

Abstract
In eukaryotic replication licensing, Cdt1 plays a key role by recruiting the MCM2-7 complex onto the origin of chromosome. The C-terminal domain of mouse Cdt1 (mCdt1C), the most conserved region in Cdt1, is essential for licensing and directly interacts with the MCM2-7 complex. We have determined the structures of mCdt1CS (mCdt1C_small; residues 452 to 557) and mCdt1CL (mCdt1C_large; residues 420 to 557) using X-ray crystallography and solution NMR spectroscopy, respectively. While the N-terminal 31 residues of mCdt1CL form a flexible loop with a short helix near the middle, the rest of mCdt1C folds into a winged helix structure. Together with the middle domain of mouse Cdt1 (mCdt1M, residues 172–368), this study reveals that Cdt1 is formed with a tandem repeat of the winged helix domain. The winged helix fold is also conserved in other licensing factors including archaeal ORC and Cdc6, which supports an idea that these replication initiators may have evolved from a common ancestor. Based on the structure of mCdt1C, in conjunction with the biochemical analysis, we propose a binding site for the MCM complex within the mCdt1C.
Keywords: mouse Cdt1, replication licensing factor, winged helix fold, mcm binding
Introduction
Eukaryotic genomic integrity requires complete and precise duplication of chromosomal DNA during replication. To maintain genomic stability, DNA replication must start once and only once per cell cycle, and eukaryotes achieve such strict regulation of replication initiation by stepwise assembly of protein complexes onto replication origins to establish “licensed” origins.1,2 The origin recognition complex (ORC) first selects origin DNA and then triggers the binding of Cdc6 and Cdt1. These two factors then recruit a putative replicative helicase, the MCM2-7 complex, to form a prereplicative complex (pre-RC) on replication origins in the G1-cell cycle phase.3–8 The MCM2-7 complex then promotes the unwinding of DNA origins, and the binding of additional factors such as Cdc45 and GINS complex at pre-RCs to initiate the DNA replication in S-phase.9–13
The licensing factor Cdt1 is essential for chromosomal replication.7,14–16 Several experiments have suggested that the interaction between Cdt1 and MCM2-7 is required to load the helicase on chromatin.7,15,16 Recent studies show that Cdt1 stimulates the binding and helicase activity of MCM4,6,7 complex in vitro.17 Overexpression of Cdt1 causes re-replication of DNA within each cell cycle, which ultimately leads to cell death and tumorigenesis.18–20 Therefore, when the S-phase starts, the activity of Cdt1 must be down-regulated. Metazoan cells achieve this down-regulation through the combined activities of cyclin-dependent kinases (CDKs) and geminin, and by the degradation of Cdt1 by proteosomes; CDK-dependent degradation by the SCF complex and PCNA-dependent degradation by the Cul4-DDB1 complex.21–26
The primary function of Cdt1 is to recruit MCM helicase onto replication origins,7,15,16 although recent study suggests that mutants of Cdt1 that do not interact with MCM2-7 can induce re-replication when overexpressed.27 The depletion of Cdt1 abolished the association between MCM2-7 and origin DNA, whereas the addition of recombinant Cdt1 fully restored pre-RC formation.28 The structure of Cdt1 can be largely divided into three parts based on sequence comparison and biochemical analyses29: the highly variable N-terminal region that binds to DNA; the middle geminin-binding region (Cdt1M), which is conserved in metazoans and which by earlier structural analysis was shown to adopt a winged helix fold30; and the C-terminal MCM-binding region (Cdt1C) which is known to directly interact with the MCM2-7 helicase and to be essential for the licensing activity of Cdt1.29,31 Mutational studies of Cdt1 from various species identified several regions and/or residues of Cdt1C that are involved in the interaction with MCM2-7.17,27,29,31 However, despite the extensive studies of Cdt1 in replication licensing, our understanding on the interactions between Cdt1C and MCM2-7 and the mechanism by which Cdt1 recruits the MCM2-7 complex to assemble the pre-RC is very limited because of the lack of the structure of Cdt1C. Here, we investigated the structure of the C-terminal region of mouse mCdt1 (mCdt1C), the most conserved region in Cdt1, and provide an insight as to how mCdt1C interacts with the MCM2-7 complex.
Results
Overall structure of mCdt1C
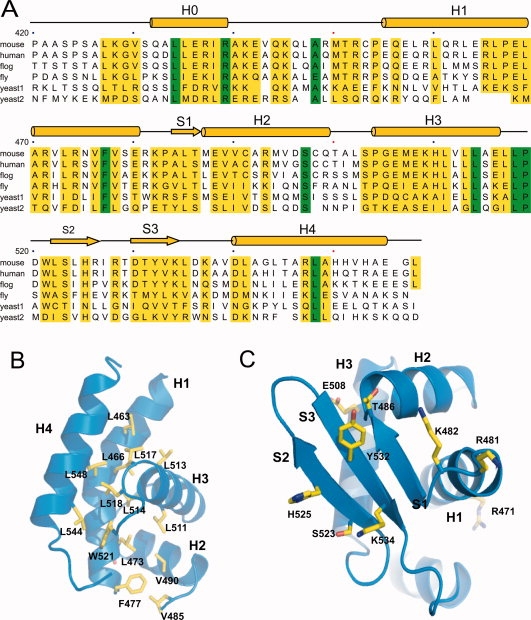
Previous studies have revealed that the MCM complex binding domain is within the C-terminal domain of mCdt1.27,29,31 Based on these studies, and our limited proteolytic digestion analysis, we produced the two mCdt1 fragments, mCdt1CL (mCdt1C_large; residues 420–557) and mCdt1CS (mCdt1C_small; residues 452–557). We then determined the solution and 1.9 Å crystal structures of mCdt1CL and mCdt1CS, respectively (Tables I and II). The mCdt1CL structure can be divided into the two parts; the first 31 residues form an N-terminal arm (residues 420 to 451), which is highly flexible [Fig. 1(A)]. The Cα and Cβ chemical shift values and the sequential and medium range NOEs of the backbone amide protons confirmed that this N-terminal region also contains a well-defined helical structure [residues 431 to 442; Fig. 1(B,C)]. Another part of mCdt1CL forms a compact core which is folded into a winged helix structure [Fig. 1(A,B,D)]. The mCdt1CL core or mCdt1CS consists of four α-helices (H1 to H4) backed on one side by three β-strands (S1 to S3). The H3 helix is packed against H1 and H4 helices in a perpendicular manner on one side and packed against the S2 and S3 strands on the other side. The middle of helix H1 is notably bent (by 40°) primarily due to Pro467, which allows its tight interaction with helix H3 [Fig. 1(B,D)]. The flexible linker between the N-terminal arm and the core of mCdt1CL allows significant movement of the N-terminal arm relative to the mCdt1 core and the end to end distance between the N-terminal ends of solution NMR structures could move more than 50 Å [Fig. 1(A)].
Table I.
