

Abstract
The intestinal epithelium restricts free passage of toxic and infectious molecules from the gut lumen while allowing selective paracellular absorption across the tight junction. Inflammatory bowel disease (IBD) patients demonstrate a loss of tight junction barrier function, increased pro-inflammatory cytokine production, and immune dysregulation; however, the relationship between these events is incompletely understood. Although tight junction barrier defects are insufficient to cause experimental IBD, mucosal immune activation is altered in response to increased epithelial permeability. Thus, an evolving model suggests that barrier dysfunction may predispose or enhance disease progression and therapies targeted to specifically restore the barrier function may provide an alternative or supplement to immunologic-based therapies.
Crohn's disease and ulcerative colitis, collectively inflammatory bowel disease (IBD), affect 1.4 million Americans. Although the exact cause of IBD remains unknown, genetic susceptibility, environmental factors, and immune dysregulation all contribute to disease pathogenesis. In addition, IBD patients demonstrate increased intestinal paracellular permeability, which reflects decreased epithelial barrier function [1,2]. While it remains unclear whether barrier dysfunction precedes disease or results from active inflammation, increased intestinal permeability is also observed in unaffected first degree relatives suggesting that a barrier defect may lead to disease progression [3,4].
An intact monolayer of intestinal epithelial cells protects the body from pathogens and other toxic luminal substances. Epithelial tight junctions maintain the intestinal barrier while regulating permeability of ions, nutrients, and water [5]. The tight junction is a multi-protein complex that forms a selectively-permeable seal between adjacent epithelial cells and demarcates the boundary between apical and basolateral membrane domains (Figure 1).
Figure 1. Cytokine regulation of epithelial barrier function.

Pro-inflammatory cytokines such as TNF, IL-1β, and LIGHT promote barrier dysfunction by inhibiting transcription of junction proteins and inducing cytoskeleton-mediated redistribution of tight junction proteins. These cytokines promote transcription of MLCK, which when activated phosphorylates myosin II, resulting in reorganization of tight junction proteins, including endocytic removal from the apical junctional complex.
Involvement of the tight junction in IBD
The tight junction is comprised of multiple proteins including transmembrane proteins such as occludin, tricellulin, claudins and junctional adhesion molecule (JAM). The intracellular portions of these transmembrane proteins interact with cytoplasmic peripheral membrane proteins, including zona occludens (ZO)-1,-2,-3 and cingulin [6]. These tight junction and cytoplasmic proteins interact with F-actin and myosin II, thereby anchoring the tight junction complex to the cytoskeleton. Once thought to be static, the association of these proteins with the tight junction is highly dynamic [7] and may play a role in epithelial barrier regulation.
Occludin was the first tight junction-associated integral membrane protein identified [8]. Although occludin knockout mice exhibit intact intestinal epithelial tight junctions and display no observable barrier defect [9,10], they have a complex disease phenotype that includes severe growth retardation, male sterility, chronic gastritis, and osteomalacia [10]. While these data have been interpreted by some to suggest the lack of an important role for occludin in tight junction integrity, in vitro studies demonstrate critical roles in tight junction assembly and maintenance [11-13]. This suggests that further analysis of occludin knockout mice under stressed condition may reveal in vivo functions of occludin and provide new insight into mechanisms of tight regulation [5].
Tricellulin is related to occludin but is preferentially localized to the tricellular junction region where three cells meet [14]. Although tricellulin is critical to maintenance of ion gradients within the inner ear [15,16], tricellulin expression in the intestine has not been described. Given the phylogenetic and structural similarities between occludin and tricellulin [14], it may be that the tricellulin accounts for normal intestinal barrier function in occludin knockout mice. This hypothesis could also be applied to inflammatory bowel disease, where intestinal epithelial occludin expression is reduced [17]. While untested the notion that tricellulin can compensate for loss of occludin expression is consistent with the observation that occludin knockdown in cultured monolayers causes tricellulin to redistribute from tricellular to bicellular junctions [18]. Thus, it will be important for future studies to assess the expression, trafficking, and function of tricellulin in IBD.
