

Abstract
Epigenetic changes in the nervous system are emerging as a critical component of enduring effects induced by early life experience, hormonal exposure, trauma and injury, or learning and memory. Sex differences in the brain are largely determined by steroid hormone exposure during a perinatal sensitive period that alters subsequent hormonal and nonhormonal responses throughout the lifespan. Steroid receptors are members of a nuclear receptor transcription factor superfamily and recruit multiple proteins that possess enzymatic activity relevant to epigenetic changes such as acetylation and methylation. Thus steroid hormones are uniquely poised to exert epigenetic effects on the developing nervous system to dictate adult sex differences in brain and behavior. Sex differences in the methylation pattern in the promoter of estrogen and progesterone receptor genes are evident in newborns and persist in adults but with a different pattern. Changes in response to injury and in methyl-binding proteins and steroid receptor coregulatory proteins are also reported. Many steroid-induced epigenetic changes are opportunistic and restricted to a single lifespan, but new evidence suggests endocrine-disrupting compounds can exert multigenerational effects. Similarly, maternal diet also induces transgenerational effects, but the impact is sex specific. The study of epigenetics of sex differences is in its earliest stages, with needed advances in understanding of the hormonal regulation of enzymes controlling acetylation and methylation, coregulatory proteins, transient versus stable DNA methylation patterns, and sex differences across the epigenome to fully understand sex differences in brain and behavior.
The term “epigenetics” is defined literally as “in addition to genetics” but in reality refers to changes in the DNA or surrounding chromatin that influence gene expression but do not change genetic composition. There are two identified ways in which meaningful epigenetic changes can occur: (1) the addition of a methyl group to a cytosine that sits just upstream of a guanine, and is referred to as a CpG island, and (2) changes to the histones that form the core of nucleosomes around which DNA is tightly packed. The dominant changes to histones are methylation and acetylation of the protruding tails, but can also include ubiquitylation, phosphorylation, and sumoylation (for review, see Goldberg et al., 2007). All these processes are enzymatically driven and therefore regulated and reversible. The methylation of DNA is tightly controlled by a family of DNA methyl transferases (DNMTs), with DNMT3A and DNMT3B inducing de novo methylation while DNMT1 predominantly maintains ongoing methylation. Changes to both the DNA and histones impact gene transcription but do so in different manners. Direct methylation of the DNA at CpG sites profoundly impacts the expression of a gene but the effect depends on the location. The promoter and upstream noncoding regions of genes are the principle regulatory sites of interest, but the sites are not predictable and vary for each gene. DNA methylation is generally repressive of gene expression, but exceptions are beginning to emerge. Conversely, changes to histones occur on specific residues but act globally to relax or tighten the chromatin surrounding a particular gene and thereby regulate access of the transcription complex. Two classes of enzymes have been the focus of investigation, the histone deacetylases (HDACs), which remove acetyl groups from lysine residues and thereby tighten the chromatin structure and reduce transcription, versus histone acetylases (HATs), which perform the opposite function, adding acetyl groups to lysine residues and weakening the electric charge between histones and DNA and relaxing the tightly wound chromatin. There is a relationship between histone and DNA modifications, with one often preceding and allowing the other and thereby further strengthening the silencing or activation of a particular gene.
The origins of epigenetics begin in early development with the single-celled zygote. The paternal genome is actively demethylated before the first cell division, followed shortly thereafter by demethylation of the maternal genome once the zygote has undergone several cleavage divisions. Proper development requires de novo methylation of the inner cell mass at the blastocyst stage and cell differentiation is dependent on maintenance of DNA methylation of large numbers of genes. Disruption of these events by genetic ablation of the DNMTs is generally embryonic lethal (Li, 2002). Moreover, a loss of maintenance DNA methylation is a frequent precipitating event in cancer as cellular signals for proliferation are released from chronic suppression (for review, see Ellis et al., 2009).
In mammals the role of epigenetics in sex differences also begins early in development with the critical process of X inactivation in females. To achieve dosage compensation, one of the two X chromosomes is largely silenced by a combination of histone modifications and DNA methylation (Avner and Heard, 2001). But not all X genes are silenced, and those that are expressed provide a potential source of sex differences in both somatic and neural cells (Chen et al., 2009). Imprinting of paternal or maternal genes is an additional source of epigenetic control of sex differences. Genetic imprinting is the systematic suppression of a portion of a chromosome from one parent via maintenance DNA methylation, leading to monoallelic expression. Many imprinted genes are associated with body growth, and this has been interpreted as indicative of the battle between the sexes, with males wanting to produce as many and as large of offspring as possible at the expense of the maternal female (Davies et al., 2008). Others see a logical inconsistency in this argument and suggest instead that imprinting serves as a coadaptation between the mother and her offspring and involves a complex interplay of imprinted gene expression in the placenta, maternal hypothalamus, and fetal brain to regulate nutrient distribution, maternal care and feeding, and the sexual behavior of adult male progeny (Keverne and Curley, 2008). Elucidating how X chromosome inactivation and/or genomic imprinting contribute to sex differences in the brain remains a poorly understood and relatively unexplored research area.
Epigenetic changes can be further divided into those that occur in the germline and are therefore heritable, versus those in somatic cells which generally persist only for the duration of a lifetime and are largely context dependent (Crews, 2008). The context may be variables in the internal or external environment, such as steroid hormones or endocrine-disrupting chemicals, respectively (Gore, 2008). Alternatively the context may be experiences as profound as early child abuse (McGowan et al., 2009), or events as mild as context-dependent learning (Lubin et al., 2008).
Similar to context-dependent epigenetic changes, sex differences in the brain are not an inherent emergent property but are instead largely determined by extrinsic factors. Chromosomal sex directs the differentiation of the bipotential gonad, and it is the steroid hormone production by the gonad that then determines the phenotype of the brain in regards to relative masculinization or feminization. Progress is being made on the cellular mechanisms of androgen- and estrogen-induced sexual differentiation of the brain and spinal cord during a perinatal sensitive window (for review, see McCarthy et al., 2009), and attention is now turning toward how epigenetics might contribute to both the establishment of, and perhaps more importantly, maintenance of sex differences in the brain. Steroids such as testosterone and estradiol are ligands for a family of nuclear transcription factors that recruit numerous coactivators and repressors into a macromolecular complex that includes HATs and HDACs. Thus it is not surprising that steroids can induce enduring epigenetic changes in the brain, a notion first elucidated almost 20 years ago by Pfaff et al. (1992) invoking “gene memory” as an explanation for the differential sensitivity of male and female brains to estradiol-induced transcription of preproenkephalin in the mediobasal hypothalamus. More recently reported are sex differences in histone acetylation in broad brain regions (Tsai et al., 2009). Many of the participants in this Society for Neuroscience mini-symposium and coauthors on this report have also been making major inroads into the epigenetic underpinnings of sex differences in the brain. But it is equally clear we are only at the beginning, and it is the goal of this mini-review to highlight both what we know so far and point the spotlight forward to what we do not know but should.
