

Abstract
Reported here are the competitive 18O/16O kinetic isotope effects (18O KIEs) on kcat/Km(O2) for three non-heme iron enzymes that activate O2 at an iron center coordinated by a 2-His-1-carboxylate facial triad: taurine dioxygenase (TauD), S-(2)-hydroxypropylphosphonic acid epoxidase (HppE), and 1-aminocyclopropyl-1-carboxylic acid oxidase (ACCO). The comparison of the measured 18O KIEs with calculated 18O equilibrium isotope effects (18O EIEs) reveals an excellent correlation with the proposed mechanisms for these enzymes. 18O KIEs of 1.0104 ± 0.0002 (TauD), 1.0120 ± 0.0002 (HppE), and 1.0215 ± 0.0005 (ACCO) suggest the formation in the rate limiting step of O2 activation of an FeIII-alkylperoxo, FeIII-OOH, and FeIV=O species, respectively. By probing only the steps from initial O2 binding up to and including the first irreversible step of O2 activation, the measured 18O KIEs can be a valuable companion to pre-steady state kinetic analyses in studying the complex catalytic mechanisms of non-heme iron enzymes.
The O2-activating, non-heme iron enzymes catalyze a wide range of oxygenation and oxidation reactions with important biological implications, such as DNA repair, hypoxic response, collagen biosynthesis, and histone demethylation.1 Most of these enzymes contain a single iron center coordinated by two His and one Asp/Glu residues in a tridentate binding motif referred to as “2-His-1-carboxylate facial triad”. Understanding the O2-activation process for these enzymes may provide key insights into the source of their divergent substrate specificity despite similarly coordinated active site metal centers.1
Several recent studies have employed the measurement of competitive 18O/16O kinetic isotope effects (18O KIEs) on kcat/Km(O2) for O2-activating metalloenzymes, in order to probe the early steps involved in O2 activation and to reveal the nature of the metal/O2 intermediate formed in the rate determining step (RDS) of kcat/Km(O2).2,3 Reported here are the 18O KIEs for three non-heme iron enzymes that activate O2 at an iron center coordinated by a 2-His-1-carboxylate facial triad: taurine dioxygenase (TauD), S-(2)-hydroxypropylphosphonic acid (S-HPP) epoxidase (HppE), and 1-aminocyclopropyl-1-carboxylic acid oxidase (ACCO). These 18O KIE measurements allow, for the first time, a direct comparison of the O2 activation process by non-heme iron enzymes employing different substrates and coreductants.1
TauD is an α-ketoglutarate (αKG)-dependent non-heme iron enzyme that catalyzes the hydroxylation of taurine in bacteria.4 Its mechanism has been extensively investigated and several Fe/O2 intermediates have been characterized, including a high-valent FeIV=O species.5–7 The RDS of kcat/Km(O2) in TauD is proposed to be the attack of an FeIII-OO•− species on αKG to form a cyclic alkylperoxo intermediate, which then undergoes oxidative decarboxylation to form the FeIV=O species. The measured 18O KIE for TauD is 1.0102 ± 0.0002 at 30 °C (Figure 1).8
Figure 1.

Isotope fractionation plots for TauD (●), HppE (■), and ACCO (◆). The fits for obtaining 18O KIEs are shown in solid, dashed, and dotted lines, respectively.8 Conditions: TauD: 0.2 μM TauD, 0.4–0.6 mM O2, 2 mM taurine, 2 mM αKG, 0.2 mM ascorbate, 50 mM BisTris pH 6.2, 30 °C; HppE: 10μM HppE, 0.4–0.6 mM O2, 1 mM S-HPP, 11 μM FMN, 1.5 mM NADH, 20 mM Tris-HCl pH 7.5, 25 °C; ACCO: 5 μM ACCO, 0.3–0.5 mM O2, 3 mM ACC, 20 mM ascorbate, 20 mM NaHCO3, 100 mM NaCl, 100 mM MOPS pH 7.2, 25 °C.
HppE is a reductase-dependent non-heme iron enzyme that catalyzes the epoxidation of S-HPP, the last step in the biosynthesis of the antibiotic fosfomycin.9 The mechanism of HppE is not as well known as for TauD, formation of an FeIII-OOH species being proposed to involve either a hydrogen atom transfer (HAT) from S-HPP or proton-coupled electron transfer (PCET) from the reductant.10 The measured 18O KIE for HppE is 1.0120 ± 0.0002 at 25 °C, using FMN in the presence of NADH as the reductant (Figure 1).8 This value is similar to the one obtained for TauD, suggesting a similar oxidation state for the Fe/O2 intermediate formed in the RDS.