The observation that barriers can develop in the absence of occludin prompted a continued search essential barrier-forming components of the tight junction [19]. This led to the identification of claudins-1 and -2 [20]. At least 24 different claudin proteins are present in mammals [21], and these proteins are the primary component of tight junction strands [22]. While the molecular anatomy of the tight junction is not yet clear, it is certain that claudins are able to form aqueous pores that permit ions and uncharged molecules to pass in a charge- and size-selective manner [23-25]. This appears to be relevant to IBD, as claudin-2, which increases paracellular conductance of sodium ions and small uncharged molecules [25,26], is increased at the tight junction in IBD [17,27]. In contrast, claudins-3, -4, -5, and -8 are removed from the tight junction in IBD patients [27,28]. The mechanisms by which claudin expression is regulated are not fully understood, although, as discussed below, cytokine signaling is one important factor. Altered transcriptional regulation and vesicular trafficking of claudins, and other tight junction proteins, in disease may be important therapeutic targets in the future.
Causes of barrier dysfunction in IBD
Barrier dysfunction includes increased paracellular permeability resulting from enhanced flux across the tight junction, but may also be caused by epithelial damage, including apoptosis, erosion, and ulceration [17,28,29]. While some data suggest that the barrier is maintained despite epithelial apoptosis [30-32], there is not uniform agreement on differing results that likely reflect the extent of apoptosis and varying experimental systems. Nonetheless, there is consensus that damage to a single cell or small group of cells, such as that induced by cytokines or inflammatory cell transmigration, barrier integrity is rapidly repaired by an actomyosin-dependent purse string mechanism [33,34]. It also seems clear that extensive epithelial damage must compromise the mucosal barrier. Improved understanding of the processes that regulate mucosal healing and development of means to accelerate epithelial repair are, therefore, important goals for treatment of inflammatory bowel disease.
In contrast to the gross barrier loss that occurs with epithelial damage, barrier dysfunction due to tight junction regulation is more selective. Therefore, it is important to discriminate between increased intestinal permeability due to epithelial loss and that which reflects tight junction-dependent changes in paracellular permeability. The latter has been carefully studied as a function of cytokine production. These studies have primarily been performed in vitro, as in vivo models may be complicated by cytokine-dependent immune cell recruitment and activation within the mucosa. The most well-studied cytokine that causes barrier dysfunction due to epithelial tight junction regulation is tumor necrosis factor (TNF) [5]. This is likely relevant to disease, as TNF is a current target of current biologic therapies for IBD [35,36], and anti-TNF therapy restores the gut barrier in Crohn's disease [37]. Therefore, our laboratory and others have focused on understanding the mechanisms by which inflammatory cytokines regulate tight junction permeability (Figure 1). Freeze-fracture electron microscopy studies showed that TNF treatment of HT29/B6 cells resulted in decreased tight junction strand number and complexity and increased frequency of strand breaks [38]. TNF also inhibits occludin promoter activity [39] and causes redistribution of occludin, ZO-1, and claudin-1 [40] (Figure 2). In vitro, the major effector responsible for TNF-induced tight junction modulation is myosin light chain kinase (MLCK), and transcription and translation of epithelial MLCK are increased by TNF in vitro and in vivo [40-42]. Moreover, MLCK inhibition corrects TNF-induced barrier defects in vitro and in vivo [43,44]. MLCK expression and activity are also enhanced in experimental models of IBD [45] and in intestinal epithelium of human IBD patients [46]. While only correlative, the further observation that the degree of MLCK upregulation in human patients parallels disease activity is consistent with the hypothesis that increased mucosal cytokine production contributes to MLCK-mediated barrier loss [46].
Figure 2. Tight junction morphology is altered in immune-mediated diarrhea.