Steroids impact DNA CpG methylation patterns
The aromatization hypothesis elucidates the principle that androgens produced by the testis lead to masculinization of the rodent brain in large part only after aromatization to estrogens, predominantly estradiol (Naftolin et al., 1975). Thus, much of our current information centers around estradiol and its receptors. There are two main isoforms of the estrogen receptor, ERα and ERβ. The dominant receptor developmentally is ERα, although ERβ is not unimportant (Kudwa et al., 2005). Overall, the molecular factors that control ER mRNA expression at the transcriptional level are not well known. One transcription factor, Stat5, has been shown to regulate ERα in the hypothalamus (Champagne et al., 2006). While the possibility of this factor regulating ERα mRNA in a similar manner in the cortex during development or after injury cannot be ruled out, it is likely that additional mechanisms of ERα mRNA regulation are involved. The regulation of gene expression by epigenetic modification is emerging as an important mechanism for controlling neuronal gene expression particularly during development. The ERα gene undergoes alterations in promoter methylation during normal as well as in pathological conditions. For example, methylation of the ER promoter has been reported to occur in the colon during aging (Issa et al., 1994), and changes in the expression of ER have been associated with the progression of breast and lung cancers (Ottaviano et al., 1994; Sasaki et al., 2002). During a neonatal sensitive period of brain development, estradiol acts upon ERα to transiently alter gene expression; however, the effects of this exposure persist throughout the individual's lifespan. One possible mechanism for a lasting difference in the brain is via an epigenetic process that creates methyl marks on DNA. Epigenetic processes begin with methylation of DNA by DNA methyltransferases. While methylation alone can interfere with protein binding and gene transcription, it is the binding of methyl-CpG-binding proteins, which subsequently recruit nuclear corepressor and histone deacetylase repressor complexes, that ultimately represses gene expression. Emerging evidence suggests sex differences in at least four related parameters: (1) DNA methylation patterns, (2) methyl transferases, (3) methyl-binding proteins, and (4) corepressor proteins, all of which can contribute to lasting differences in the brain (Fig. 1).
Figure 1.
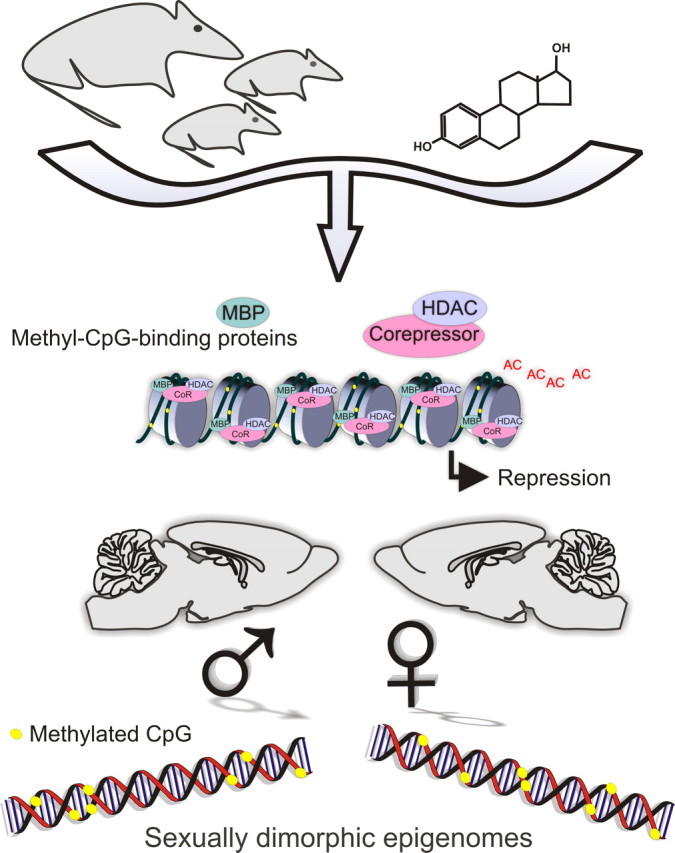
Epigenetics of sex differences in the brain. Emerging evidence suggests sex differences in at least four related parameters: (1) DNA methylation patterns, (2) methyl transferases, (3) methyl-binding proteins, and (4) corepressor proteins, all of which can contribute to lasting differences in the brain and behavior.
Brain sex differences and DNA methylation patterns
Previous research has indicated that the early social/maternal environment can modify sex differences in behavior, and recent evidence suggests that it may do so in an epigenetic manner. Variations in maternal care during the early neonatal period has enduring effects on adult sexual behavior (Moore, 1992; Rhees et al., 2001; Cameron et al., 2008) and on ERα expression by altering ERα promoter methylation (Champagne et al., 2006). As rat mothers typically groom the anogenital region of males more than females, it is possible that variations in maternal care may be contributing to sex differences in behavior by epigenetically modifying the genome. This would suggest that the mom's behavior may place marks on DNA that later code for male versus female gene transcription rates. The level of ERα expression in the preoptic area is sexually dimorphic, with males exhibiting lower levels during development (DonCarlos and Handa, 1994; Yokosuka et al., 1997; Yamamoto et al., 2006), and this difference appears to persist into adulthood (Brown et al., 1988; Lauber et al., 1991; Maerkel et al., 2007). As the ERα promoter region is subject to epigenetic modification by maternal–pup interactions, Anthony Auger and colleagues examined whether sex differences occur in ERα promoter CpG methylation within the developing preoptic area (POA). They also examined whether estradiol or somatosensory stimuli associated with maternal grooming contribute to this difference. This would suggest that sex differences organized by a transient hormone surge or early social experience may be maintained through epigenetic modification of DNA. There is a sex difference in methylation of the ERα promoter within the developing rat preoptic area. Specifically, males exhibit more ERα promoter methylation than females, and the increase in methylation correlates with the decreased ERα expression in males, thereby providing evidence that estradiol exposure and somatosensory stimuli associated with maternal grooming may partly establish this sex difference. These data suggest that early social interaction, similar to hormones, may converge at the genome to organize typical sex differences in the brain via epigenomic differentiation. These data also suggest how early neonatal experiences may be stored on DNA and ultimately shape lasting sex differences in brain and behavior.
