

Abstract
Domain 4 of the anthrax protective antigen (PA) plays a key role in cellular receptor recognition as well as in pH-dependent pore formation. We present here the 1.95 Å crystal structure of domain 4, which adopts a fold that is identical to that observed in the full-length protein. We have also investigated the structural properties of the isolated domain 4 as a function of pH, as well as the pH-dependence on binding to the von Willebrand factor A domain of capillary morphogenesis protein 2 (CMG2). Our results provide evidence that the isolated domain 4 maintains structure and interactions with CMG2 at pH 5, a pH that is known to cause release of the receptor on conversion of the heptameric prepore (PA63)7 to a membrane-spanning pore. Our results suggest that receptor release is not driven solely by a pH-induced unfolding of domain 4.
Keywords: anthrax, CMG2, domain, pH, protective antigen, toxin
Introduction
Bacillus anthracis secretes an 83 kDa, four domain protein called the protective antigen (PA), which binds one of two host cell receptors that have been identified to date: anthrax toxin receptor 1/tumor endothelial marker 8 (ATR/TEM8 – ANTXR1)1,2 or capillary morphogenesis protein 2 (CMG2 – ANTXR2).3,4 On binding to the receptor, PA is cleaved by a cell-surface furin-like protease into two fragments, a 20 kDa fragment which dissociates into the extracellular milieu, and a 63 kDa fragment that remains bound. The 63 kDa fragment spontaneously oligomerizes into a heptameric (PA63)7 structure called the prepore. Formation of the prepore creates binding sites for edema factor (EF) and or lethal factor (LF), two enzymatic moieties which along with PA constitute the anthrax toxin.5 Receptor-mediated endocytosis carries the toxin into the cell, where it is trafficked to late endosomes that eventually become acidic.6 Bound to the cell receptor and at a pH of ∼5–6, the prepore undergoes a major structural change to form a membrane spanning β-barrel pore, which allows EF and LF to transit through the pore into the cell.7,8 The manifestation of enzymatic activities of EF and LF, once inside the cell cytosol, results in disease pathogenesis.
The receptor plays a key role in this process, as it is needed for both entry of the toxin into the cell, and preventing premature pore formation until the toxin reaches the correct cellular compartment.9,10 When bound to the von Willebrand factor A (vWA) domain of ATR/TEM8, the pH threshold of pore formation is pH 6, whereas when bound to the vWA domain of CMG2 the pH threshold for pore formation is lowered to pH 5.10 Each of the receptors utilize a conserved metal-ion-dependant-adhesion (MIDAS) site within the vWA domain for binding to domain 4 of PA, which along with residues in the receptor, coordinates a magnesium ion that is critically important for binding.2 Domain 4, comprising residues 595–735, was shown as an isolated domain fused to glutathione S-transferase (GST) to be sufficient for binding to the vWA domain of CMG2, although interactions from domain 2 are also important.11–13 Indeed, recent mutagenesis studies suggest that residues within the vWA domain that interact with domain 2 determine the pH threshold for pore formation.14
At the acidic pH of the endosome, the domain 2 β2–β3 strands peel away from domain 2 and insert into the membrane, forming a 14-stranded beta barrel pore.15,16 For this event to occur in the presence of the receptor, domain 2 must partially dissociate from the receptor and lose potential stabilizing interactions with domains 3 and 4. Indeed, recent studies using immunoprecipitation have shown that both ATR/TEM8 and CMG2 dissociate from the prepore at pH values required for pore formation,10 the latter corroborated with data from NMR studies.17 Although not well-understood, we hypothesized that the large structural change from the prepore to the pore is likely to induce receptor dissociation, through a pH-induced unfolding of domain 4. In this study, we use X-ray crystallography to determine the structure of domain 4, and in addition we have investigated the structural properties of the isolated domain 4, whether it undergoes a structural change (unfolding) or loss of binding to CMG2 at pH values known to induce pore formation.
Results
Previous experiments have shown that a GST-domain 4 fusion is capable of binding to CMG2,1 and because we use the same gene construct (encoding residues 595–735) for our studies, we assume that the structural properties are also the same. An SDS-PAGE of the proteins used in this study are shown in Figure 1, and include the GST-domain 4 fusion, the vWA domain of CMG2 and, after cleavage of GST from domain 4 with thrombin, the isolated domain 4. To determine the extent to which the isolated domain 4 assumes the correct tertiary structure, we have characterized the structure using X-ray crystallography, fluorescence, circular dichroism, and NMR methods, as well as association to the vWA domain of CMG2 at pH 8 and 5.