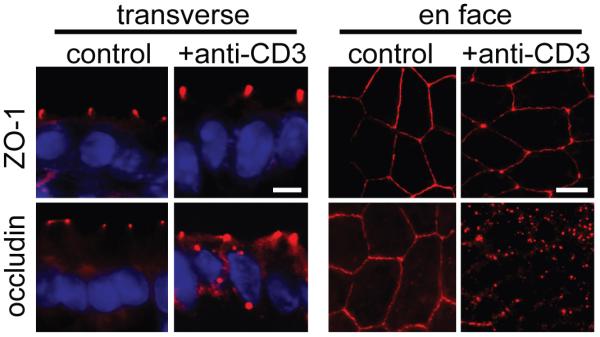
Mice were injected intraperitoneally with anti-CD3 antibody to induce acute immune-mediated diarrhea which is accompanied by increased TNF and IFNγ production. Immunofluorescence localization of ZO-1 and occludin in small small intestinal epithelium was assessed before or 3 hours after anti-CD3 treatment. While ZO-1 distribution appears unchanged in transverse sections, when viewed en face, ZO-1 staining at the junction appears thinner and concentrated more at tricellular junctions with anti-CD3 treatment. Similarly, occludin internalization into vesicles is seen in both transverse and en face sections following treatment with anti-CD3 [44]. (From Clayburgh et al. J Clin Invest 115: 2702-15, 2005, with permission.)
Other pro-inflammatory cytokines may mediate barrier function through modulation of MLCK activity. For example, LIGHT, another TNF family member, also promotes MLCK-induced tight junction disruption [47]. Although TNF and LIGHT signal through different epithelial receptors [45,47], they both likely contribute to IBD [48]. While the details are less well-defined, it also appears that IL-1β enhances paracellular permeability via MLCK [49]. Thus, MLCK represents a common effector used by multiple cytokines to modulate paracellular permeability and is an important target for future therapies to restore barrier function during active disease.
Roles of barrier regulation in colitis
First-degree relatives of Crohn's disease patients are at increased risk of developing IBD. The presence of barrier defects in some healthy relatives suggests that barrier loss may contribute to disease progression [1,2]. This hypothesis is supported by the demonstration in multiple studies that increased intestinal paracellular permeability during remission is a marker of impending disease reactivation [50]. Despite this, no mutations in tight junction proteins or defined regulators of barrier function have been reported in IBD [51]. However, the genetic linkage of certain NOD2/CARD15 mutations to barrier defects [4] suggests that immune activation may be responsible for the early barrier defects observed in healthy relatives and in patients prior to disease reactivation. Given the essential role of MLCK in immune-mediated tight junction regulation, our laboratory generated a transgenic mouse expressing constitutively-active MLCK (CA-MLCK) exclusively within the an intestinal epithelium [52]. Although CA-MLCK transgenic mice display chronic increased epithelial permeability, these mice did not develop disease [52]. However, subclinical mucosal inflammation was present in these mice, as demonstrated by increased CD4+ lamina propria mononuclear cells (LPMC) and enhanced migration of CD11c+ positive dendritic cells to the superficial lamina propria. Mucosal expression of TNF and IFNγ was increased at six weeks of age, and these increases were not strictly dependent on mature lymphocytes, as findings were similar in Rag1−/−/CA-MLCK mice. Moreover, adoptive transfer of CD4+CD45Rbhi cells into Rag1−/−/CA-MLCK mice resulted in accelerated disease development as well as more severe colitis compared to Rag1−/− mice (Figure 3). Thus, barrier dysfunction may predispose or contribute to progression of immune-mediated intestinal damage. Consistent with this, one recent study has shown that an antagonist against zonulin, which regulates intracellular tight junction disassembly, may limit mucosal immune activation in IL-10 −/− mice [53]. Development of therapeutic approaches to correct the molecular defects that give rise to barrier dysfunction, therefore, has great potential as a non-immunologic therapy for inflammatory bowel disease.
