

Abstract
Ataxia with oculomotor apraxia type 2 (AOA2) is caused by a diversity of mutations within the coding region of the senataxin gene. Recently, rare noncoding senataxin mutations affecting RNA processing have been identified in AOA2. Here, we report the case of an 18-year-old woman, with classic clinical features of AOA2, who was found to harbor a mutation within senataxin intron 16. This mutation disrupts the local 5′ splice site architecture via a novel intronic frameshift mechanism, causing skipping of exon 16 with predicted disruption of the conserved DNA/RNA helicase domain. RNA processing mutations expand the growing complexity of pathogenic senataxin mutations.
Keywords: AOA2, Ataxia, Helicase, RNA splicing, Senataxin
Introduction
Ataxia with oculomotor apraxia type 2 (AOA2) is an autosomal recessive hereditary ataxia caused by mutation of the senataxin gene (SETX) on chromosome 9q34 [1–4]. The disease is characterized primarily by vermian cerebellar atrophy with prominent gait ataxia, a peripheral sensorimotor neuropathy, areflexia, and elevated levels of α-fetoprotein [1–3, 5–7]. Eye findings commonly include saccadic ocular pursuit with nystagmus; however, despite the name, oculomotor apraxia is not found in many patients [1–3, 5–7]. AOA2 is thought to be one of the most common autosomal recessive ataxias after Friedreich’s ataxia [2, 4, 7]. The clinical phenotype associated with SETX mutations also has an interesting, although rare, variability. Notably, an autosomal dominant phenotype of juvenile-onset amyotrophic lateral sclerosis (ALS4) has been associated with mutations in this gene distinct from those which cause AOA2 [8–10].
The senataxin gene is quite polymorphic, and a variety of homozygous and compound heterozygous coding mutations have been reported which cause AOA2 including missense [1, 3, 5–7, 9, 11, 12], nonsense [1, 3, 6, 7, 13], frameshift [1, 3, 5–7, 12, 14–16], gene rearrangement [5], deletion [7] or duplication [14]. One coding mutation at the terminus of exon 15 has also been reported to disrupt RNA splicing [16]. In contrast to these many coding mutations, only two pathogenic noncoding mutations have been reported which result in AOA2 [5, 7, 17]. Here, we report a patient from a large consanguineous family who was found to possess a novel homozygous mutation in intron 16 which results in aberrant splicing of the SETX pre-mRNA and clinical AOA2.
Case Report
The patient is an 18-year-old woman who presented with complaints of imbalance, falls, and difficulty walking. Onset was at age 14, and course was slowly progressive. Past medical history included polycystic ovarian syndrome diagnosed at age 16. Neurological examination was notable for a mild appendicular and gait ataxia, slowing of vertical saccades, and a mild dysarthria. Deep tendon reflexes were absent, and there was reduced sensation in the feet to pinprick, temperature, and vibration, which was most severely impaired. There was no oculomotor apraxia. Laboratory studies were notable for elevated alpha-fetoprotein (7.8 ng/mL, normal less than 6.7 ng/mL). Normal studies included serum electrolytes, renal function testing, liver function testing, vitamin B12, folate, homocysteine, methylmalonic acid, copper, ceruloplasmin, creatinine kinase, thyroid function studies, serum protein electrophoresis with immunofixation, erythrocyte sedimentation rate, antinuclear antibodies, and tests for GAD-65, thyroglobulin, gliadin, and Yo autoantibodies. Rapid plasma reagin and HTLV I/II screening were nonreactive. A sensorimotor neuropathy autoantibody panel (Athena Diagnostics, Inc.) was negative. Previous tests for follicle-stimulating hormone, luteinizing hormone, prolactin, estradiol, progesterone, testosterone, and cortisol were normal. Magnetic resonance imaging showed cerebellar atrophy. A nerve conduction study showed a sensorimotor axonal neuropathy. Echocardiogram was normal. The family is from Pakistan, and consanguinity was reported (Fig. 1), but there were no other individuals in the family identified with ataxia.
Fig. 1.