Statistics of Data Collection and Refinement
| Data sets | Native | Hg-SAD |
|---|---|---|
| Diffraction data | ||
| Wavelength (Å) | 1.0000 | 1.0064 |
| Resolution (Å) | 50–1.9 | 50–2.2 |
| Measured reflections | 313,879 | 361,298 |
| Unique reflections | 11,404 | 7,433 |
| Completeness (%) | 100 (100) | 99.9 (99.7) |
| Average (I/σ) | 67.1 (12.1) | 15.4 (2.2) |
| Rsym (%) | 10.1 (36.9) | 10.6 (34.7) |
| Phasing | ||
| Overall figure of merit (50–2.4 Å) | 0.30 | |
| Refinement | ||
| Resolution range (Å) | 30–1.9 | |
| Number of reflections | 11,126 | |
| Rworking (%) | 21.7 | |
| Rfree (%) | 23.8 | |
| Number of water molecules | 94 | |
| RMS deviation bond length (Å) | 0.006 | |
| RMS deviation bond angles (Å) | 1.1 | |
Rsym = ΣhΣi|Ih,i – Ih|/ΣhΣIIh,i, where Ih is the mean intensity of the i observations of symmetry-related reflections of h. R = Σ|Fobs – Fcalc|/ΣFobs, where Fobs = Fpi and Fcalc is the calculated protein structure factor from the atomic model (Rfree was calculated with 5% of the reflections). RMS deviation in bond lengths and angles are the deviations from ideal values, and the RMS deviation in B factors is calculated between bonded atoms.
Table II.
NMR Structural Statistics and Atomic RMS Differences
| Parameters | mCdt1C (420–557) |
|---|---|
| Structure statistics | |
| Completeness bb/sc assign,a % | 99/99 |
| Consensus NOE assign,b % | 64 |
| Total NOE peaks assigned,c (N/Caliphatic/Caromatic) | 3513 (1389/2019/105) |
| NOE constraints | 3295 |
| Short range | 2451 |
| Medium range | 473 |
| Long ranged | 371 |
| No. dihedral angle constraints,e φ/ψ | 94/98 |
| No. hydrogen bond constraints | 86 |
| CYANA target function, Å2 | 2.04 ± 0.28 |
| RMSDs backbone RMSD to mean,f Å | 0.55 ± 0.14 |
| RMSDs heavy atom RMSD to mean,f Å | 0.99 ± 0.11 |
| CNS solve calculation: | |
| RMSDs backbone RMSD to mean,f Å | 0.41 ± 0.07 |
| RMSDs heavy atom RMSD to mean,f Å | 0.92 ± 0.05 |
| Ramachandran plot | |
| Residues in most favored regions, % | 82.7 |
| Residues in additional allowed regions, % | 13.7 |
| Residues in generously allowed regions, % | 2.7 |
| Residues in disallowed regions, % | 0.9g |
For backbone (bb); the assignment yields was calculated by excluding N-terminal 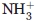 , Pro 15N, and 13C′ shifts of residues preceding Pro residues. For side-chains (sc); excluding side-chain OH, 13C′ and aromatic quaternary 13C shifts, and Lys , Arg NH2.
, Pro 15N, and 13C′ shifts of residues preceding Pro residues. For side-chains (sc); excluding side-chain OH, 13C′ and aromatic quaternary 13C shifts, and Lys , Arg NH2.
CYANA32 assigned results.
Short-range (|i – j| ≤ 1), medium-range (1 < |i – j| ≤ 5), long-range (|i – j| ≥ 5) assignment with NOE connecting residues i and j.
Calculated by using TALOS.34
RMSDs for well-folded domain, residues 469 to 544.
This result produced with the residues from flexible part.
Figure 1.
Overall structure of mCdt1CL. (A) Backbone (Cα, N, and CO) superposition of 20 low-energy calculated structures of mCdt1CL. (B) Ribbon representation of a selected structure of mCdt1CL, which is closest to the mean. (C) Primary sequence of mCdt1CL with the summary of the NOE data. For the short- and medium-range NOEs, the line thickness is approximately proportional to NOE intensity (classified as weak, medium, or strong). The red square marked for the residues from helix H0 in the flexible N-terminal part. (D) Crystal structure of mCdt1CS in two different views. (E) Stereodiagram of the superimposed structures of mCdt1CS (blue) and mCdt1CL (orange). N-terminal region (420 to 450) of mCdt1CL was removed for clarity.
The structure of the mCdt1CL core was found to be similar to that of mCdt1CS, with an RMS deviation values of 2.4–2.8 Å for 107 aligned Cα atoms. However, despite the overall structural similarities between mCdt1CL and mCdt1CS, the two structures are different in several regions [Fig. 1(E)]. First, the N-terminal half of helix H1 is directed toward helix H3 in mCdt1CS, whereas the equivalent region is bent in the opposite direction and points away from helix H3 in mCdt1CL. In the crystal structure, Gln456 forms hydrogen bonds with the side chains of Glu480 and Arg493 from symmetry-related molecules. Thus, the structural differences between the N-terminal half of the H1 helix of Cdt1CL (NMR) and Cdt1CS (crystal) could be due to the crystal packing. Second, in mCdt1CS, the C-terminal end forms a part of the H4 helix and is packed in an anti-parallel manner against the N-terminal region of helix H1 through hydrophobic contacts, whereas the equivalent region in mCdt1CL is loosely folded and directed to helix H3.
Structural similarities with archaeal licensing factors
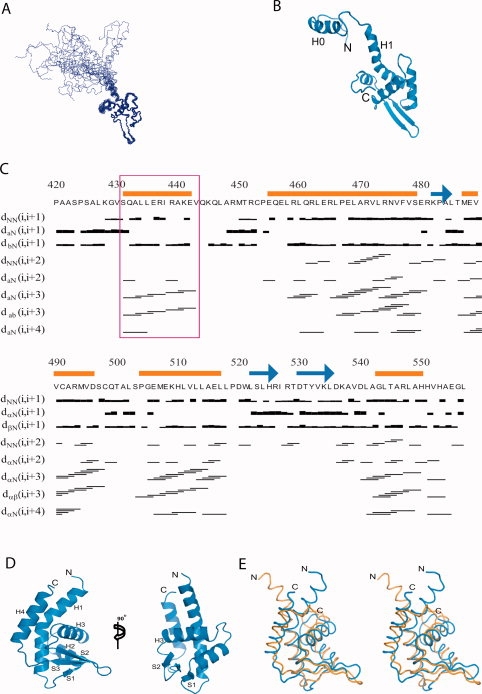
A database search using DALI35 revealed that the structure of mCdt1CS is remarkably similar to the structures of two archaeal homologues of the eukaryotic replication initiator Cdc6 and Orc2; Pyrobaculum aerophilum Cdc6 (PaCdc6, Z = 7.3) with an RMSD value of 4.4 Å for 88 Cα atoms and Aeropyrum pernix Orc2 (ApOrc2, Z = 6.5) with an RMSD value of 5.1 Å for 92 Cα atoms, although no significant sequence similarity (10 to 11%) was observed between mCdt1C and these proteins (Fig. 2).36,37 These two mammalian licensing factor homologues possess an AAA+ ATPase domain and a winged helix domain at their N-terminal and C-terminal ends, respectively. The AAA+ domains of Orc2 and Cdc6 hydrolyze ATP to load the MCM2-7 complex onto origin DNA during pre-RC assembly, whereas their winged helix domains bind to DNA.36,37 The presence of winged helix domains in the C-terminal regions of mCdt1, archaeal PaCdc6, and ApOrc2 provide additional support for the conservation of the replication licensing factors. Moreover, the fact that the presence of winged helix domains in archaeal Orc2 and Cdc6 homologues extends to Cdt1, which is unique to eukaryotes, suggests that the origin selection systems have diverged to perform more complicated functions during evolution. The structure of mCdt1C also similar to that of mCdt1M (Z = 7.1, RMSD of 3.4 Å for 91 Cα atoms), which shares only 9% sequence identity.30 Thus, a large part of mCdt1 is composed of a tandem repeat of winged helix domain (Fig. 2).