ACCO is an ascorbate-dependent non-heme iron enzyme that catalyzes the last step in ethylene biosynthesis, an important plant hormone.11 Recent steady-state kinetic studies of ACCO suggest that substrate oxidation occurs after the RDS of O2 activation, which most probably involves the formation of an FeIV=O species.12 The 18O KIE for ACCO is 1.0215 ± 0.0005 at 25 °C (Figure 1), one of the largest values measured for O2-activating metalloenzymes.13
Competitive 18O KIEs on kcat/Km(O2) reflect changes in the oxygen bond order that occur in all steps from initial O2 binding up to and including the first irreversible step.19 To help interpret the measured 18O KIEs, calculated 18O equilibrium isotope effects (18O EIEs) can be obtained from vibrational frequencies of reactants and products,14 following the formalism developed by Bigeleisen and Mayer.20 These calculated 18O EIEs can be used as upper limits for the measured 18O KIEs (assuming a negligible isotope effect contribution from the reaction coordinate frequency),21 allowing a direct comparison between model reactions and experimental values. Using known frequencies for FeIII-OO•−,15 FeIII-OOH,1 and FeIV=O species,7 the relevant Fe/O2 intermediates for the studied enzymes,1,22 we calculated 18O EIEs of 1.0080, 1.0172, and 1.0287, respectively (Table 1).8 In addition, 18O EIEs of 1.0187 and 1.0143 were calculated for an Fe-alkylperoxo species, FeIII-OOtBu,16,17 and an Fe-peroxocarbonate species,18 respectively, the latter being similar to a proposed intermediate in TauD.6
Table 1.
Vibrational frequencies (cm−1) of Fe/O2 species, the corresponding calculated 18O EIEs, and experimental 18O E/KIEs.
| molecule | Frequency (cm−1) |
18O EIE (calc)a | 18O EIE (expt)b | 18O KIE (expt)c | ||
|---|---|---|---|---|---|---|
| mode | ν16-16 | ν18-16 | ||||
| FeIII-OO•− | Fe-Od | 555 | 526 | 1.0080 | 1.0054 (Mb) | NDe |
| O-Od | 1136 | 1100f | ||||
| FeIII-OOH | Fe-Og | 621 | 599 | 1.0172 | 1.0113 (Hr) | 1.0120 (HppE) |
| O-Og | 844 | 820f | ||||
| O-Hh | 3539 | 3527 | ||||
| O-O-Hh | 1205 | 1199 | ||||
| FeIII-OOtBu | Fe-Oi | 637 | 612 | 1.0187 | ND | ND |
| O-Oj | 860 | 829f | ||||
| O-tBuj | 746 | 738 | ||||
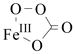 |
Fe-Ok | 547 | 524 | 1.0143 | ND | 1.0102 (TauD) |
| O-Ok | 884 | 863f | ||||
| O-Ck | 965 | 946 | ||||
| O-C-Ok | 728 | 710 | ||||
| FeIV=O | Fe-Ol | 821 | 787 | 1.0287 | ND | 1.0215 (ACCO) |
The 18O EIEs were calculated using the Bigeleisen-Mayer equation and known frequencies (Ref 8).
Measured 18O EIEs for O2-binding proteins myoglobin (Mb) and hemerythrin (Hr) (Ref 14).
This work.
Ref 15.
Not determined.
ν18-16 was calculated as follows: ν18-16 = (ν16-16 ν 18-18)½ (Ref 8).
Ref 1.
Ref 14.
Ref 16.
Ref 17.
Ref 18.
Ref 7.
The comparison of the measured 18O KIEs with calculated 18O EIEs reveals an excellent correlation with the proposed mechanisms for these enzymes (Scheme 1). In TauD, the formation of the cyclic alkylperoxo intermediate in the rate determining step is supported by the measured 18O KIE of 1.0102 ± 0.0002, higher than the calculated 18O EIE for formation of an FeIII-OO•− species (1.0080) and the measured 18O EIE for Mb (1.0054 ± 0.0006, Table 1),14 but lower than the calculated 18O EIE for an Fe-peroxocarbonate species (1.0143). The 18O KIE of 1.0120 ± 0.0002 for HppE is similar to the measured 18O EIE of 1.0113 ± 0.0005 for Hr14 and lower than the calculated 18O EIE of 1.0172 for an FeIII-OOH species (Table 1), suggesting a rate determining formation of an FeIII-OOH species.23 The relatively small measured 18O KIEs in TauD and HppE could have been diminished vs. calculated 18O EIEs due to the presence of a partially rate limiting O2 binding step. This appears not to be the case for HppE, where preliminary data support a 18O/16O discrimination free from kinetic complexity.23 For TauD, a kinetically significant O2 binding step cannot be ruled out,6 yet the excellent agreement between the measured 18O KIE and the calculated 18O EIE (Table 1) strongly suggests a rate limiting formation of the cyclic alkylperoxo intermediate. Thus, in both TauD and HppE the observed 18O KIEs are concluded to be mechanistic in origin.
Scheme 1.

Proposed mechanisms of O2 activation for TauD, HppE, and ACCO (RDS = proposed rate determining step of kcat/Km(O2), R-H = taurine, Asc = ascorbate).