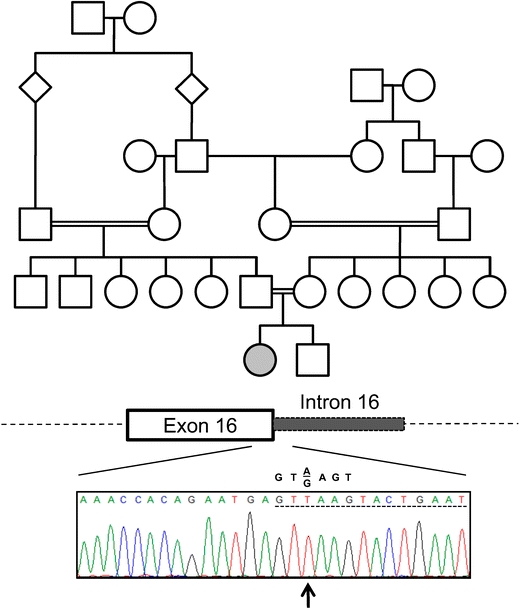
A homozygous intronic point mutation in the senataxin gene in a patient with ataxia with oculomotor apraxia type 2. The pedigree of the family described in this report is shown. The affected proband is indicated by a filled circle. No other individuals were reported to have ataxia. A schematic of the senataxin exon/intron junction is shown for exon 16 with the corresponding results of DNA sequencing for the proband. Intronic sequence is denoted by a dashed underline. The identified mutation, a thymidine insertion at position +2 of the intron, is indicated by an arrow. The consensus sequence of a 5′ splice site is shown above the patient’s sequence
Genetic testing included normal sequencing of the aprataxin gene (APTX) and the alpha-tocopherol transfer protein gene (TTPA). No repeat expansions were detected in the frataxin gene (FXN). Sequencing of the senataxin gene revealed a homozygous insertion of a thymidine at position +2 of intron 16 (IVS16 + 2insT; Fig. 1).
Materials and Methods
Genomic DNA was prepared from blood using Autopure LS (Qiagen) according to the manufacturer’s instructions. Sequencing of senataxin coding exons and the neighboring intronic regions was performed from genomic DNA using a 3730 DNA Analyzer (Applied Biosystems). Primer sequences utilized were as described in Criscuolo et al. [5].
Total RNA from peripheral blood was extracted using the PAXgene Blood RNA Kit (Qiagen) per the manufacturer’s instructions. RNA quality control was performed using an Agilent Bioanalyzer 2100 (Agilent Technologies). Human fetal and adult brain were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders (University of Maryland, Baltimore, MD, USA). Total RNA was obtained using an RNeasy Mini Kit (Qiagen) per the manufacturer’s instructions. Reverse transcription (RT) was performed using total RNA primed with oligo-dT in the SuperScript III First-Strand Synthesis System (Invitrogen). Polymerase chain reaction (PCR) was performed using a high-fidelity proofreading polymerase (Accuprime Pfx, Invitrogen) and approximately 10–50 ng of cDNA. PCR cycle settings were 94°C for 30 s, 55°C for 30 s, and 72°C for 30–45 s for a total of 35–40 cycles. For nested PCR, dilutions of the initial PCR reaction (ranging from 0.08% to 6%) were used in a second reaction with primers internal to the first set. All PCR products were confirmed by sequencing as above. All primer sequences are available on request. Gel images were captured using a BioSpectrum AC Imaging System (UVP).
Exon numbering is based on the 26 exons known to be expressed by the SETX gene (NM_015046). The SETX coding region is represented by exons 3–26 (with exons 14–26 encoding the DNA/RNA helicase domain). The novel exon reported here is designated exon 25b to match with current exon numbering.
Results
Sequencing of intron 16 in this patient identified a homozygous insertion of one base pair, thymidine (T), at position +2 of intron 16 (Fig. 1). The patient’s mother was found to be heterozygous for this mutation (data not shown). The father was not available for testing so we cannot determine if the second mutation was inherited (as predicted by the pedigree) or arose de novo.
As senataxin is expressed in lymphoblasts, we were able to investigate the functional consequences of this mutation, which we hypothesized would disrupt normal RNA splicing. Total RNA from the proband’s mother was converted to cDNA for use in nested RT-PCR with primers spanning exons 15 and 17 of senataxin. This demonstrated the presence of the expected band representing exons 15–17 as well an additional band corresponding to the skipping of exon 16 (Fig. 2). In the patient, only the misspliced product was detected (Fig. 2). The new protein product generated from this splicing event is predicted to lack a functional DNA/RNA helicase domain.
Fig. 2.