Figure 2.
Domain structure of mCdt1, indicating functional regions (top). Overall structure of mCdt1C, which is compared with three homologs: mCdt1M (PDB 2ZXX),30 PaCdc6 (1FNN),36 and ApOrc2 (1W5T).37
Binding sites for the MCM complex within Cdt1C
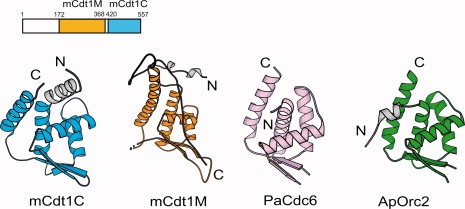
Sequence comparison analysis of the Cdt1C domains from various species reveals that conserved residues are clustered in both the N-terminal arm and mCdt1 core regions [Fig. 3(A–C)]. In N-terminal arm, most of the conserved residues are clustered in the H0 helix and the loop connecting the H0 and H1 helices (H0-H1 loop), which are fully exposed to solvent. Previous yeast two hybrid and GST pull down analyses with mouse Cdt1 suggested that residues between 407 and 477 of mCdt1 are important for binding to MCM6.29 In particular, mutation of Lys441 and Lys445 significantly reduced the affinity toward MCM4,6,7 and fails to stimulate DNA binding and helicase activities of the MCM4,6,7 complex (Table III).17 Lys441 and Lys445 are located in the H0 helix and the H0-H1 loop, respectively [Fig. 4(A)]. In addition to these two lysine residues, several conserved residues in the H0 helix and the H0-H1 loop are charged, including Glu436, Arg437, and Arg439 [Fig. 3(A)].
Figure 3.
(A) Primary and secondary structure of mCdt1C showing identity with orthologues from human, frog, fly, worm, and yeast (1: Schizosaccharomyces pombe and 2: Saccharomyces cerevisiae). Highly conserved residues are colored with yellow bar and absolutely conserved residues are colored with green. (B) Conserved hydrophobic residues in mCdt1C are shown in yellow stick. (C) Conserved exposed residues in the mCdt1C core are clustered in three β-strands.
Table III.
Summary of the Point Mutant Proteins Used in This and Other Studiesa
| Residues | MCM binding | Re-replication |
|---|---|---|
| K441A, K445Ab | − | NDc |
| P504T,E508A,H510A,L514Ad | − | + |
| R465A,E468A,L469A,A470S, R471Ae | − | + |
| R368K,P467H,L473E,V476A, F477Af | − | + |
| R481S (K543S)g | + | + |
| K482S (K544S)g | − | + |
| T486A (T548A)g | + | + |
| R481S, K482S, T486Ah | − | - |
| S523Ag | + | + |
| H525Sg | + | + |
| R528Sg | + | + |
| S523A, H525S, R528Sh | − | − |
| V512Ag | + | ND |
| L513Ag | + | ND |
| E516Ag | + | ND |
Residues number is based on mCdt1.
From You and Masai.17
Not determined.
Originally residues of human Cdt1 (hCdt1) were mutated, but mCdt1 residues equivalent to hCdt1 (P492T, E496A, H498A, L502A) were listed for convenience.27
mCdt1 residues equivalent to hCdt1 (R453A, E456A, L457A, A458S, R459A) were listed for convenience.27
mCdt1 residues equivalent to hCdt1 (R356K, P455H,L461E,V464A, F465A) were listed for convenience.27
Mutations from this study. Residue number inside parenthesis is for the Xenopus Cdt1.
Mutations from this study. Both mCdt1 and Xenopus Cdt1 mutant proteins were used.
Figure 4.
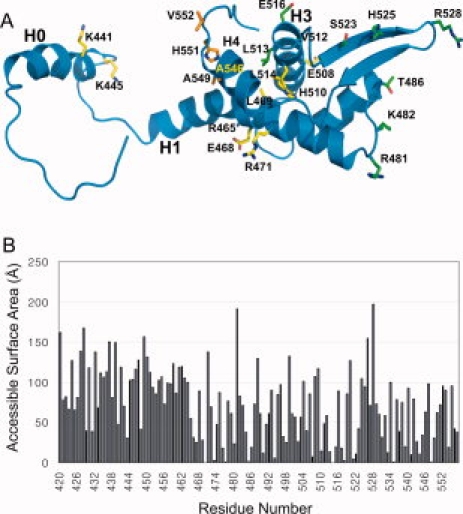
MCM complex binding interface of mCdt1C. (A) Residues that affected the MCM binding in this and previous studies17,27 are shown in green and yellow, respectively. The putative residues of Cdt1C which we proposed to interact with MCM are shown in orange. Please see Table III for the details of each mutated residues. (B) Bar graphs showing the accessible surface area of each residue of mCdt1CL.
Figure 4(B) shows the plot of the accessible surface area of each residues of Cdt1CL. The mCdt1C core is primarily formed with two hydrophobic patches, in which a number of conserved residues are tightly packed [Fig. 3(B)]. One patch is formed with Leu463, Leu466, Leu473 (helix H1), Leu514 and Leu517 (H3), Leu518 and Trp521 (loop H3-S2), and Leu544 and Leu548 (H4). Another hydrophobic patch consists of Leu473, Phe477, Val485, Val490, and Leu511. While the conserved hydrophobic residues are clustered in the helices to provide the stability to the mCdt1C core, majority of conserved polar residues are at the surface of the three-stranded sheet [Fig. 3(A,C)]. These include Arg481 and Lys482 (loop H1-S1), Thr486 (strand S1), Ser523 and His525 (S2), Tyr532 and Lys534 (S3). In addition, central region of the H1 helix and the H3 helix contain a few conserved polar residues including Arg465, Arg471, Glu508, and His510 [Figs. 3(A) and 4(A)].
Previous mutational studies showed that replacement of some of these conserved residues abrogates the interaction between Cdt1 and the MCM complex (Table III).27 For instance, the human Cdt1 (hCdt1) multiple mutant protein, in which Glu496, His498, and Leu502 (equivalent to Glu508, His510, and Leu514 of mCdt1) in the H3 helix or Arg453, Glu456, Leu457, Ala458, and Arg459 (Arg465, Glu468, Leu469, Ala470, and Arg471 of mCdt1) in the H1 helix are simultaneously mutated, fails to interact with the MCM complex. The positions of these residues are shown in Figure 4(A).