Figure 3. Increased paracellular permeability accelerates immune-mediated colitis.

CD4+CD45Rbhi (triangles) or CD4+CD25Rblo (circles) T-cells were adoptively transferred into RAG1−/− mice (red symbols) or RAG1−/− mice expressing constitutively active MLCK (yellow symbols). RAG1−/−/CA-MLCK recipients transferred with CD4+CD45Rbhi T-cells exhibited increased weight loss and mortality post-transfer compared to RAG1−/− recipients [52]. (From Su et al. Gastroenterol 136: 551-63, 2009, with permission.)
The absence of ‘spontaneous’ disease in CA-MLCK transgenic mice might prompt one to conclude that the barrier defect induced by the transgene was insufficient. However, the magnitude of barrier loss observed was similar to that in healthy relatives of Crohn's disease patients and, as noted above, the transgenic mice had increased susceptibility to adoptive transfer colitis [52]. Interestingly, increased colonic mucosal IL-10 expression was present the CA-MLCK transgenic mice [52]. This suggests that, in immunologically ‘normal’ individuals, barrier defects may trigger immunoregulatory responses that prevent inappropriate immune activation. It is important to recognize that these barrier defects are quantitatively and qualitatively different from the massive barrier loss induced by severe, widespread epithelial destruction and mucosal ulceration, as occurs in some enteric infections as well as the DSS model of colitis.
Consistent with the hypothesis that limited barrier defects may activate immunoregulatory processes, one recent study has shown that transient barrier defects can protect mice from future insults [54]. In this study, barrier function was disrupted by local epithelial damage after intrarectal ethanol administration. In addition to increased intestinal permeability, these treatments enhanced IFNγ and IL-10 production by LPMC and caused expansion of a regulatory CD4+ T-cell population expressing latency-associated peptide (LAP). While expansion of CD4+LAP+ LPMCs was required for protection, adoptive transfer of these cells was insufficient to ameliorate colitis, suggesting that other factors contribute to the observed prevention of disease [54].
Taken together, both of these studies show that barrier dysfunction alone is not sufficient to promote disease, but can alter susceptibility to colitis through regulation of mucosal immunity. While much work is needed to define the mechanisms of this immunoregulation, the increased exposure of the mucosa to luminal microbiota or their products by Toll-like receptors, including TLR2, may be involved [54]. These studies highlight the importance of the subtle interplay between epithelial barrier function and mucosal immune regulation that may represent a future target for disease prevention.
Conclusions
Epithelial barrier dysfunction and inflammation are major contributors to the pathogenesis intestinal disease; however, much remains unknown about how these two processes contribute independently to disease initiation. Cytokine-induced barrier dysfunction is known to exacerbate colitis, perhaps due to increased translocation of microbial products. While barrier dysfunction alone is insufficient to cause disease, it can lead to subclinical activation of immune responses that may affect disease development at later times. Careful study of tight junction regulation and its contribution to disease initiation will likely provide new targets for the development of IBD therapeutics.