The work of Jaclyn Schwarz and Bridget Nugent suggests that there may be a more complex pattern of early epigenetic marks in the developing and adult POA. In contrast to Auger's findings, Schwarz and Nugent saw increased methylation in 1- to 2-d-old pups in the female POA, when compared to males and estradiol-treated females, at two specific CpG sites on the ERα promoter. Analysis at PN60 determined that the percentage of methylation at many CpG sites along the ERα promoter of adults was nearly double that of the levels seen at PN1, suggesting a developmental regulation of ERα methylation regardless of sex. The effect of sex and neonatal hormone exposure at PN1 persisted into adulthood, however, at only one site along the ERα promoter. The individual CpG sites exhibiting changes during development and in adulthood were not the same, revealing yet another level of complexity. As adults, females have nearly 30% greater methylation at a CpG site at which there was no effect of sex or hormone in the neonate. How and why the pattern of CpG methylation of the ERα promoter changes across development is not known, but these data indicate that during the critical period of sexual differentiation, estradiol exposure can epigenetically modulate the expression of its own receptor, ERα. In contrast, there was no effect of sex or hormone on the methylation levels detected on the ERβ promoter. The POA is a brain region important to the expression of male sex behavior, and therefore methylation on the ERα promoter may be increased in adult females in order to silence estradiol-responsive sites relevant to male sex behavior.
The hypothalamus also exhibits sex differences in neuronal morphology and is a brain region critical to the expression of female sexual behavior, making it an important site of estradiol-induced defeminization in the male brain. A concurrent analysis to that done in the POA reveals that again 1-d-old females have significantly higher levels of methylation than males at two CpG sites along the ERα promoter. In contrast to the POA, treatment of females with estradiol 24 h before analysis only decreased methylation at these sites to intermediate levels, not significantly different from either males or females. Analysis of the progesterone receptor (PR) promoter region revealed a similar effect. Females had significantly greater levels of methylation than males at one site along the PR promoter, and treatment of females with estradiol intermediately reduced methylation at this same site to levels not significantly different from males or females. The induction of PR by estradiol treatment is a well established primary effect of the steroid. These results expand the epigenetic impact of estradiol beyond its own receptor to include, at least, progesterone receptor. On the PR promoter, the sex difference seen neonatally was again reversed in adulthood. Specifically, males had a significantly greater level of methylation than females at the same CpG site modulated in the neonate. In addition, neonatal treatment of females with estradiol significantly increased methylation at this same CpG site to levels seen in males at adulthood. Thus this specific CpG site on the PR promoter may be an important site of epigenetic modification by estradiol during development and adulthood.
The POA and hypothalamus are canonical brain regions rich in ER and subjected to the organizational/activational hypothesis of steroid-induced sexual differentiation. Both areas exhibit profound and robust sex differences in neuronal and glial morphology and/or number and both areas are critical to the expression of sexual behavior in adulthood. However ER of both isoforms are also found in regions outside of the POA and hypothalamus and with distinct patterns of expression in different brain regions and with varying levels of expression during different stages of development (DonCarlos, 1996; Shughrue et al., 1997). High levels of estrogen receptor protein expression in extrahypothalamic areas including the cortex and hippocampus are observed at birth, but decline as animals approach puberty (Pfaff and Keiner, 1973). ER mRNA expression changes correlate with the changes in protein expression in the hippocampus and cortex (Prewitt and Wilson, 2007), suggesting the developmental changes are in transcriptional regulation and not due to posttranscription or translational processes. To determine whether methylation correlates with the decline in ER mRNA expression in the cortex during early postnatal development, Wilson and colleagues examined the methylation status of several of the ER promoters and found at least one of the six promoters of the mouse ERα gene becomes progressively methylated beginning at postnatal day 10 in both male and female mice, which corresponds with the age at which the decline in ER mRNA expression in the cortex begins.
In addition to the changes in expression in ERα mRNA observed during development, ERα expression is also dramatically regulated following brain injury. Low, physiological levels of estradiol prevent cell death in the cortex following middle cerebral artery occlusion (MCAO) and in this model, the estradiol-mediated neuroprotection requires ERα (Dubal et al., 2001). As discussed above, ERα expression is very low in the adult cortex, however, following unilateral MCAO, ERα levels in the cortex are rapidly increased on the injured side of the brain (Dubal et al., 2006). This upregulation is independent of exogenous estradiol treatment. Furthermore, the increase in ERα mRNA expression only occurs in females, as males do not show a similar increase in the absence or presence of estradiol (Westberry et al., 2008). When Wilson et al. examined the methylation status of the ER promoter in male and female rats following MCAO they observed that ischemia decreased methylation of the ERα promoter in the ischemic cortex in females, while there was no change in methylation in the ERα promoter in males. Again this difference was independent of exogenous estradiol treatment, suggesting an intrinsic gender difference in ERα promoter methylation and mRNA expression.
The data obtained from both the POA and the hypothalamus indicate that estradiol epigenetically regulates genes underlying sex differences in the brain and behavior, whereas the data from the cortex suggest intrinsic factors, i.e., genetic sex, can also mediate epigenetic changes in ERα. Moreover there are both site- and region-specific developmental changes in the methylation pattern in the ERα promoter, and there is plasticity in response in the adult as demonstrated by changes following injury. Thus the estrogen receptor seems to represent a particularly complex yet facile target for epigenetic regulation. This conclusion is both expected and surprising. Estrogens and estrogen receptors are proving to be far more mechanistically multifaceted than ever imagined, with rapid actions at the membrane as well as the nucleus, local intraneuronal synthesis, and the capacity to activate kinases and directly modulate ion channel function and hence neuronal excitability. We can now add epigenetic regulation of the promoter to the growing list of ways in which this receptor both modulates and is modulated. Connecting these epigenetic changes to estrogen-mediated functional endpoints is the more daunting challenge. While there are many effects of ER that are directly relevant to neuronal function, there are many more where ER serves the purpose of regulating a downstream mediator that subsequently alters neuronal morphology or function (McCarthy, 2008). Thus it could be argued that the important epigenetic changes are ones induced by ER activation on other genes, not the ER itself. To date, other than PR, there have been no such effects identified.