We used cross-saturation transferred measurements to identify the interface between mCdt1CL and the MCM complex [Fig. 5(A)]. In this method, saturation in the MCM complex caused by selective irradiation is transferred to the residues of bound mCdt1CL at the interface. The contact residues of mCdt1CL are identified by observing decreases in the peak intensities of 1H–15N shift correlation spectra. A plot of the intensity ratios of peaks determined during the cross-saturation transferred experiment are shown in Figure 5(A). We also show the surface of Cdt1, in which the residues found to interact with MCM by the cross saturation in Figure 5(B). The transferred cross-saturation experiment showed that the H3 helix of mCdt1C is involved in the interaction with the MCM2-7 complex, which supports the mutational analysis of Teer and Dutta.27 Residues near Cys491 and Cys498 in the H2 helix were not accounted in our cross-saturation transferred analysis due to a possible artifact caused by the irradiation of protons of cysteine residues. Previously studies using human and Xenopus Cdt1 proteins showed that deletion of the C-terminal region (equivalent to residues 510 to 557 in mCdt1) containing the S2 and S3 strands and the parts of H4 and H5 helices prevents the interaction between Cdt1C and MCM, which suggests that the region spanning residues 510 to 557 of mCdt1 is required for the interaction with the MCM2-7 complex.27,31 Although this C-terminal region does not contain many conserved polar residues, Ser523, His525, and Asp540 are well conserved and Arg529, Tyr532, Arg547, and His550 are conserved in metazoan Cdt1 proteins. In addition, Yanagi et al. reported that deletion of residues 407 to 477 of mCdt1 abolished the MCM binding activity of mCdt1. Together, these data implicate that the MCM binding region of mCdt1 may not be confined to the limited region, and instead more broad regions of mCdt1C are involved in the MCM binding.
Figure 5.
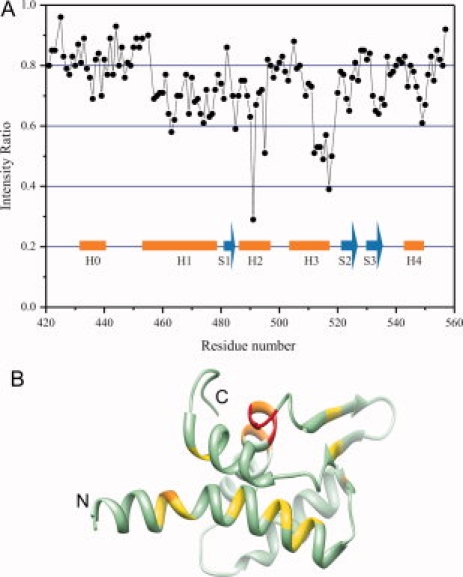
Plots of the intensity ratios of the peaks in the transferred cross-saturation experiments. (A) The intensity ratios of the peaks originating from the backbone amide groups with irradiation to those without irradiation measured in 10% H2O/90% D2O. Residues near Cys491 and Cys498 in helix H2 were not accounted due to the possible artifact from the irradiation of γ protons of the cysteine residues. The saturation time used was 3 s. (B) A ribbon diagram of mCdt1C which are colored based on the cross saturation experiment. Residues of the H1 and H3 helices with the best signal are colored with orange and the residues with the second best signal are colored with yellow.
Based on our structural information including accessible surface area of mCdt1C, sequence conservation, and cross-saturation transferred analysis, we also generated a series of Cdt1 mutant proteins, and then analyzed whether these mutations on Cdt1M+C (residues 177-557) affect interactions between the Cdt1 proteins and MCM2,4,6,7 complex [Figs. 3(B,C) and 4(A)]. The surface around the three strands in mCdt1C contains several conserved polar residues, and thus, we selectively mutated these residues in the three β-strands and surrounding loops in mCdt1 to either alanine or serine. Because we wanted to observe clear mutational effects, we mutated single or multiple residues in mCdt1. First, we simultaneously replaced residues Arg481, Lys482, and Thr486 in M1 mutant, and mutated residues Ser523, His525, and Arg528 in M2 mutant [Figs. 3(C) and 4(A)]. We also construct three additional mutants in helix H3, that is, V512A (M3), L513A (M4), and E516S (M5).
We then tested if these mutant proteins bind to MCM using GST-pull down assay. Figure 6(A) shows that while GST-mCdt1 M1 mutant protein (residues 177–557) failed to interact with the MCM complex, GST-mCdt1 M2 protein interacted weakly. We also assayed mouse Cdt1-mediated MCM loading on the chromatin by Western blotting. Cdt1-depleted Xenopus egg extract and sperm chromatin were incubated with mutant proteins, and MCM4 loading was then detected [Fig. 6(B)]. While M1 mutant protein of mCdt1C failed to recruit the MCM complex to chromatin as compared with wild-type Cdt1, the M2 mutant protein loaded only a small amount of the MCM complex onto chromatin. In contrast to M1 and M2 mutant proteins, the binding affinities of the M3, M4, and M5 mutants on MCM complex were comparable to that of wild type Cdt1. We wished to identify which specific residues of the six mutated residues from M1 and M2 proteins are involved in the MCM-loading and DNA replication. We have used Xenopus Cdt1 (residues 239–620) instead of mCdt1M+C to analyze the activities of the single point mutant proteins as Xenopus Cdt1 protein might more accurately reflect the activities of Cdt1 in Xenopus egg extract system (Table III). These point mutant proteins of Xenopus Cdt1 include Lys543Ser (M11, equivalent to Arg481 of mCdt1), Lys544Ser (M12, Lys482 of mCdt1), Thr548Ala (M13, Thr486 of mCdt1), Lys543Ser-Lys544Ser-Thr548Ala (M1), Ser585Ala (M21, Ser523 of mCdt1), His587Ser (M22, His525 of mCdt1), Arg590Ser (M23, Arg528 of mCdt1), and Ser585Ala-His587Ser-Arg590Ser (M2). The mutated residues of the M1 and M2 proteins of Xenopus Cdt1 (residues 239–620) are equivalent to those of mCdt1M+C. Again, we have used Cdt1-depleted system for the MCM-loading and replication activity analyses [Fig. 6(C,D)]. Interestingly, as shown in Figure 6(C,D), all of the single point mutant proteins of Xenopus Cdt1 (residues 239–620) showed MCM-loading and replication activities similar to that of the wild type, while the M1 and M2 triple mutant proteins showed significantly reduced activities. These data suggest that the decreased activities of the M1 and M2 triple mutant protein are due to the additive effects of each single point mutated residues, and the MCM binding interface might not be restricted to certain confined region within mCdt1. We analyzed if the decreased MCM-loading and DNA synthesis activities of the M1 and M2 mutant proteins of Cdt1C are due to the overall structural perturbation of triple mutation. Because GST-M1 and M2 mutant proteins of mCdt1M+C could not be purified more than 95% purity, we used M1 and M2 mCdt1CL mutant proteins (residues 420 to 557) for circular dichroism analysis [Fig. 6(E)]. The M1 and M2 mutant proteins of mCdt1CL exhibit virtually identical curves on circular dichroism analysis compared to that of wild-type mCdt1CL, which suggests that the mutational effects on MCM-loading and DNA synthesis activities of the mCdt1C mutant proteins are unlikely due to the perturbation of the overall structure of mCdt1C. Overall, our studies suggest that the strand region appears to be important for MCM complex binding, and together with previous studies, residues in the H0-H1 loop, the H0, H1 and H3 helices, and the S2 and S3 strands from mCdt1 contribute to the interactions with the MCM complex.