For ACCO, the large 18O KIE of 1.0215 ± 0.0005 (lower than only one calculated 18O EIE of 1.0287, Table 1) implies a significant change in the oxygen bond order and points toward FeIV=O species formation as the RDS of O2 activation (Scheme 1).13 In ACCO, the oxidation of the FeIII-O2•− species to FeIII-OOH by ascorbate is proposed to be reversible, similar to the reversible O2 binding observed in hemerythrin.14 Interestingly, the initial inner sphere reduction of O2 and formation of an Fe-OO•− species does not appear to be rate limiting for any of these enzymes, in accordance with a recently proposed reversible O2 binding as a requisite for the specific reactivity of non-heme iron enzymes.24 This is in contrast to a rate limiting and irreversible outer sphere activation of O2 by glucose oxidase.2,3
The studies reported herein reveal the ability of 18O KIE measurements to dissect the complex catalytic mechanisms of non-heme iron enzymes. By probing only the steps from O2 binding to the first irreversible step of O2 activation, these measurements offer a valuable companion to pre-steady state kinetic analyses, which investigate the nature of the intermediate immediately preceding the RDS. In the present case, measured 18O KIEs for three non-heme iron enzymes that use different reductants in the course of O2 activation have been directly related to each enzyme’s distinct chemical mechanism.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health through a grant to JPK (GM 25765) and HWL (GM 40451) and a National Research Service Award postdoctoral fellowship to LMM (GM 078802).
Footnotes
Supporting Information Available: Protein expression and purification procedures, 18O KIE experimental details, and 18O EIE calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 2.Roth JP, Klinman JP. In: Isotopes Effects in Chemistry and Biology. Limbach H-H, editor. Marcel Dekker; New York: 2004. p. 645. [Google Scholar]
- 3.Roth JP. Curr Opin Chem Biol. 2007;11:142. doi: 10.1016/j.cbpa.2007.01.683. [DOI] [PubMed] [Google Scholar]
- 4.Eichhorn E, vanderPloeg JR, Kertesz MA, Leisinger T. J Biol Chem. 1997;272:23031. doi: 10.1074/jbc.272.37.23031. [DOI] [PubMed] [Google Scholar]
- 5.Price JC, Barr EW, Tirupati B, Bollinger JM, Krebs C. Biochemistry. 2003;42:7497. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 6.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM. Biochemistry. 2005;44:8138. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 7.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 8.See Supporting Information.
- 9.Liu PH, Murakami K, Seki T, He XM, Yeung SM, Kuzuyama T, Seto H, Liu HW. J Am Chem Soc. 2001;123:4619. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- 10.Yan F, Munos JW, Liu PH, Liu HW. Biochemistry. 2006;45:11473. doi: 10.1021/bi060839c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.John P. Physiol Plant. 1997;100:583. [Google Scholar]
- 12.Thrower JS, Mirica LM, McCusker KP, Klinman JP. Biochemistry. 2006;45:13108. doi: 10.1021/bi061097q. [DOI] [PubMed] [Google Scholar]
- 13.Mirica LM, Klinman JP. Proc Natl Acad Sci U S A. submitted. [Google Scholar]
- 14.Tian GC, Klinman JP. J Am Chem Soc. 1993;115:8891. [Google Scholar]
- 15.Das TK, Couture M, Ouellet Y, Guertin M, Rousseau DL. Proc Natl Acad Sci U S A. 2001;98:479. doi: 10.1073/pnas.98.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehnert N, Ho RYN, Que L, Solomon EI. J Am Chem Soc. 2001;123:12802. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 17.Lehnert N, Fujisawa K, Solomon EI. Inorg Chem. 2003;42:469. doi: 10.1021/ic020496g. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto K, Nagatomo S, Fujinami S, Furutachi H, Ogo S, Suzuki M, Uehara A, Maeda Y, Watanabe Y, Kitagawa T. Angew Chem, Int Ed. 2002;41:1202. doi: 10.1002/1521-3773(20020402)41:7<1202::aid-anie1202>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 19.Tian GC, Berry JA, Klinman JP. Biochemistry. 1994;33:14650. doi: 10.1021/bi00167a030. [DOI] [PubMed] [Google Scholar]
- 20.Bigeleisen J, Mayer MG. J Chem Phys. 1947;15:261. [Google Scholar]
- 21.Lanci MP, Brinkley DW, Stone KL, Smirnov VV, Roth JP. Angew Chem, Int Ed. 2005;44:7273. doi: 10.1002/anie.200502096. [DOI] [PubMed] [Google Scholar]
- 22.Burger RM, Tian GC, Drlica K. J Am Chem Soc. 1995;117:1167. [Google Scholar]
- 23.Detailed KIE mechanistic studies of HppE are underway.
- 24.Bollinger JM, Krebs C. Curr Opin Chem Biol. 2007;11:151. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.