Brain sex differences and methyl-binding proteins
Differences in methylation are only one step in gene repression. Methylated DNA is then bound by methyl-binding proteins, one of which is called methyl-CpG-binding protein 2 (MeCP2). Upon binding to methylated DNA, methyl-binding proteins, such as MeCP2, recruit corepressor proteins and HDACs to modify chromatin and repress gene transcription (Nan et al., 1998). Interestingly, mutations of the MECP2 gene appear to cause Rett syndrome (Amir et al., 1999), while subtle reductions of MeCP2 expression are speculated to be associated with male dominated social and neurodevelopmental disorders (Nagarajan et al., 2006). To determine whether MeCP2 expression is critical for social development and sexual differentiation of behavior, we used siRNA to target local and transient reduction of MeCP2 protein in the developing amygdala. We then assessed the impact of this reduction on juvenile social play behavior, a behavior which occurs at higher rates in juvenile males contrasted to females (Olioff and Stewart, 1978). Interestingly, neonatal treatment with MeCP2 siRNA disrupted juvenile social play behavior in males but not females. In contrast, reduced Mecp2 expression did not appear to alter juvenile sociability or adult anxiety-like behavior, suggesting that this disruption may be associated with subtle behavioral modification (Kurian et al., 2008). These data support that epigenetic processes, such as methyl-binding proteins, likely contribute to differentiation of brain and social behavior. They also suggest that molecules reading DNA marks are also important factors for typical social development.
Brain sex differences and corepressors
The next step to gene repression following binding of methyl-binding proteins to methylated DNA is the recruitment of corepressor complexes. Corepressors decrease gene transcription through their association with HDACs (Tsai and O'Malley, 1994). One of the first corepressors to be identified was nuclear receptor corepressor (NCoR), via its interaction with thyroid hormone receptors (Hörlein et al., 1995). It was then found to be a corepressor molecule for androgen and estrogen receptors (Lavinsky et al., 1998; Yoon and Wong, 2006). NCoR also represses gene expression via its direct or indirect interactions with methyl-binding proteins, such as Kaiso (Yoon et al., 2003) and possibly MeCP2 (Kokura et al., 2001; Cukier et al., 2008). Auger and colleagues have identified a sex difference in the expression of NCoR within the developing amygdala, and that estradiol exposure may be partly responsible for this sex difference. The corepressor molecule, NCoR, acts in the brain to blunt sex differences in juvenile social play behavior in males, and appears critical for appropriate anxiety-like behavior in both juvenile males and females. As NCoR participates in DNA methylation-induced gene repression, it is possible that abnormal functioning of NCoR may underlie increased anxiety in some individuals with epigenetic neural developmental disorders associated with NCoR dysfunction, such as Rett syndrome or Huntington's disease. As nuclear receptors are involved in numerous processes within an organism, it is crucial to understand how these processes govern nuclear receptor activity. The modification of DNA, methyl-binding proteins, and coregulatory proteins are likely a mechanism for how relatively few molecules can impact a large number of transcriptional responses or provide a mechanism for how DNA is transcribed in response to changes in steroid hormones or the social environment.
Brain sex differences and DNMT activity
The observation of sex differences in CpG methylation within 24 h of birth or after exogenous estradiol treatment prompted an investigation by Schwarz and colleagues into activity of the DNMT enzymes which transfer methyl groups to cytosines in CpG dinucleotides. Overall DNMT activity in the POA of 1-d-old pups was significantly greater in males than in females, and increased in females by exogenous estradiol treatment consistent with the direction of effects observed on CpG methylation. Protein levels of DNMT1 and DNMT3, the two isoforms important for maintenance and de novo methylation respectively, were not affected by sex or estradiol treatment, suggesting the primary mode of regulation is activity, not enzyme synthesis. The gratifyingly consistent relationship between DNMT activity and levels of methylation of ERα promoter in the POA was not maintained for the mediobasal hypothalamus were 1-d-old males also had significantly higher levels of DNMT activity than females, and treatment of females with estradiol significantly increased the activity of DNMTs within 24 h. These results are contrary to the methylation levels observed on the two genes described above, ERα and PR, and indicate that there may be an estradiol-mediated increase in methylation levels of other genes in the MBH that have not yet been explored. Indeed, this is more in line of what would be expected, as discussed above, given that estradiol predominantly acts to induce other signal transduction systems to alter neuronal function. Nonetheless, establishing how estradiol increases DNMT activity will be an important future direction. Connecting steroid-induced changes in DNMT activity to specific genes will also greatly advance our understanding of the enduring consequences of hormone exposure during sensitive developmental periods.
Brain sex differences and histone modifications
Morphological sex differences in the nervous system are established by exposure to testosterone and its aromatized end-product, estradiol, during critical periods in development. In most cases the mechanism(s) through which testosterone and estradiol direct these neural changes are not known. Effects of early exposure to steroids last a lifetime, and there is often a delay between the hormone exposure and the appearance of a morphological sex difference. For example, the bed nucleus of the stria terminalis (BNST) is a limbic forebrain region that exhibits several sex differences. The most prominent region of the BNST, the principal nucleus (BNSTp), is larger in volume and contains more cells in males than in females. This has been established in mice, rats, guinea pigs, and humans (Hines et al., 1985, 1992; Guillamón et al., 1988; Forger et al., 2004). In mice and rats, this sex difference is due to greater developmental cell death in females. The sex difference in cell death is not apparent until approximately postnatal day 6, but is determined by testosterone exposure on the day of birth (Chung et al., 2000; Gotsiridze et al., 2007; Hisasue et al., 2007).
Elaine Murray and colleagues hypothesized that sexual differentiation of BNSTp cell number requires orchestrated changes in histone acetylation following testosterone exposure. As discussed above, several of the best-known steroid hormone receptor coactivators have HAT activity or recruit HATs to the transcription complex whereas corepressors often have HDAC activity (Spencer et al., 1997; Kishimoto et al., 2006; Kininis et al., 2007). To test whether sex differences in the BNSTp require changes in histone acetylation/deacetylation, Murray and colleagues administered an HDAC inhibitor, valproic acid (VPA), to neonatal mice during the critical period for sexual differentiation. They found that treatment with the HDAC inhibitor transiently increased histone H3 acetylation level in the brain of newborn mice (Murray et al., 2009a). In addition, when examined at 3 weeks of age (i.e., after the period of sexual differentiation), males and androgenized females treated with VPA had female-like volume and cell number in the BNSTp. Thus, inhibition of HDAC activity prevented masculinization of cell number in the BNSTp. Importantly, these effects were dependent on hormone status, as HDAC inhibition had no effect in females not treated with testosterone. Valproic acid also had no effect on volume and cell number in two control brain regions that were not sexually dimorphic. These findings are consistent with the hypothesis that testosterone acts via epigenetic processes, in particular the regulation of histone acetylation, to direct sexual differentiation of the brain that is determined by sex differences in cell death. In normal cell populations treatment with an HDAC inhibitor alters the expression of a relatively small number of genes. The genes affected, however, are often related to the cell cycle or apoptosis (Glaser et al., 2003; Menegola et al., 2007). The sex difference in cell number in the BNSTp requires bax, a prodeath member of the bcl-2 family of apoptosis related genes (Forger et al., 2004; Gotsiridze et al., 2007).Therefore, a disruption of the hormonal regulation of bax or other bcl-2 family genes is a potential mechanism of valproic acid action.