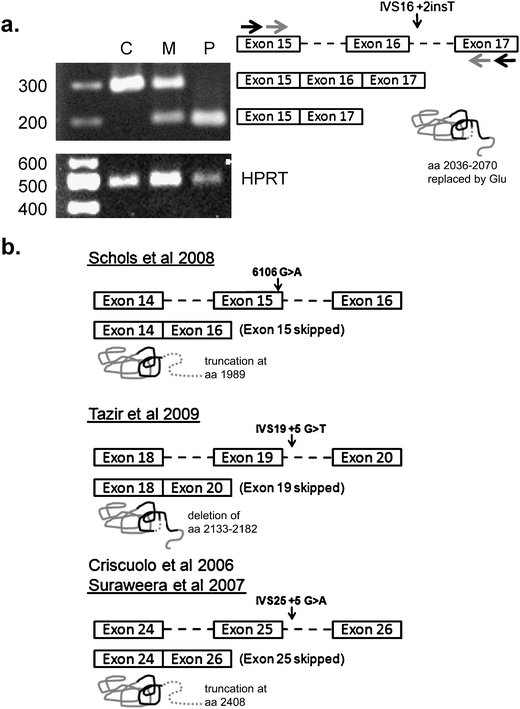
Aberrant splicing of senataxin in patients with AOA2. a Total peripheral blood RNA from the proband (P) and her mother (M) as well as an unrelated normal control individual (C) was used for RT-PCR with primers amplifying across exons 15 and 17 (black arrows) followed by internal nested primers (gray arrows). Exons are shown in genomic context with intronic sequences indicated by dashed lines. The detected spliced products (shown) either include or exclude exon 16 (291 or 189 base pairs, respectively). HPRT was amplified as a positive control (489 base pair product). Size markers are indicated in base pairs. The predicted effect on senataxin protein is shown graphically with the DNA/RNA helicase domain (aa 1931–2456) indicated by a black line and an in-frame deletion shown by the dashed segment. b Previously published senataxin mutations which affect RNA splicing are shown with the corresponding mutation (arrow) in the genomic context. The abnormal splicing event is indicated by boxes, and the resulting effect on the protein is shown graphically as in part a. Protein truncation is shown by a dashed line
Previously, senataxin has not been thought to undergo significant alternative splicing [9]. However, review of the Alternative Splicing and Transcript Diversity database [18] identifies a total of four additional human senataxin transcripts supported by expressed sequence tag (EST) data which include the DNA/RNA helicase domain (data not shown), and therefore may potentially have functional significance (see “Discussion”). The largest alternate transcript includes an additional 3′ exon (which we term exon 25b) which adds 29 amino acids at the C-terminal portion of the DNA/RNA helicase domain after position 2429 (Fig. 3). The remaining isoforms are smaller due to utilization of alternative transcription start sites (in exon 10) and/or alternative splicing involving exon 25b or exon 24 (which removes 33 amino acids in the DNA/RNA helicase domain after position 2366; Fig. 3). We find support for these splicing patterns from multiple unique ESTs (Fig. 3). Furthermore, using RT-PCR, we detected transcripts featuring the alternative use of both exon 25b and exon 24 in both human fetal and adult brain (Fig. 3).
Fig. 3.
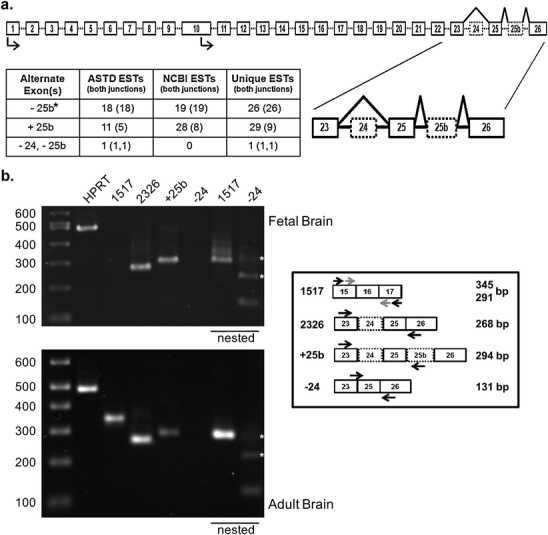
Alternative splicing of the senataxin gene. a The genomic structure of the senataxin gene is shown. Exons are boxed and introns are shown by a dashed line. Predicted transcription start sites are indicated by arrows. Suspected alternative exons are indicated by dotted boxes and the alternative splicing pattern is shown by a black line. The region of exons 23–26 is expanded to highlight the details of this alternative splicing. Supporting data from ESTs is shown in the table. Data compiled from the Alternative Splicing and Transcript Diversity database and the National Center for Biotechnology Information EST database. The number of ESTs with at least one splice junction represented is shown with the number of ESTs covering both splice junctions shown in parentheses. For events involving multiple exons, the number of transcripts including both junctions is shown for each exon. Asterisk: the pattern excluding exon 25b is the established pattern of senataxin splicing in the literature. b Confirmation of senataxin alternative splicing in human fetal and adult brain. Total RNA from human fetal (upper panel) or adult (lower panel) brain was used for RT-PCR with primers (black arrows) designed to amplify the suspected alternative splice products shown in the diagram. HPRT (489 bp) was used as a positive control. Products not detected in the initial PCR reaction were re-evaluated using a nested PCR reaction (underlined lanes) to detect lower abundance messages. Due to the size of the senataxin message, product 1517 can require nested PCR (gray arrows) to detect with oligo-dT primed cDNA. All PCR products were confirmed by sequencing. Molecular weight markers are indicated in base pairs. Asterisk: these additional bands are PCR artifacts (data not shown) produced by mispriming of the primer used to detect the exon 23—exon 25 splice junction
Discussion
Senataxin is a large 2,677 amino acid protein whose function still remains relatively unknown. Although multiple regions of the protein appear to be important for disease, an attractive target is the conserved C-terminal DNA/RNA helicase domain found at amino acids 1931–2456 (encoded by exons 14–26) [1, 8, 12]. Mutations that alter or disrupt this domain cause AOA2, presumably through a loss of function [1, 3, 8, 9, 12]. Recent studies strengthen the relevance of the DNA/RNA helicase domain by providing evidence for a role of senataxin in DNA damage response [17] and potential roles in transcription and RNA processing based on work with the yeast ortholog [19].