Figure 6.
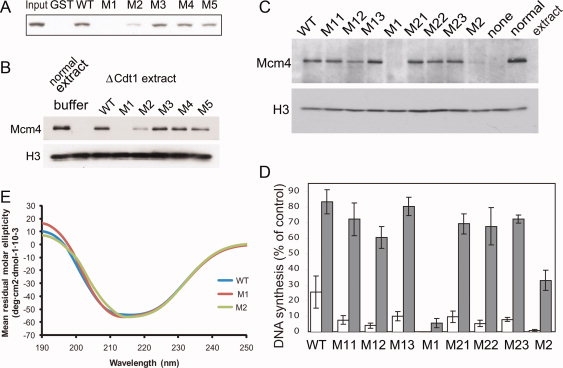
Interaction between mCdt1 and the MCM complex. (A) The association between mCdt1 and MCM complex. Purified MCM2,4,6,7 complex was incubated with various GST-mCdt1 mutant proteins (residues 177–557), and detected by Western blot using anti-MCM6. In M1 and M2 mutants, clusters of three residues (Arg481, Lys482, Thr486) and (Ser523, His525, Arg528) were simultaneously mutated, respectively. For M3, M4, and M5, the mutatons are V512A, L513A, and E516S, respectively. (B) The MCM loading activities of mCdt1 mutant proteins (M1 to M5: see text) onto chromatin were measured at 23°C, and compared to that of wild-type mCdt1. Normal extracts were used instead of Cdt1-depleted extracts for a sample applied on the lane “normal extract.” The data includes results of three independent experiments. Histone H3 was used for a loading control to show that equal amounts of chromatin were recovered in all lanes. (C) The MCM loading activities of Xenopus Cdt1 mutant proteins. The protocols are basically same as those in (B). M11, Lys543Ser; M12, Lys544Ser; M13, Thr548Ala; M1, Lys543Ser-Lys544Ser-Thr548Ala; M21, Ser585Ala; M22, His587Ser; M23, Arg590Ser; M2, Ser585Ala-His587Ser-Arg590Ser. (D) DNA synthesis activities of various Cdt1 mutant proteins. Values are indicated as percentages compared to that using buffer and normal extracts instead of Cdt1-mutant fractions and geminin extracts, respectively, after every values are subtracted by background value when buffer was used for Cdt1-fraction. Error bars indicated standard error of three independent experiments. (E) Analysis of conformational differences between the WT and mutant mCdt1CL proteins by circular dichroism. The spectra for mutant proteins, M1, and M2, are compared with that of a WT Cdt1CL protein. Because of the difficulties of obtaining mutant proteins with high purity (over 95%), we used Cdt1CL M1 and M2 (residues 420–557) mutant proteins to compare the conformational difference with the wild-type Cdt1CL.
Discussion
In this study, we determined the atomic structure of the mCdt1C by X-ray crystallography and NMR spectroscopy and identified the MCM binding site within mCdt1C. Our analyses in conjunction with previous studies show that the primary licensing factors required to recruit the MCM complex, that is, Orc1, Orc2, Cdc6, and Cdt1,30,36–39 all possess the winged helix fold, which suggests that these proteins may have evolved from a common ancestor, and that during evolution the winged helix domain may have fused with other domains to form distinct replication licensing factors in eukaryotes.
Because the initiation of DNA replication is a crucial event in life, it is not surprising that the mechanistic elements of the replication initiation process are conserved in prokaryotes and eukaryotes. Nevertheless, available structural evidence on the conservation of the replication machinery is limited to the AAA+ domain, which is shared by bacterial DnaA, archaeal Cdc6 and Orc2 homologues, and eukaryotic ORC.36–40 In this study, we provide an additional evidence of conservation in eukaryotic licensing factors by showing that the winged helix domain is present in the C-terminal region of archaeal Cdc6 and Orc2 homologues and eukaryotic Cdt1.
During evolution, bacterial genes whose products function as dimers are frequently duplicated and fused to form single-chain multidomain proteins in eukaryotes. Moreover, each domain is specialized to diverge in sequence and possibly function.41 In addition, it has been suggested that duplicated proteins evolve more quickly than nonduplicated proteins.42 It is possible that the winged helix domain in archaea was used as a prototype fold in the early replication system, and that it was duplicated and fused to result in two repetitive domains that may have evolved to gain novel functions, such as geminin binding (by the Cdt1M domain) and the MCM complex binding (by the Cdt1C domain), and ultimately to control replication licensing. Our hypothesis that the duplication of a winged helix domain was an early event that was followed by later development for geminin binding on Cdt1M is supported by the facts that (i) simple eukaryotes such as yeasts do not contain geminin; (ii) the yeast Cdt1 sequence differs remarkably from mammalian Cdt1 (∼20% sequence identity); (iii) in the geminin-mCdt1M structure, the primary geminin binding site is formed by residues 177 to 190, which lie outside the winged helix domain in mCdt1M.30
The winged helix protein family is a structural motif commonly used to bind specific sequence of DNA, with the recognition helix inserted into the major groove and the wing reaching across adjacent minor groove43 [Fig. 7(A)]. Previous studies have shown that Cdt1 can associate with single stranded or double stranded DNA.29 Interestingly, the N-terminal (residues 1–130) and the geminin-binding (176–293) regions of Cdt1 contribute to DNA binding, whereas the Cdt1C domain does not bind to DNA.29,30 This is interesting because (i) the helix-turn-helix (HTH) and β-hairpin motifs of mCdt1C generate highly basic surface as shown in Figure 7(B) and the basic surface around these motifs appears to interact with the MCM2-7 complex rather than binding to DNA. The structure of mCdt1C explains why the winged helix domain of Cdt1C does not interact with DNA. In general, the orientations of helices H1 and H3 in the winged helix domain are known to critical for DNA binding.43 In Cdt1C, helix H1 is significantly longer and the middle portion of H1 is notably kinked. As a result, when the structure of Cdt1C was superimposed onto the winged helix domains of other structures, such as, Orc1-DNA, Cdc6, and E2F1-DNA, the H1 helix of mCdt1C would collide with DNA [Fig. 7(A)].36–39,44 Moreover, the H3 helix which is a DNA recognition helix in the winged helix domain has only one basic residue, while others have typically more than three basic residues.
Figure 7.
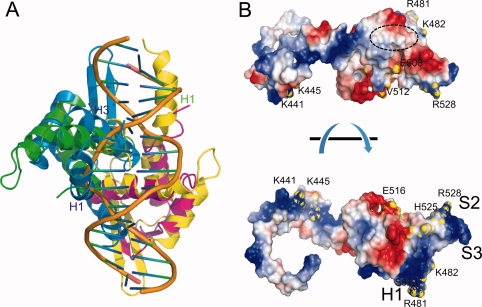
(A) Structural superposition of Cdt1CS (blue and yellow) onto the E2F-DP1-DNA complex (PDB 1CF7). Residues from the H1 and H3 helices of Cdt1CS (or E2F or DP1) form a primary binding to DNA. These regions would collide with the modeled DNA. (B) Surface potential of mCdt1C. The positively and negatively charged surface areas are colored blue and red, respectively. Some of the MCM complex binding residues are marked and labeled. The region equivalent to the DNA binding region in the E2F-DNA complex is marked with a dotted circle.