Figure 1.

Proteins used in this study. Purified proteins were subjected to SDS-PAGE using a 4–20% gradient gel (Bio-Rad) and stained using Coomassie blue.
Refined domain 4 structure
Plate-shaped crystals of domain 4 were obtained using sitting drop vapor diffusion, which diffracted to a resolution of 1.95 Å. The structure solution was carried out by molecular replacement using domain 4 (residues 595–735) of the full-length PA (PDB:1ACC).18 The asymmetric unit contained two molecules (A and B) of domain 4 which are structurally similar to domain 4 within the full-length PA. The overall root mean square (RMS) difference between Cα atoms of domain 4 in the full-length PA and that of the isolated domain 4 is 0.73 Å and 0.54 Å between chains A and B, respectively. The final model included residues R595 and G735, but residues T706 through T716 of molecule B were disordered and could not be fit to the observed electron density maps. Additionally, residues G593 and S594, which remained from the thrombin cleavage, could be traced at the N-terminus of each molecule. The noncrystallographic dimer of domain 4 is shown in Figure 2, along with a figure of an overlay between domain 4 of the full-length PA and the isolated domain 4 molecules A and B. A summary of the relevant data collection and refinement statistics are shown in Table I.
Figure 2.
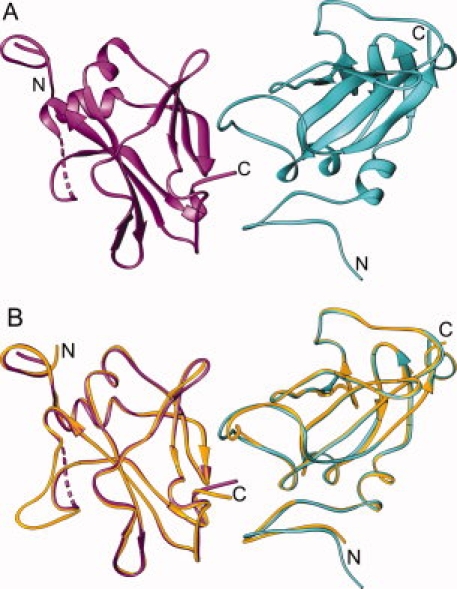
Structure of domain 4. A: Noncrystallographic dimer showing subunit A (turquoise) and subunit B (magenta). The terminal ends of each subunit are indicated by N and C. Disordered residues in subunit B are represented by the dashed line. B: Domain 4 from full-length PA (yellow) superimposed onto each subunit of the noncrystallographic dimer from (A).
Table I.
Crystallographic Data for Protective Antigen Domain 4 Refined to 1.95 Å Resolution
| Protective antigen domain 4 | |
|---|---|
| Data collection | |
| Unit-cell parameters (Å, °) | a = 62.74, b = 35.85, c = 129.09, β = 99.3 |
| Space group | C2 |
| Resolution (Å) | 20.0–1.95 |
| Wavelength (Å) | 1.5418 |
| Temperature (K) | 93 |
| Observed reflections | 65,480 |
| Unique reflections | 20,594 |
| 〈I/σ(I)〉a | 25.1 (4.3) |
| Completeness (%)a | 97.7 (87.1) |
| Redundancya | 3.2 (2.5) |
| Rmerge (%)a,b | 6.8 (29.1) |
| Refinement | |
| Resolution (Å) | 20.0–1.95 |
| Reflections (working/test) | 19,523/1,050 |
| Rfactor/Rfree (%)c | 19.9/25.8 |
| No. of atoms (protein (A:B)/water) | 1166:1081/111 |
| Model quality | |
| R.m.s deviations | |
| Bond lengths (Å) | 0.021 |
| Bond angles (°) | 1.930 |
| Average B factor (Å2) | |
| All atoms | 29.9 |
| Protein (A/B) | 27.1/32.4 |
| Water | 34.2 |
| Coordinate error based on Rfree (Å) | 0.179 |
| Ramachandran Plot | |
| Favored (%) | 99.3 |
| Allowed (%) | 0.7 |
Values in parentheses are for the 2.02 to 1.95 Å resolution shell.
Rmerge = ΣhklΣi |Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl), where Ii(hkl) is the intensity measured for the ith reflection and 〈I(hkl)〉 is the average intensity of all reflections with indices hkl.