Acknowledgements
The authors are supported by the National Institutes of Health T32HL007237 and F32DK084859 (KLE), R01RK061931 and R01DK068271 (JRT), and the Crohn's and Colitis Foundation of America (JRT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 2.May GR, Sutherland LR, Meddings JB. Is small intestinal permeability really increased in relatives of patients with Crohn's disease? Gastroenterology. 1993;104:1627–1632. doi: 10.1016/0016-5085(93)90638-s. [DOI] [PubMed] [Google Scholar]
- 3.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, Krugliak P, Rotter JI. Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology. 1989;97:927–931. doi: 10.1016/0016-5085(89)91499-6. [DOI] [PubMed] [Google Scholar]
- 4.Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, Kuechler I, Krueger S, Schmidt HH, Lochs H. Genetic basis for increased intestinal permeability in families with Crohn's disease: role of CARD15 3020insC mutation? Gut. 2006;55:342–347. doi: 10.1136/gut.2005.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. 1998;60:121–142. doi: 10.1146/annurev.physiol.60.1.121. [DOI] [PubMed] [Google Scholar]
- 7**.Shen L, Weber CR, Turner JR. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol. 2008;181:683–695. doi: 10.1083/jcb.200711165. Tight junction protein interactions have been thought to be relatively static. This study used FRAP techniques to show that tight junction proteins are highly dynamic and are continuously trafficked between the tight junction and other sites as well as within the tight junction. Notably, the mechanisms and kinetics of this exchange differ for specific proteins, thereby demonstrating the transient nature of the tight junction protein complex and, potentially, explaining the rapid alterations in structure induced by inflammatory stimuli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, Saitou M, Tsukita S, Fromm M. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669:34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu AS, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Lynch RD, Schneeberger EE. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol. 2005;288:C1231–1241. doi: 10.1152/ajpcell.00581.2004. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, Desiderio D, Guntaka R, Rao R. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A. 2009;106:61–66. doi: 10.1073/pnas.0802741106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elias BC, Suzuki T, Seth A, Giorgianni F, Kale G, Shen L, Turner JR, Naren A, Desiderio DM, Rao R. Phosphorylation of Tyr-398 and Tyr-402 in Occludin Prevents Its Interaction with ZO-1 and Destabilizes Its Assembly at the Tight Junctions. J Biol Chem. 2009;284:1559–1569. doi: 10.1074/jbc.M804783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171:939–945. doi: 10.1083/jcb.200510043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chishti MS, Bhatti A, Tamim S, Lee K, McDonald ML, Leal SM, Ahmad W. Splice-site mutations in the TRIC gene underlie autosomal recessive nonsyndromic hearing impairment in Pakistani families. J Hum Genet. 2008;53:101–105. doi: 10.1007/s10038-007-0209-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, Ramzan K, Khan SN, Chattaraj P, Friedman PL, Anderson JM, et al. Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet. 2006;79:1040–1051. doi: 10.1086/510022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 18*.Ikenouchi J, Sasaki H, Tsukita S, Furuse M. Loss of Occludin Affects Tricellular Localization of Tricellulin. Mol Biol Cell. 2008 doi: 10.1091/mbc.E08-05-0530. This is the first study demonstrating that occludin preferentially displaces tricellulin at bicellular junctions, suggesting that tricellulin may share some functional redundancy with occludin in tight junction assembly and function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M, Takano H, Noda T, Tsukita S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998;141:397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 22.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143:391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology (Bethesda) 2004;19:331–338. doi: 10.1152/physiol.00027.2004. [DOI] [PubMed] [Google Scholar]
- 24.Van Itallie CM, Fanning AS, Anderson JM. Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am J Physiol Renal Physiol. 2003;285:F1078–1084. doi: 10.1152/ajprenal.00116.2003. [DOI] [PubMed] [Google Scholar]
- 25**.Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci. 2008;121:298–305. doi: 10.1242/jcs.021485. By characterizing tight junction permeability in relation to solute size, the authors demonstrate that claudin-2 expression can regulate the density of small tight junction pores without affecting flux across large pores. The increase in claudin-2 expression seen in IBD may, therefore, result in increased density of small pores and reduced tight junction barrier function. [DOI] [PubMed] [Google Scholar]
- 26.Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, Fromm M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115:4969–4976. doi: 10.1242/jcs.00165. [DOI] [PubMed] [Google Scholar]