Our NMR and mutation analyses in conjunction with previous studies by other groups provide insights into the binding site for the MCM complex. According to the recent mutational studies on the MCM-Cdt1C interface, the H0 and H1 helices and intervening loop, and the H3 and H4 helices of mCdt1C would form a binding surface of MCM, and our transferred cross-saturation experiment supports the involvement of the H3 helix of Cdt1 in MCM binding17,27,31 [Fig. 5(A,B)]. The cluster of the conserved and charged residues in the H0 helix and the H0-H1 loop suggest that H-bonds and/or electrostatic interactions might play an important role for the interaction between the N-terminal arm of Cdt1C and MCM2-7. Our structure reveals that Ala546, Ala549, His551, and Val552 from the H4 helix are directed to this binding surface, which suggests that these residues might be involved in the interaction with MCM [Fig. 4(A)]. In addition, our finding on the significantly reduced MCM-binding activity of M1 and M2 triple mutants of mCdt1 proteins suggests that the binding site for MCM within mCdt1C is unlikely to limited to a confined region. The M1 and M2 sites are formed with Arg481 and Lys482 (loop H1-S1), Thr486 (strand S1), Ser523 and His525 (strand S2), and Arg528 (loop S2-S3) [Fig. 7(B)]. As the linker that connects the N-terminal arm and the mCdt1C core is highly flexible, it is possible that binding of the MCM protein could significantly alter the position of the N-terminal arm such that the N-terminal arm could form a MCM-binding site with the β-sheet region. The two putative MCM binding sites formed by several helices and β-sheet are over 20 Å apart. However, because the MCM complex is expected to form a large hexameric ring (∼300 kD), it is possible that the MCM binding covers these two regions in Cdt1C through bipartite interactions.
Previously, we showed that mCdt1M most resembles Bacillus subtilus replication terminating protein (BsRTP).30 mCdt1C also shares high structural similarity with BsRTP, although the degree of similarity decreased compared to that of mCdt1M (Z = 5.4, RMSD of 6.5 Å for 87 Cα atoms). In particular, helices H2 and H3 and the three stranded β-sheet are very similar in mCdt1C and BsRTP, whereas the lengths and orientations of their N- and C-terminal helices are different [Fig. 8(A,B)]. In addition to the structural similarity, mCdt1 and BsRTP share some similar features: (i) both proteins interact with the ring-shaped replicative helicases, MCM complex and bacterial DnaB that function via hexamerization (Kaplan and Odonnell).45 RTP arrests DNA unwinding by specifically interacting and inhibiting the DnaB helicase.46 (ii) Both mCdt1 and BsRTP share the conserved binding sites for the replicative helicase—a three-stranded β-sheet. In BsRTP, Tyr33 from the S1 strand plays a critical role in maintaining a contrahelicase activity by binding DnaB.46 Although no tyrosine residue is positioned in strand S1 in Cdt1, a conserved Tyr532 residue in strand S2 is closely located and might replace the role of Tyr 33 of BsRTP in recruiting replicative helicase [Fig. 8(B)]. Thus, it is possible that the winged helix domain may have evolved to form bacterial replication termination factors, and mCdt1 may bind to the MCM2-7 complex using a similar strategy, whereby BsRTP interacts with DnaB helicase.47
Figure 8.
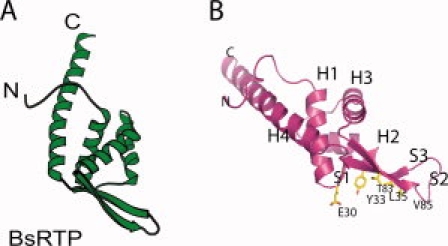
Structure of BsRTP. (A) Overall structure of BsRTP is shown in same orientation as that of mCdt1C in Figure 2. (B) DnaB binding region of BsRTP. The residues in the three-stranded sheet are important for DnaB binding are colored in yellow.
In summary, by demonstrating that Cdt1 is formed with two winged helix repeats, we provide additional structural and biochemical evidence that eukaryotic licensing factors have evolved from the common ancestor. Interestingly, the winged helix motif of the mCdt1C is used to interact with the MCM complex rather than DNA. Detailed understanding of the molecular mechanism for the Cdt1-mediated MCM recruitment would require the Cdt1-MCM complex structure. Nevertheless, the X-ray and solution NMR structure of mCdt1C presented here would allow more systematic approaches to elucidate the interaction between Cdt1 and the MCM2-7 complex.
Materials and Methods
Cloning, expression, and purification
His-tagged mCdt1CS and mCdt1CL were synthesized by PCR, and products were digested with NdeI-XhoI and inserted into a PET28a vector. Each protein was purified using a nickel column followed by a cation exchange (Mono-S column) and gel filtration (Superdex 75 column) equilibrated with 50 mM Tris-HCl, 200 mM NaCl, 5 mM dithiothreitol (DTT) at pH 6.8. Mouse MCM2,4,6,7 complex was purified from High5 insect cell (Invitrogen) using baculovirus system at >95% homogeneity as previously described.48
Crystallization and structure determination
Crystals of mCdt1CS were grown at room temperature by hanging drop vapor diffusion from 3.8M sodium chloride, 100 mM sodium citrate, 5 mM dithiothreitol (DTT), pH 5.5. The crystals formed in space group P41212 with a = 52.57, b = 52.57, c = 96.04 Å and contained one molecule in an asymmetric unit. Diffraction data were collected at −170°C with crystals flash frozen in crystallization buffer containing 30% glycerol. A single-wavelength data (SAD) set was collected using a Hg-bound crystal on beamline 4A at Pohang Acceleratory Laboratory (PAL). Integration, scaling, and merging of the diffraction data were done with the HKL2000 suite of programs. Two sites and initial phases were determined using the CNS program.49 After density modification, the electron density map calculated to 2.5 Å was of excellent quality and allowed us to trace most of the chains. Successive rounds of model building using the program Coot,50 simulated annealing refinement with the CNS49 program and phase combination allowed the complete building of the structure.
NMR spectroscopy and structure calculation
NMR measurements were performed at 25°C on 1 mM 13C, 15N-labeled mCdt1C samples in 20 mM MES hydrate (pH 6.7), 80 mM NaCl, 10 mM β-mercaptoethanol with 10% D2O on DRX800 and DRX900 spectrometers (Bruker). All NMR spectra were processed with NMRPipe and analyzed with SPARKY 3.110. 1H, 15N, and 13C resonance assignments were obtained from the following 3D heteronuclear correlation experiments: CBCA(CO)NH, CBCANH, HN(CA)CO, HNCO, HN(CO)CA, HNCA, HBHA(CO)NH, H(CCO)NH, C(CCO)NH, HCCH-COSY, CCH-TOCSY, and HCCH-TOCSY. Steady-state 15N {1H}-nuclear Overhauser effects (NOEs) of 13C, 15N-labeled mCdt1C were measured following the methods.51 3D 15N-heteronuclear single quantum coherence (HSQC)-NOESY (100 ms mixing time), 3D 13C-HSQC-NOESY for aliphatic (100 ms mixing time) and aromatic carbons (120 ms mixing time) were recorded for obtaining interproton distance restraints. Spectra were referenced by external calibration on 2,2-dimethyl-silapentane-5-sulfonic acid (DSS). The chemical shift assignments for mCdt1C were obtained with the aid of the fully automated NMR structure analysis system FLYA,52 and by interactive methods. The program CYANA32 was used for the automated assignment of NOE distance restraints33 and for the structure determination using torsion angle dynamics. For residues located in regular secondary structure segments, φ and ψ backbone dihedral angle restraints were derived from chemical shifts using the program TALOS.34 Restraints for 21 hydrogen bonds were used. CYANA structure calculations were started with 100 random conformers. The 20 conformers with the lowest target function values were selected. Restrained energy minimization was performed with the program OPALp using the AMBER force field.