Although an HDAC inhibitor blocked masculinization in the BNSTp, it is unlikely that a simple formula such as “more acetylation equals more masculinization” will apply to all brain areas. For example, males of many species have greater expression of the neuropeptide vasopressin in the lateral septum than do females (De Vries and Panzica, 2006). Preliminary findings suggest that the same valproic acid treatment that blocks masculinization of the BNSTp may masculinize vasopressin expression in female mice (Murray et al., 2009b). Upon reflection, this is not surprising. It is unlikely that development of a masculine phenotype depends on global increases in histone acetylation or that female development requires uniformly less acetylation. Rather, gonadal steroid hormones and their receptors, in conjunction with coactivators and corepressors with HAT or HDAC activity, respectively, likely orchestrate orderly patterns of acetylation of histones associated with specific genes. This would lead to increased expression of some genes in males and others in females, as is in fact what is seen (Speert et al., 2007).
Understanding the epigenetic regulation of cell number in the BNSTp and in other brain regions may have important clinical implications. The BNSTp plays a vital role in modulating emotional and stress responses (Choi et al., 2007, 2008) and sex differences in stress responsivity are well established (Bale, 2006). Alterations in histone proteins and DNA methylation have been identified in patients with schizophrenia, bipolar disorder, and other psychiatric diseases, and several of the changes seen in patient populations are sex specific (Connor and Akbarian, 2008; Akbarian and Huang, 2009). Thus, understanding how gonadal steroids alter the neuronal epigenome is likely to be important for explaining sex differences in the susceptibility to psychiatric and neurological disease.
Sex differences in brain and epigenetic changes to the germline
The discussion thus far has focused on epigenetic changes that would be considered context dependent, meaning they occur only within a single lifespan and may be transient within that lifespan. This is indeed appropriate for changes that are opportunistically established in response to experience or hormone exposure. Emerging evidence now indicates germline epigenetic changes can also influence brain and behavior sex differences.
Environmental endocrine-disrupting compounds which either mimic or disrupt steroid hormone signaling are an increasing source of concern for numerous reasons. This list has been made even longer by the observations of Andrea Gore, David Crews, and colleagues of transgenerational changes in brain and behavior induced by agricultural chemicals such as vinclozolin, a fungicide with antiandrogenic activity (Skinner et al., 2008). A wide ranging analysis of the transcriptome indicated that the expression of 92 genes in the hippocampus and 276 genes in the amygdala were transgenerationally altered in males exposed to vinclozolin. In the females, the expression of 1301 genes in the hippocampus and 172 genes in the amygdala were transgenerationally altered. Examination of the F3 generation indicated opposite effects on anxiety-like behaviors in males versus females. Relating transgenerational changes in the transcriptome to these behavioral effects, and why and where they are different in males and females presents a difficult but critical hurdle.
Along with the alarming increases in environmental chemicals of unknown action, industrialized nations have also witnessed rapid and profound elevations in obesity rates and, to a lesser extent, height over the extraordinarily brief evolutionary time scale of approximately five generations (James, 2008). While stochastic Mendelian inheritance is not likely to explain such rapid escalations, changes in available nutrition can clearly shape these traits. However, putative epigenetic mechanisms by which nutritive factors program future generations to inherit either an obese or tall phenotype have not been well characterized. In response to maternal high fat diet during gestation and nursing, Tracy Bale and Greg Dunn previously reported that two generations of offspring exhibit increased body length, reduced insulin sensitivity, and reduced leptin levels. Both male and female first-generation offspring exposed to maternal high fat diet transmit this phenotype to second-generation animals. That the paternal lineage transmits the body length and insulin insensitivity traits is compelling evidence of a germline-based epigenetic mechanism of inheritance. This result avoids confounding variables associated with maternal transmission where potentially altered uterine environment, maternal behavior, or metabolism and not an intergenerational epigenetic effect could influence the first- to second-generation transmission. Additionally, their studies provide evidence for a sex-dependent mechanism in both first- and second-generation female offspring where female insulin-like growth factor 1 (IGF-1) levels were masculinized by maternal high-fat diet. Female-specific elevations in IGF-1 are accompanied by increased expression of the growth hormone-secretion stimulating ghrelin receptor (GHSR). These alterations correlated with significantly decreased expression of a GHSR-associated transcriptional repressor, AF5q31, exclusively in female offspring. Transcriptional alterations in GHSR are associated with reduced DNA methylation at the GHSR CpG island, providing a candidate epigenetic basis for the transmission of the phenotype.
Though Bale and Dunn are the first to report increased body length in response to maternal diet, studies in a variety of model organisms have detected the perpetuation of obesity, metabolic syndrome, liver dysfunction, and cardiovascular disease through at least the first generation (Férézou-Viala et al., 2007; Gniuli et al., 2008; Parente et al., 2008). Exposure to altered maternal diet and its physiological consequences can program offspring for disease risk via a variety of mechanisms including leptin or insulin dysregulation, intrauterine growth restriction (IUGR), an altered supply of methyl donor molecules such as folic acid, or gestational diabetes. However, acute exposure to altered maternal diet may result in some traits that terminate with the first generation, whereas others may be epigenetically inherited by the second generation. Moreover, sex differences exist in both the capacity to transmit traits as well as to inherit them. For example, recent evidence supports sex dependency in the transmission of obesity and glucose intolerance resulting from maternal caloric restriction, as reduced birth weight persisted in both male and female second-generation offspring solely through the paternal lineage whereas obesity transmitted only through the maternal line in ICR strain mice (Jimenez-Chillaron et al., 2009). Embryo transfer from pregnant IUGR rats to control rats resulted in hyperglycemia, hyperinsulinemia, and increased hepatic weight in second-generation offspring, but only in females (Thamotharan et al., 2007). However, repeatedly fasting adult male Swiss mice resulted in the transmission of a reduced serum glucose phenotype to both male and female offspring, suggesting a germline-based effect that is distinct from the phenotypes transmitted by calorie restricted females (Anderson et al., 2006).