In this report, we identified a novel intronic mutation resulting in aberrant splicing of the SETX mRNA in a patient with clinical AOA2. Three previous mutations that affect splicing of the senataxin pre-mRNA have been reported [5, 7, 16, 17] (Fig. 2). One of these results from a G>A transition at c.6106, the last nucleotide of exon 15, resulting in skipping of the exon [16], with a frameshift mutation and truncation at amino acid 1989. The other two mutations are intronic—one, IVS25+5G>A, results in skipping of exon 25 leading to a frameshift [5, 17] with protein truncation at amino acid 2408, and another, IVS19+5G>T, which skips exon 19, resulting in an in-frame deletion of amino acids 2133–2182 [7]. The mutation reported here, IVS16+2T, results in skipping of exon 16 (Fig. 2). The mechanism of this mutation is novel relative to the other known SETX splicing mutations, as the inserted T at position +2 induces an intronic frameshift in the neighborhood of the intron 16 5′ splice site sequence which would be predicted to dramatically reduce splice site utilization [20]. The resultant skipping of exon 16 identified in Fig. 2 would result in the in-frame replacement of amino acids 2036–2070 with a glutamic acid residue. Interestingly, all these RNA processing mutations affect the DNA/RNA helicase domain, further implicating the importance of this region in AOA2 pathogenesis [12].
Additionally, it is also of considerable interest that despite the wide variety of mutations seen in AOA2, only a few distinct heterozygous missense mutations have thus far been identified in motor neuron disease [8–10]. This observation would be consistent with the proposed gain-of-function hypothesis in ALS4. It is worth noting that one of the SETX splicing mutations seen in AOA2, IVS19+5 G>T [7], removes residue 2136 which has been detected as mutated in cases of ALS4 (R2136H) [8, 9]. As the patients clinically have AOA2 [7], this demonstrates that deletion of this residue is insufficient to produce a motor neuron phenotype even when homozygous, suggesting that an intact DNA/RNA helicase domain is required for the development of ALS4.
In addition to AOA2, the patient reported here also carries a diagnosis of polycystic ovarian syndrome. This may be relevant, as early menopause and premature ovarian failure have been reported in other cases of AOA2 [2, 5, 13], suggesting a germ line effect, at least in affected women. While the genetic risk for polycystic ovarian syndrome is complex [21], given the above findings, it is interesting to speculate that senataxin mutations (or perhaps a specific subset thereof) may alter the risk of this associated condition in affected women.
Finally, we have demonstrated the presence of alternatively spliced senataxin transcripts in human fetal and adult brain. It is of considerable interest that the variability in these transcripts resides in the C-terminal portion of DNA/RNA helicase domain. The role of these (and potentially other) isoforms of senataxin in endogenous function and in the pathogenesis of AOA2 and ALS4 is an important new area of study. In conjunction with the known patient data, these observations highlight RNA processing errors as a newly recognized class of SETX mutations in AOA2 and adds to the growing diversity of pathogenic mutations seen in this gene.
Acknowledgements
We thank Dr. Dan Geschwind, Director of the UCLA Neurogenetics program, which supported this work, for his critical comments and guidance. We also wish to thank Dr. Giovanni Coppola for reagents, technical advice, and critical comments regarding this manuscript. BLF is a recipient of a Clinical Research Training Fellowship from the American Academy of Neurology Foundation.