Transferred cross-saturation experiment between Cdt1 and MCM complex
NMR experiments were carried out at 298 K on a Bruker DRX900 spectrometer. After purification of mouse MCM2,4,6,7 complex, we mixed it with mCdt1CL (1:50 molar ratio) at the low concentration (1 mg/mL of the MCM complex and 2.5 mg/mL of mCdt1CL) and concentrated to 0.5 mM mCdt1CL and 10 μM MCM complex in the buffer containing 10% H2O/90% D2O, 20 mM MES, 80 mM NaCl, 10 mM β-mercaptoethanol, pH 6.8. Measurements were made using a sample of 0.5 mM 2H, 15N-labeled mCdt1CL in complex with the unlabeled 10 μM MCM complex. The cross-saturation experiments were performed using the pulse scheme made by Takahashi.53 Saturation of the aliphatic protons of the MCM fragment was done using the HypSec decoupling scheme. The saturation frequency was set at 1.5 ppm with ±1 ppm saturation spectrum width. The measurement times were 23 h with 3.0 s for the saturation time. All spectra were processed and analyzed with the program nmrPipe and Sparky.
The MCM complex binding assay
Four hundred nanograms of GST-tagged mCdt1CL proteins and 100 ng of full-length MCM2,4,6,7 complex were mixed with glutathione-Sepharose (Amersham Biosciences) in 100 μL of NET-gel buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.1% Nonidet P-40, 0.25% gelatin, and protease inhibitor cocktail (Roche) for 4 h at 4°C. After washing with NET-gel buffer, precipitates were dissolved with 30 μL of 2× Laemmli sample buffer and then subjected to SDS-PAGE and Western blotting using anti-MCM6 (Santa Cruz Biotechnology) antibody.
MCM complex loading assay
Xenopus egg extracts, Xenopus demembranated sperm nuclei, and Cdt1-depleted extracts were prepared as previously described.23 GST-tagged mCdt1M+C or Xenopus Cdt1 (residues 239–620) proteins were used for the reaction. Cdt1 fraction (1 μL, final conc. 10 nM) and sperm nuclei (75 ng) were incubated for 20 min at 23°C with 5 μL of Cdt1-depleted extracts. Chromatin fractions were then isolated and Mcm4 in the fractions was detected by immunoblotting. Histone H3 was also visualized for a loading control.
Replication activity assay
Wild-type or mutant Xenopus Cdt1 proteins (1 μL) were added into Cdt1-depleted extracts (1 μL) containing sperm nuclei (18 ng) and incubated for 20 min at 23°C. Concentration of Xenopus Cdt1 is 1 nM (white) or 10 nM (gray) at this stage. Then, geminin-extracts (6 μL) was added and further incubated for 90 min to allow DNA replication. Values are indicated as percentages compared to that using buffer and normal extracts instead of Cdt1-mutant fractions and geminin extracts, respectively, after every values are subtracted by background value when buffer was used for Cdt1-fraction.
Structural changes of mutant proteins
Structural changes of mutant proteins (10 μM) compared with wild type were monitored by a circular dichroism (CD) spectrometer (Jasco J-715) at various wavelengths (200–250 nm). All the samples used here were prepared in the buffer containing 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 7 mM β-mercaptoethanol.
Acknowledgments
The authors thank Y. Ishimi for kindly providing baculovirus expression vectors for mouse MCM2,4,6,7 complex. They also thank Y. Kim and J.M. Choi for initiating the project and helping in crystallographic work. Crystallographic and NMR coordinates and structure factors have been deposited in the RCSB Protein Data Bank with accession code 3A4C and 2KLO, respectively.
References
- 1.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 2.Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 3.Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- 4.Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- 5.Leatherwood J, Lopez-Girona A, Russell P. Interaction of Cdc2 and Cdc18 with a fission yeast ORC2-like protein. Nature. 1996;379:360–363. doi: 10.1038/379360a0. [DOI] [PubMed] [Google Scholar]
- 6.Klemm RD, Austin RJ, Bell SP. Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell. 1997;88:493–502. doi: 10.1016/s0092-8674(00)81889-9. [DOI] [PubMed] [Google Scholar]
- 7.Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000;404:625–628. doi: 10.1038/35007110. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka S, Diffley JF. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat Cell Biol. 2002;4:198–207. doi: 10.1038/ncb757. [DOI] [PubMed] [Google Scholar]
- 9.Sclafani RA, Jackson AL. Cdc7 protein kinase for DNA metabolism comes of age. Mol Microbiol. 1994;11:805–810. doi: 10.1111/j.1365-2958.1994.tb00358.x. [DOI] [PubMed] [Google Scholar]
- 10.Masai H, Miyake T, Arai K. hsk1+, a Schizosaccharomyces pombe gene related to Saccharomyces cerevisiae CDC7, is required for chromosomal replication. EMBO J. 1995;14:3094–3104. doi: 10.1002/j.1460-2075.1995.tb07312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wohlschlegel JA, Dhar SK, Prokhorova TA, Dutta A, Walter JC. Xenopus Mcm10 binds origins DNA replication after Mcm2–7 stimulates origin binding Cdc45. Mol Cell. 2002;9:233–240. doi: 10.1016/s1097-2765(02)00456-2. [DOI] [PubMed] [Google Scholar]
- 12.Gregan J, Lindner K, Brimage L, Franklin R, Namdar M, Hart EA, Aves SJ, Kearsey SE. Fission yeast Cdc23/Mcm10 functions after pre-replicative complex formation to promote Cdc45 chromatin binding. Mol Biol Cells. 2003;14:3876–3887. doi: 10.1091/mbc.E03-02-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takayama Y, Kamimura Y, Okawa M, Muramatsu S, Sugino A, Araki H. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 2003;17:1153–1165. doi: 10.1101/gad.1065903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofmann JF, Beach D. Cdt1 is an essential target of the Cdc10/Sct1 transcription factor: requirement for DNA replication and inhibition of mitosis. EMBO J. 1994;13:425–434. doi: 10.1002/j.1460-2075.1994.tb06277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maiorano D, Moreau J, Mechali M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature. 2000;404:622–625. doi: 10.1038/35007104. [DOI] [PubMed] [Google Scholar]
- 16.Whittaker AJ, Royzman I, Orr-Weaver TL. Drosophila double parked: a conserved, essential replication protein that colocalizes with the origin recognition complex and links DNA replication with mitosis and the down-regulation of S phase transcripts. Genes Dev. 2000;14:1765–1776. [PMC free article] [PubMed] [Google Scholar]