Additional research aims to parse out sex differences in heritability for maternal diet exposure phenotypes. A recent study in rats detected increased body mass in second-generation males but impairments in glucose metabolism in both sexes in response to in utero protein restriction, supporting an inheritance dichotomy (Pinheiro et al., 2008). Furthermore, the timing of protein restriction during development mediates sex-dependent inheritance in second-generation offspring, as male rats developed insulin resistance in response to restriction during the nursing period while females developed sensitivity following gestational restriction (Zambrano et al., 2005). In humans, examination of the relationship between food supply and disease mortality across generations in the Swedish parish of Overkalix revealed similar sex differences in transmission and inheritance (Pembrey et al., 2006). Links between grandmaternal diet during pregnancy and granddaughters' mortality risk were found in addition to associations between grandpaternal diet during the slow growth period before puberty and mortality risk only in grandsons (Pembrey et al., 2006). Excessive caloric intake in males during the slow growth period also resulted in an increased risk for diabetes-associated mortality in grandchildren (Kaati et al., 2002). The authors suggest that the sex specificity of transmission observed in these studies is due to the respective windows of sensitivity for developing germ cells in males and females.
Exposure to both excessive and restricted consumption by either parent results in sex-specific outcomes that vary in transmissibility. The findings of Bale and Dunn provide compelling support for an adaptive, sex-dependent epigenetic mechanism linking body size with maternal diet. As enhanced body size can augment the fitness of an organism under certain circumstances, an increase in body length in response to maternal high fat diet provides an epigenetic contribution that may explain increases in human height over the last century. These results are a novel but not surprising addition to the body of evidence identifying the consequences of maternal diet on the sex-specific inheritance and transmission of traits. Genome-wide transcriptional variance underwrites sexual dimorphisms observed between males and females as a result of differential gonadal steroids, sex chromosomes, and imprinting.
Summary and conclusions
The relative importance of epigenetic modifications to the establishment and maintenance of sex differences in brain and behavior remains an open question, but that there is some role cannot be denied. The promoter of the ERα gene appears to be a particularly robustly and reliably regulated site of epigenetic modification, but whether this is due to looking where the light is good, or a genuine reflection of the essential biology, remains to be seen. Histones and their acetylation are another source of sex-specific responses and the observation that this includes effects on cell death speaks to its fundamental importance. Expanding our field of view beyond the classic brain regions involved in sexual differentiation of sex behavior reveals a rich array of additional effects that include the response to brain injury, effects of endocrine-disrupting compounds, and the consequences of maternal diet to the primary offspring and subsequent generations.
Together, these results suggest that despite the supremacy of gonadal steroid hormones, the battle of the sexes is ultimately fought upon the DNA. It is the combination of chromosomes, steroid hormones, and early neonatal experiences that guide this DNA scuffle. Ultimately we require a thorough understanding of regulation of enzymes controlling acetylation and methylation, coregulatory proteins, transient versus stable DNA methylation patterns, steroid receptor cycling on/off DNA, and sex differences across the epigenome to fully understand sex differences in brain and behavior.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R01 MH52716 and R01 NS050525 to M.M.M., NIH Grant R01 MH072956 to A.P.A., National Science Foundation Grant IOS-0919944 to M.E.W., NIH Grants R01 MH068482 and K02 MH072825 to N.G.F., NIH Grant R01 MH047538 to G.J.D., and pilot funding from the University of Pennsylvania Diabetes Center NIH Grant DK019525 to T.L.B.
References
- Akbarian S, Huang HS. Epigenetic regulation in human brain-focus on histone lysine methylation. Biol Psychiatry. 2009;65:198–203. doi: 10.1016/j.biopsych.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Anderson LM, Riffle L, Wilson R, Travlos GS, Lubomirski MS, Alvord WG. Preconceptional fasting of fathers alters serum glucose in offspring of mice. Nutrition. 2006;22:327–331. doi: 10.1016/j.nut.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet. 2001;2:59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- Bale TL. Stress sensitivity and the development of affective disorders. Horm Behav. 2006;50:529–533. doi: 10.1016/j.yhbeh.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Brown TJ, Hochberg RB, Zielinski JE, MacLusky NJ. Regional sex differences in cell nuclear estrogen-binding capacity in the rat hypothalamus and preoptic area. Endocrinology. 1988;123:1761–1770. doi: 10.1210/endo-123-4-1761. [DOI] [PubMed] [Google Scholar]
- Cameron N, Del Corpo A, Diorio J, McAllister K, Sharma S, Meaney MJ. Maternal programming of sexual behavior and hypothalamic-pituitary-gonadal function in the female rat. PLoS ONE. 2008;3:e2210. doi: 10.1371/journal.pone.0002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147:2909–2915. doi: 10.1210/en.2005-1119. [DOI] [PubMed] [Google Scholar]
- Chen X, Grisham W, Arnold AP. X chromosome number causes sex differences in gene expression in adult mouse striatum. Eur J Neurosci. 2009;29:768–776. doi: 10.1111/j.1460-9568.2009.06610.x. [DOI] [PubMed] [Google Scholar]
- Choi DC, Furay AR, Evanson NK, Ostrander MM, Ulrich-Lai YM, Herman JP. Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic-pituitary-adrenal axis activity: implications for the integration of limbic inputs. J Neurosci. 2007;27:2025–2034. doi: 10.1523/JNEUROSCI.4301-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DC, Evanson NK, Furay AR, Ulrich-Lai YM, Ostrander MM, Herman JP. The anteroventral bed nucleus of the stria terminalis differentially regulates hypothalamic-pituitary-adrenocortical axis responses to acute and chronic stress. Endocrinology. 2008;149:818–826. doi: 10.1210/en.2007-0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WC, Swaab DF, De Vries GJ. Apoptosis during sexual differentiation of the bed nucleus of the stria terminalis in the rat brain. J Neurobiol. 2000;43:234–243. [PubMed] [Google Scholar]
