Chem Soc. 2009;131:14579–14583. doi: 10.1021/ja906122g.See, also: Schmittel M, Burghart A, Malisch W, Reising J, Sollner R. J Org Chem. 1998;63:396–400.Schmittel M, Haeuseler A. J Organomet Chem. 2002;661:169–179.
- 3.Hwu JR, Chen CN, Shiao SS. J Org Chem. 2002;60:856–862. [Google Scholar]
- 4.(a) Zhang Y, Raines AJ, Flowers RA. Org Lett. 2003;5:2363–2365. doi: 10.1021/ol034763d. [DOI] [PubMed] [Google Scholar]; (b) Devery JJ, III, Mohanta PK, Casey BM, Flowers RA., II Synlett. 2009;2009:1490–1494. doi: 10.1055/s-0029-1217163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phenols have been shown to undergo a related oxidative allylation, see: Sabot C, Commare B, Duceppe MA, Nahi S, Guérard KC, Canesi S. Synlett. 2008;2008:3226–3230.
- 6.Hirao and coworkers have reported that oxidative allylation of 1,3-dicarbonyl compounds with allylsilanes can be achieved using VO(OEt)Cl2 as the oxidant, see: Hirao T, Sakaguchi M, Ishikawa T, Ikeda I. Synth Commun. 1995;25:2579–2585.
- 7.The exact timing of the process from intermediate 3 onwards is unclear. It seems likely that cleavage of the Si–C bond preceeds the second oxidation and Si–O bond cleavage. X in Figure 1 is most likely nitrate, although it is known that cleavage of Si–C bonds in radical cations is accelerated by nucleophilic solvents such as acetonitrile, see: Dinnocenzo JP, Farid S, Goodman JL, Gould IR, Mattes SL, Todd WP. J Am Chem Soc. 2002;111:8973–8975.
- 8.Zhang Y, Raines AJ, Flowers RA., II J Org Chem. 2004;69:6267–6272. doi: 10.1021/jo048955d. [DOI] [PubMed] [Google Scholar]
- 9.Chandra A, Pigza JA, Han JS, Mutnick D, Johnston JN. J Am Chem Soc. 2009;131:3470–3471. doi: 10.1021/ja900536d.Narasaka and coworkers reported two examples of an alkene adding to a vinylogous amide under the action of CAN, see: Narasaka K, Okauchi T, Tanaka K, Murakami M. Chem Lett. 1992:2099–2102.
- 10.Hudson CM, Marzabadi MR, Moeller KD, New DG. J Am Chem Soc. 1991;113:7372–7385. [Google Scholar]
- 11.Beeson TD, Mastracchio A, Hong JB, Ashton K, MacMillan DWC. Science. 2007;316:582–585. [PubMed] [Google Scholar]
- 12.See Supporting Information for full experimental details.
- 13.For a systematic analysis and discussion of such relative nucleophilicities, see: Mayr H, Bug T, Gotta MF, Hering N, Irrgang B, Janker B, Kempf B, Loos R, Ofial AR, Remennikov G, Schimmel H. J Am Chem Soc. 2001;123:9500–9512. doi: 10.1021/ja010890y.
- 14.(a) Corey EJ, Sneen RA. J Am Chem Soc. 1956;78:6269–6278. [Google Scholar]; (b) Velluz L, Valls J, Nominé G. Angew Chem Int Ed. 1965;4:181–200. [Google Scholar]; (c) Evans DA. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; Orlanda, FL: 1984. p. 2. [Google Scholar]
- 15.Prepared by a modification of the procedure for axial alkylation reported by Lightner DA, Bouman TD, Vincent Crist B, Rodgers SL, Knobeloch MA, Jones AM. J Am Chem Soc. 1987;109:6248–6259.
- 16.Resubjection of the 1:1 mixture of diastereomers of 8c to the reaction conditions led to equilibration to the favored anti isomer.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.