- 27.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on Claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85:1139–1162. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 28.Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Downregulation of epithelial apoptosis and barrier repair in active Crohn's disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–1302. doi: 10.1136/gut.2003.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulzke JD, Bojarski C, Zeissig S, Heller F, Gitter AH, Fromm M. Disrupted barrier function through epithelial cell apoptosis. Ann N Y Acad Sci. 2006;1072:288–299. doi: 10.1196/annals.1326.027. [DOI] [PubMed] [Google Scholar]
- 30.Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol. 2001;11:1847–1857. doi: 10.1016/s0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- 31.Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, McKenna M, Rose T, Montrose MH. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902–912. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Gunzel D, Florian P, Richter JF, Troeger H, Schulzke JD, Fromm M, Gitter AH. Restitution of single-cell defects in the mouse colon epithelium differs from that of cultured cells. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1496–1507. doi: 10.1152/ajpregu.00470.2005. [DOI] [PubMed] [Google Scholar]
- 33.Russo JM, Florian P, Shen L, Graham WV, Tretiakova MS, Gitter AH, Mrsny RJ, Turner JR. Distinct temporal-spatial roles for rho kinase and myosin light chain kinase in epithelial purse-string wound closure. Gastroenterology. 2005;128:987–1001. doi: 10.1053/j.gastro.2005.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Florian P, Schoneberg T, Schulzke JD, Fromm M, Gitter AH. Single-cell epithelial defects close rapidly by an actinomyosin purse string mechanism with functional tight junctions. J Physiol. 2002;545:485–499. doi: 10.1113/jphysiol.2002.031161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baert FJ, D'Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, Geboes K, Rutgeerts PJ. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn's ileocolitis. Gastroenterology. 1999;116:22–28. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 36.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 37.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, Rutgeerts P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. 2002;97:2000–2004. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 38.Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, Bode H, Epple HJ, Riecken EO, Schulzke JD. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci. 1999;112:137–146. doi: 10.1242/jcs.112.1.137. [DOI] [PubMed] [Google Scholar]
- 39.Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, Schulzke JD. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000;113:2085–2090. doi: 10.1242/jcs.113.11.2085. [DOI] [PubMed] [Google Scholar]
- 40.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, Lin A, Turner JR. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem. 2006;281:26205–26215. doi: 10.1074/jbc.M602164200. [DOI] [PubMed] [Google Scholar]
- 42.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 43.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 44.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131:1153–1163. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 47.Schwarz BT, Wang F, Shen L, Clayburgh DR, Su L, Wang Y, Fu YX, Turner JR. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology. 2007;132:2383–2394. doi: 10.1053/j.gastro.2007.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mackay F, Browning JL, Lawton P, Shah SA, Comiskey M, Bhan AK, Mizoguchi E, Terhorst C, Simpson SJ. Both the lymphotoxin and tumor necrosis factor pathways are involved in experimental murine models of colitis. Gastroenterology. 1998;115:1464–1475. doi: 10.1016/s0016-5085(98)70025-3. [DOI] [PubMed] [Google Scholar]
- 49.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. Journal of Immunology. 2007;178:4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet. 1993;341:1437–1439. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 51.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52**.Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, Abraham C, Turner JR. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136:551–563. doi: 10.1053/j.gastro.2008.10.081. Although mice expressing constitutively-active MLCK in the intestinal epithelium do not develop active colitis, compromised barrier function results in subclinical mucosal inflammation and increases susceptibility to experimental colitis. This study suggests that increased paracellular permeability may function as a “first hit” in the development of disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53**.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. This is the first study to suggest that enhanced small intestinal barrier function may be of therapeutic benefit in experimental IBD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Boirivant M, Amendola A, Butera A, Sanchez M, Xu L, Marinaro M, Kitani A, Di Giacinto C, Strober W, Fuss IJ. A Transient Breach in the Epithelial Barrier Leads to Regulatory T-Cell Generation and Resistance to Experimental Colitis. Gastroenterology. 2008;135:1612–1623. doi: 10.1053/j.gastro.2008.07.028. The authors show that intrarectal administration of ethanol or zonula occludens toxin antagonist disrupts barrier function and protects against TNBS-induced colitis through activation of CD4+LAP+ LPMCs, highlighting the role of barrier dysfunction in mucosal immune activation. [DOI] [PubMed] [Google Scholar]