- Connor CM, Akbarian S. DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics. 2008;3:55–58. doi: 10.4161/epi.3.2.5938. [DOI] [PubMed] [Google Scholar]
- Crews D. Epigenetics and its implications for behavioral neuroendocrinology. Front Neuroendocrinol. 2008;29:344–357. doi: 10.1016/j.yfrne.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier HN, Perez AM, Collins AL, Zhou Z, Zoghbi HY, Botas J. Genetic modifiers of MeCP2 function in Drosophila. PLoS Genet. 2008;4:e1000179. doi: 10.1371/journal.pgen.1000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies W, Lynn PM, Relkovic D, Wilkinson LS. Imprinted genes and neuroendocrine function. Front Neuroendocrinol. 2008;29:413–427. doi: 10.1016/j.yfrne.2007.12.001. [DOI] [PubMed] [Google Scholar]
- De Vries GJ, Panzica GC. Sexual differentiation of central vasopressin and vasotocin systems in vertebrates: different mechanisms, similar endpoints. Neuroscience. 2006;138:947–955. doi: 10.1016/j.neuroscience.2005.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DonCarlos LL. Developmental profile and regulation of estrogen receptor (ER) mRNA expression in the preoptic area of prenatal rats. Brain Res Dev Brain Res. 1996;94:224–233. doi: 10.1016/0165-3806(96)00067-3. [DOI] [PubMed] [Google Scholar]
- DonCarlos LL, Handa RJ. Developmental profile of estrogen receptor mRNA in the preoptic area of male and female neonatal rats. Brain Res Dev Brain Res. 1994;79:283–289. doi: 10.1016/0165-3806(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Rau SW, Shughrue PJ, Zhu H, Yu J, Cashion AB, Suzuki S, Gerhold LM, Bottner MB, Dubal SB, Merchanthaler I, Kindy MS, Wise PM. Differential modulation of estrogen receptors (ERs) in ischemic brain injury: a role for ERalpha in estradiol-mediated protection against delayed cell death. Endocrinology. 2006;147:3076–3084. doi: 10.1210/en.2005-1177. [DOI] [PubMed] [Google Scholar]
- Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- Férézou-Viala J, Roy AF, Sérougne C, Gripois D, Parquet M, Bailleux V, Gertler A, Delplanque B, Djiane J, Riottot M, Taouis M. Long-term consequences of maternal high-fat feeding on hypothalamic leptin sensitivity and diet-induced obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1056–R1062. doi: 10.1152/ajpregu.00117.2007. [DOI] [PubMed] [Google Scholar]
- Forger NG, Rosen GJ, Waters EM, Jacob D, Simerly RB, de Vries GJ. Deletion of Bax eliminates sex differences in the mouse forebrain. Proc Natl Acad Sci U S A. 2004;101:13666–13671. doi: 10.1073/pnas.0404644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. 2003;2:151–163. [PubMed] [Google Scholar]
- Gniuli D, Calcagno A, Caristo ME, Mancuso A, Macchi V, Mingrone G, Vettor R. Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J Lipid Res. 2008;49:1936–1945. doi: 10.1194/jlr.M800033-JLR200. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Gore AC. Developmental programming and endocrine disruptor effects on reproductive neuroendocrine systems. Front Neuroendocrinol. 2008;29:358–374. doi: 10.1016/j.yfrne.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotsiridze T, Kang N, Jacob D, Forger NG. Development of sex differences in the principal nucleus of the bed nucleus of the stria terminalis of mice: role of Bax-dependent cell death. Dev Neurobiol. 2007;67:355–362. doi: 10.1002/dneu.20353. [DOI] [PubMed] [Google Scholar]
- Guillamón A, Segovia S, del Abril A. Early effects of gonadal steroids on the neuron number in the medial posterior region and the lateral division of the bed nucleus of the stria terminalis in the rat. Brain Res Dev Brain Res. 1988;44:281–290. doi: 10.1016/0165-3806(88)90226-x. [DOI] [PubMed] [Google Scholar]
- Hines M, Davis FC, Coquelin A, Goy RW, Gorski RA. Sexually dimorphic regions in the medial preoptic area and the bed nucleus of the stria terminalis of the guinea pig brain: a description and an investigation of their relationship to gonadal steroids in adulthood. J Neurosci. 1985;5:40–47. doi: 10.1523/JNEUROSCI.05-01-00040.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines M, Allen LS, Gorski RA. Sex differences in subregions of the medial nucleus of the amygdala and the bed nucleus of the stria terminalis of the rat. Brain Res. 1992;579:321–326. doi: 10.1016/0006-8993(92)90068-k. [DOI] [PubMed] [Google Scholar]
- Hisasue S, Seney ML, Immerman E, Forger NG. Cell death and the development of sex differences in the bed nucleus of the stria terminalis in mice. Soc Neurosci Abstr. 2007;33:294–23. [Google Scholar]
- Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Söderström M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- James WP. The epidemiology of obesity: the size of the problem. J Intern Med. 2008;263:336–352. doi: 10.1111/j.1365-2796.2008.01922.x. [DOI] [PubMed] [Google Scholar]
- Jimenez-Chillaron JC, Isganaitis E, Charalambous M, Gesta S, Pentinat-Pelegrin T, Faucette RR, Otis JP, Chow A, Diaz R, Ferguson-Smith A, Patti ME. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes. 2009;58:460–468. doi: 10.2337/db08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents' and grandparents' slow growth period. Eur J Hum Genet. 2002;10:682–688. doi: 10.1038/sj.ejhg.5200859. [DOI] [PubMed] [Google Scholar]
- Keverne EB, Curley JP. Epigenetics, brain evolution and behaviour. Front Neuroendocrinol. 2008;29:398–412. doi: 10.1016/j.yfrne.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC, Clark AG, Kraus WL. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol. 2007;27:5090–5104. doi: 10.1128/MCB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto M, Fujiki R, Takezawa S, Sasaki Y, Nakamura T, Yamaoka K, Kitagawa H, Kato S. Nuclear receptor mediated gene regulation through chromatin remodeling and histone modifications. Endocr J. 2006;53:157–172. doi: 10.1507/endocrj.53.157. [DOI] [PubMed] [Google Scholar]
- Kokura K, Kaul SC, Wadhwa R, Nomura T, Khan MM, Shinagawa T, Yasukawa T, Colmenares C, Ishii S. The Ski protein family is required for MeCP2-mediated transcriptional repression. J Biol Chem. 2001;276:34115–34121. doi: 10.1074/jbc.M105747200. [DOI] [PubMed] [Google Scholar]
- Kudwa AE, Bodo C, Gustafsson JA, Rissman EF. A previously uncharacterized role for estrogen receptor beta: defeminization of male brain and behavior. Proc Natl Acad Sci U S A. 2005;102:4608–4612. doi: 10.1073/pnas.0500752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian JR, Bychowski ME, Forbes-Lorman RM, Auger CJ, Auger AP. Mecp2 organizes juvenile social behavior in a sex-specific manner. J Neurosci. 2008;28:7137–7142. doi: 10.1523/JNEUROSCI.1345-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber AH, Mobbs CV, Muramatsu M, Pfaff DW. Estrogen receptor messenger RNA expression in rat hypothalamus as a function of genetic sex and estrogen dose. Endocrinology. 1991;129:3180–3186. doi: 10.1210/endo-129-6-3180. [DOI] [PubMed] [Google Scholar]
- Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, Hilsenbeck SG, Osborne CK, Glass CK, Rosenfeld MG, Rose DW. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A. 1998;95:2920–2925. doi: 10.1073/pnas.95.6.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerkel K, Durrer S, Henseler M, Schlumpf M, Lichtensteiger W. Sexually dimorphic gene regulation in brain as a target for endocrine disrupters: developmental exposure of rats to 4-methyl benzylidene camphor. Toxicol Appl Pharmacol. 2007;218:152–165. doi: 10.1016/j.taap.2006.10.026. [DOI] [PubMed] [Google Scholar]
- McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–124. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, de Vries GJ, Forger NG. Sexual differentiation of the brain: mode, mechanisms, and meaning. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, brain and behavior. Ed 2. Vol 3. San Diego: Academic; 2009. pp. 1707–1744. [Google Scholar]
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegola J, Steppe M, Schapoval EE. Dissolution test for citalopram in tablets and comparison of in vitro dissolution profiles. Eur J Pharm Biopharm. 2007;67:524–530. doi: 10.1016/j.ejpb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Moore CL. The role of maternal stimulation in the development of sexual behavior and its neural basis. Ann N Y Acad Sci. 1992;662:160–177. doi: 10.1111/j.1749-6632.1992.tb22859.x. [DOI] [PubMed] [Google Scholar]
- Murray EK, Hien A, de Vries GJ, Forger NG. Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology. 2009a;150:4241–4247. doi: 10.1210/en.2009-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray EK, Fernandez JL, Forger NG, De Vries GJ. Effect of perinatal HDAC inhibitor administration on sexual differentiation of the vasopressin innervation in the mouse brain. Soc Neurosci Abstr. 2009b;35:666–3. [Google Scholar]
- Naftolin F, Ryan KJ, Davies IJ, Reddy VV, Flores F, Petro Z, Kuhn M, White RJ, Takaoka Y, Wolin L. The formation of estrogens by central neuroendocrine tissues. Recent Prog Horm Res. 1975;31:295–319. doi: 10.1016/b978-0-12-571131-9.50012-8. [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Olioff M, Stewart J. Sex differences in the play behavior of prepubescent rats. Physiol Behav. 1978;20:113–115. doi: 10.1016/0031-9384(78)90060-4. [DOI] [PubMed] [Google Scholar]
- Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–2555. [PubMed] [Google Scholar]
- Parente LB, Aguila MB, Mandarim-de-Lacerda CA. Deleterious effects of high-fat diet on perinatal and postweaning periods in adult rat offspring. Clin Nutr. 2008;27:623–634. doi: 10.1016/j.clnu.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjöström M, Golding J. Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet. 2006;14:159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- Pfaff D, Keiner M. Atlas of estradiol concentrating cells in the central nervous system of the female rat. J Comp Neurol. 1973;151:121–158. doi: 10.1002/cne.901510204. [DOI] [PubMed] [Google Scholar]
- Pfaff DW, Brooks PJ, Funabashi T, Pfaus JG, Mobbs CV. Gene memory in neuroendocrine and behavioural systems. Ciba Found Symp. 1992;168:165–183. doi: 10.1002/9780470514283.ch11. discussion 183–186. [DOI] [PubMed] [Google Scholar]
- Pinheiro AR, Salvucci ID, Aguila MB, Mandarim-de-Lacerda CA. Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin Sci (Lond) 2008;114:381–392. doi: 10.1042/CS20070302. [DOI] [PubMed] [Google Scholar]
- Prewitt AK, Wilson ME. Changes in estrogen receptor-alpha mRNA in the mouse cortex during development. Brain Res. 2007;1134:62–69. doi: 10.1016/j.brainres.2006.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhees RW, Lephart ED, Eliason D. Effects of maternal separation during early postnatal development on male sexual behavior and female reproductive function. Behav Brain Res. 2001;123:1–10. doi: 10.1016/s0166-4328(00)00381-8. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Tanaka Y, Perinchery G, Dharia A, Kotcherguina I, Fujimoto S, Dahiya R. Methylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancer. J Natl Cancer Inst. 2002;94:384–390. doi: 10.1093/jnci/94.5.384. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Skinner MK, Anway MD, Savenkova MI, Gore AC, Crews D. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One. 2008;3:e3745. doi: 10.1371/journal.pone.0003745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speert DB, Konkle AT, Zup SL, Schwarz JM, Shiroor C, Taylor ME, McCarthy MM. Focal adhesion kinase and paxillin: novel regulators of brain sexual differentiation? Endocrinology. 2007;148:3391–3401. doi: 10.1210/en.2006-0845. [DOI] [PubMed] [Google Scholar]
- Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- Thamotharan M, Garg M, Oak S, Rogers LM, Pan G, Sangiorgi F, Lee PW, Devaskar SU. Transgenerational inheritance of the insulin-resistant phenotype in embryo-transferred intrauterine growth-restricted adult female rat offspring. Am J Physiol Endocrinol Metab. 2007;292:E1270–1279. doi: 10.1152/ajpendo.00462.2006. [DOI] [PubMed] [Google Scholar]
- Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- Tsai HW, Grant PA, Rissman EF. Sex differences in histone modifications in the neonatal mouse brain. Epigenetics. 2009;4:47–53. doi: 10.4161/epi.4.1.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westberry JM, Prewitt AK, Wilson ME. Epigenetic regulation of the estrogen receptor alpha promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience. 2008;152:982–989. doi: 10.1016/j.neuroscience.2008.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Carter CS, Cushing BS. Neonatal manipulation of oxytocin affects expression of estrogen receptor alpha. Neuroscience. 2006;137:157–164. doi: 10.1016/j.neuroscience.2005.08.065. [DOI] [PubMed] [Google Scholar]
- Yokosuka M, Okamura H, Hayashi S. Postnatal development and sex difference in neurons containing estrogen receptor-alpha immunoreactivity in the preoptic brain, the diencephalon, and the amygdala in the rat. J Comp Neurol. 1997;389:81–93. doi: 10.1002/(sici)1096-9861(19971208)389:1<81::aid-cne6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Yoon HG, Wong J. The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist- and antagonist-regulated transcription by androgen receptor. Mol Endocrinol. 2006;20:1048–1060. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Reynolds AB, Qin J, Wong J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell. 2003;12:723–734. doi: 10.1016/j.molcel.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Zambrano E, Martínez-Samayoa PM, Bautista CJ, Deás M, Guillén L, Rodríguez-González GL, Guzmán C, Larrea F, Nathanielsz PW. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J Physiol. 2005;566:225–236. doi: 10.1113/jphysiol.2005.086462. [DOI] [PMC free article] [PubMed] [Google Scholar]