- 17.You Z, Masai H. Cdt1 forms a complex with the minichromosome maintenance protein (MCM) and activates its helicase activity. J Biol Chem. 2008;283:24469–24477. doi: 10.1074/jbc.M803212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arentson E, Faloon P, Seo J, Moon E, Studts JM, Fremont DH, Choi K. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene. 2002;21:1150–1158. doi: 10.1038/sj.onc.1205175. [DOI] [PubMed] [Google Scholar]
- 19.Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene. 2002;21:6624–6632. doi: 10.1038/sj.onc.1205910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, Wagle N, Hwang DS, Dutta A. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11:997–1008. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 21.Mcgarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998;93:1043–1053. doi: 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- 22.Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science. 2000;290:2309–2312. doi: 10.1126/science.290.5500.2309. [DOI] [PubMed] [Google Scholar]
- 23.Tada S, Li A, Maiorano D, Mechali M, Blow JJ. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat Cell Biol. 2001;3:107–113. doi: 10.1038/35055000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003;5:1008–1015. doi: 10.1038/ncb1061. [DOI] [PubMed] [Google Scholar]
- 25.Li A, Blow JJ. Non-proteolytic inactivation of geminin requires CDK-dependent ubiquitination. Nat Cell Biol. 2004;6:260–267. doi: 10.1038/ncb1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem. 2006;281:6246–6252. doi: 10.1074/jbc.M512705200. [DOI] [PubMed] [Google Scholar]
- 27.Teer JK, Dutta A. Human Cdt1 lacking the evolutionarily conserved region that interacts with MCM2–7 is capable of inducing re-replication. J Biol Chem. 2008;283:6817–6825. doi: 10.1074/jbc.M708767200. [DOI] [PubMed] [Google Scholar]
- 28.Randell JC, Bowers JL, Rodriguez HK, Bell SP. Sequential ATP hydrolysis by Cdc6 and ORC directs loading of the Mcm2–7 helicase. Mol Cell. 2006;21:29–39. doi: 10.1016/j.molcel.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 29.Yanagi K, Mizuno T, You Z, Hanaoka F. Mouse geminin inhibits not only Cdt1-MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J Biol Chem. 2000;277:40871–40880. doi: 10.1074/jbc.M206202200. [DOI] [PubMed] [Google Scholar]
- 30.Lee C, Hong B, Choi JM, Kim Y, Watanabe S, Ishimi Y, Enomoto T, Tada S, Kim Y, Cho Y. Structural basis for inhibition of the replication licensing factor Cdt1 by geminin. Nature. 2004;430:913–917. doi: 10.1038/nature02813. [DOI] [PubMed] [Google Scholar]
- 31.Ferenbach A, Li A, Brito-Martins M, Blow JJ. Functional domains of the Xenopus replication licensing factor Cdt1. Nucleic Acids Res. 2005;33:316–324. doi: 10.1093/nar/gki176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmerman DE, Kulikowski CA, Huang Y, Feng W, Tashiro M, Shimotakahara S, Chien C, Powers R, Montelione GT. Automated analysis of protein NMR assignments using methods from artificial intelligence. J Mol Biol. 1997;269:592–610. doi: 10.1006/jmbi.1997.1052. [DOI] [PubMed] [Google Scholar]
- 33.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 34.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 35.Holm L, Sander C. Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 1997;25:231–234. doi: 10.1093/nar/25.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Smith CL, Deryckere D, Deangelis K, Martin GS, Berger JM. Structure and function of Cdc6/Cdc18: implications for origin recognition and checkpoint control. Mol Cell. 2000;6:637–648. doi: 10.1016/s1097-2765(00)00062-9. [DOI] [PubMed] [Google Scholar]
- 37.Singleton MR, Morales R, Grainge I, Cook N, Isupov MN, Wigley DB. Conformational changes induced by nucleotide binding in Cdc6/ORC from Aeropyrum pernix. J Mol Biol. 2004;343:547–557. doi: 10.1016/j.jmb.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 38.Dueber EL, Corn JE, Bell SD, Berger JM. Replication origin recognition and deformation by a heterodimeric archaeal Orc1 complex. Science. 2007;317:1210–1213. doi: 10.1126/science.1143690. [DOI] [PubMed] [Google Scholar]
- 39.Gaudier M, Schuwirth BS, Westcott SL, Wigley DB. Structural basis of DNA replication origin recognition by an ORC protein. Science. 2007;317:1213–1216. doi: 10.1126/science.1143664. [DOI] [PubMed] [Google Scholar]
- 40.Erzberger JP, Mott ML, Berger JM. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- 41.Andreeva A, Murzin AG. Evolution of protein fold in the presence of functional constraints. Curr Opin Struct Biol. 2006;16:399–408. doi: 10.1016/j.sbi.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D. A census of protein repeats. J Mol Biol. 1999;293:151–160. doi: 10.1006/jmbi.1999.3136. [DOI] [PubMed] [Google Scholar]
- 43.Gajiwala KS, Burley SK. Winged helix proteins. Curr Opin Struct Biol. 2000;10:110–116. doi: 10.1016/s0959-440x(99)00057-3. [DOI] [PubMed] [Google Scholar]
- 44.Zheng N, Fraenkel E, Pabo CO, Pavletich NP. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 1999;13:666–674. doi: 10.1101/gad.13.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey S, Eliason WK, Steitz TA. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science. 2007;318:459–463. doi: 10.1126/science.1147353. [DOI] [PubMed] [Google Scholar]
- 46.Bussiere DE, Bastia D, White SW. Crystal structure of the replication terminator protein from B. subtilis at 2.6 Å. Cell. 1995;80:651–660. doi: 10.1016/0092-8674(95)90519-7. [DOI] [PubMed] [Google Scholar]
- 47.Manna AC, Pai KS, Bussiere DE, Davies C, White SW, Bastia D. Helicase-contrahelicase interaction and the mechanism of termination of DNA replication. Cell. 1996;87:881–891. doi: 10.1016/s0092-8674(00)81995-9. [DOI] [PubMed] [Google Scholar]
- 48.You Z, Komamura Y, Ishimi Y. Biochemical analysis of the intrinsic Mcm4-Mcm6-mcm7 DNA helicase activity. Mol Cell Biol. 1999;12:8003–8015. doi: 10.1128/mcb.19.12.8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Farrow NA, Zhang O, Forman-Kay JD, Kay LE. A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J Biomol NMR. 1994;4:727–734. doi: 10.1007/BF00404280. [DOI] [PubMed] [Google Scholar]
- 52.Lo´Pez-Me´Ndez B, Guntert P. Automated protein structure determination from NMR spectra. J Am Chem Soc. 2006;128:13112–13122. doi: 10.1021/ja061136l. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi H, Nakanishi T, Kami K, Arata Y, Shimada I. A novel NMR method for determining the interfaces of large protein-protein complexes. Nat Struct Biol. 2000;7:220–223. doi: 10.1038/73331. [DOI] [PubMed] [Google Scholar]