Disclosures
The authors have reported no conflicts of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 2.Le Ber I, Bouslam N, Rivaud-Pechoux S, Guimaraes J, Benomar A, Chamayou C, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759–767. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- 3.Duquette A, Roddier K, McNabb-Baltar J, Gosselin I, St-Denis A, Dicaire MJ, et al. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol. 2005;57:408–414. doi: 10.1002/ana.20408. [DOI] [PubMed] [Google Scholar]
- 4.Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007;6:245–257. doi: 10.1016/S1474-4422(07)70054-6. [DOI] [PubMed] [Google Scholar]
- 5.Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Sacca F, Grieco GS, et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006;66:1207–1210. doi: 10.1212/01.wnl.0000208402.10512.4a. [DOI] [PubMed] [Google Scholar]
- 6.Anheim M, Fleury MC, Franques J, Moreira MC, Delaunoy JP, Stoppa-Lyonnet D, et al. Clinical and molecular findings of ataxia with oculomotor apraxia type 2 in 4 families. Arch Neurol. 2008;65:958–962. doi: 10.1001/archneur.65.7.958. [DOI] [PubMed] [Google Scholar]
- 7.Tazir M, Ali-Pacha L, M’Zahem A, Delaunoy JP, Fritsch M, Nouioua S, et al. Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci. 2009;278:77–81. doi: 10.1016/j.jns.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen YZ, Hashemi SH, Anderson SK, Huang Y, Moreira MC, Lynch DR, et al. Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol Dis. 2006;23:97–108. doi: 10.1016/j.nbd.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Zhao ZH, Chen WZ, Wu ZY, Wang N, Zhao GX, Chen WJ, et al. A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;10(2):118–122. doi: 10.1080/17482960802572673. [DOI] [PubMed] [Google Scholar]
- 11.Asaka T, Yokoji H, Ito J, Yamaguchi K, Matsushima A. Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX. Neurology. 2006;66:1580–1581. doi: 10.1212/01.wnl.0000216135.59699.9b. [DOI] [PubMed] [Google Scholar]
- 12.Fogel BL, Perlman S. Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology. 2006;67:2083–2084. doi: 10.1212/01.wnl.0000247661.19601.28. [DOI] [PubMed] [Google Scholar]
- 13.Lynch DR, Braastad CD, Nagan N. Ovarian failure in ataxia with oculomotor apraxia type 2. Am J Med Genet. 2007;143A:1775–1777. doi: 10.1002/ajmg.a.31816. [DOI] [PubMed] [Google Scholar]
- 14.Arning L, Schols L, Cin H, Souquet M, Epplen JT, Timmann D. Identification and characterisation of a large Senataxin (SETX) gene duplication in ataxia with ocular apraxia type 2 (AOA2) Neurogenetics. 2008;9:295–299. doi: 10.1007/s10048-008-0139-z. [DOI] [PubMed] [Google Scholar]
- 15.Nicolaou P, Georghiou A, Votsi C, Middleton LT, Zamba-Papanicolaou E, Christodoulou K. A novel c.5308_5311delGAGA mutation in Senataxin in a Cypriot family with an autosomal recessive cerebellar ataxia. BMC Med Genet. 2008;9:28. doi: 10.1186/1471-2350-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schols L, Arning L, Schule R, Epplen JT, Timmann D. “Pseudodominant inheritance” of ataxia with ocular apraxia type 2 (AOA2) J Neurol. 2008;255:495–501. doi: 10.1007/s00415-008-0707-z. [DOI] [PubMed] [Google Scholar]
- 17.Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177:969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koscielny G, Le Texier V, Gopalakrishnan C, Kumanduri V, Riethoven JJ, Nardone F, et al. ASTD: the alternative splicing and transcript diversity database. Genomics. 2009;93:213–220. doi: 10.1016/j.ygeno.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Ursic D, Chinchilla K, Finkel JS, Culbertson MR. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res. 2004;32:2441–2452. doi: 10.1093/nar/gkh561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korzinov OM, Astakhova TV, Vlasov PK, Roitberg MA. Statistical analysis of DNA sequences nearby splicing sites. Mol Biol (Mosk) 2008;42:150–162. [PubMed] [Google Scholar]
- 21.Goodarzi MO, Azziz R. Diagnosis, epidemiology, and genetics of the polycystic ovary syndrome. Best Pract Res Clin Endocrinol Metab. 2006;20:193–205. doi: 10.1016/j.beem.2006.02.005. [DOI] [PubMed] [Google Scholar]