Rfactor = Σhkl ||Fobs (hkl) | − |Fcalc (hkl) ||/Σhkl |Fobs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
Fluorescence experiments as a function of pH
The fluorescence excitation and emission spectra in a buffer comprised of Bis-Tris/HEPES/cacodylic acid at pH 8 and 5 are shown in Figure 3. Domain 4 contains no disulfides and no tryptophans, but there are eight tyrosines and four phenylalanines, and the fluorescence spectra (excitation and emission) are consistent with the presence of these residues (λex = 277 nm, λem = 305 nm) {Fig. 3(A)]. At pH 5, there is a small increase in the fluorescence emission, but we observe no change in the wavelength maximum, indicating no gross structural changes at pH 5. We chose pH 5 because at this pH, in the presence of the vWA domain of CMG2 the prepore is known to undergo a structural transition to a membrane spanning pore.14,19 To determine the extent to which pH influences domain 4 structure, we monitored the emission intensity (305 nm) as a function of pH using a buffer system consisting of Bis-Tris/HEPES/cacodylic acid and citric acid. We carried out two separate experiments, one with a protein incubated overnight (4°C) at pH 2 and the other incubated overnight (4°C) at pH 8, and then added the protein to buffers (10 mM BisTris/HEPES/cacodylic acid/citric acid) ranging from pH 2 to 8. The pH unfolding process is reversible and indicates that the structure adopted in solution is relatively stable to pH. The data were fit using nonlinear least squares analysis to a two state Henderson-Hasselbalch protonation equilibrium, yielding an apparent pKa of 4.96 ± 0.07. We note that the transitions could not be fit well at lower pH values, suggesting the possibility of a second, independent transition at low pH.
Figure 3.
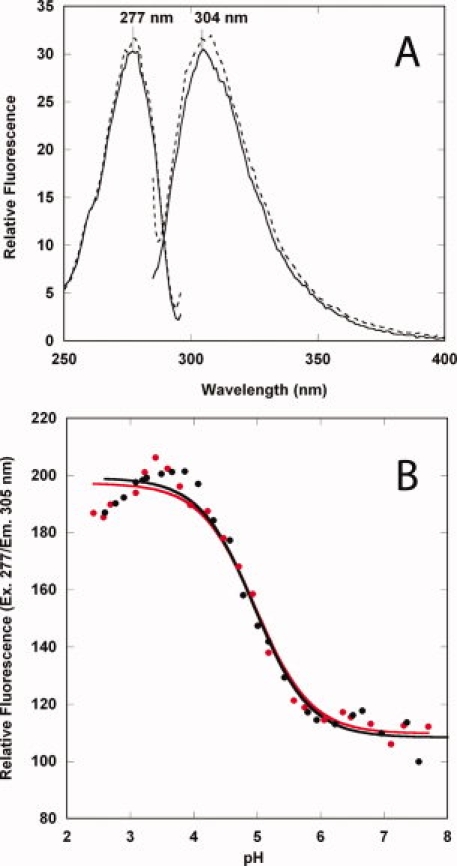
Fluorescence properties of domain 4. A: Fluorescence excitation and emission spectra of domain 4 in 10 mM Bis-Tris/HEPES/cacodylic acid at pH 8 (dashed line) and pH 5 (solid line). Spectra were recorded at 20°C, 1 μM final protein concentration. Excitation spectra were recorded with the emission wavelength set at 305 nm, and the emission spectra were recorded with an excitation wavelength of 277 nm. B: pH-dependence of the emission intensity at 305 nm (Ex. 280). Data were recorded at 20°C, 1 μM final concentration in buffers containing 10 mM each of Bis-Tris/HEPES/cacodylic acid/citric acid, spanning the range from pH 2 to 8. Data starting with protein at pH 2 ( in red color) and pH 8 (•) are shown. The transitions were fit to a two state protonation equilibrium based on the Henderson–Hasselbalch equation.
in red color) and pH 8 (•) are shown. The transitions were fit to a two state protonation equilibrium based on the Henderson–Hasselbalch equation.
Circular dichroism—Stability to temperature at pH 8 and 5
We also used circular dichroism (CD) spectroscopy to measure secondary structural content at pH 8 and 5, as well as the stability and reversibility of folding to temperature at these pH values (Fig. 4). The far-UV CD of domain 4 is unusual, in that there is a single minimum at ∼198 nm (approximately −18,000 degree cm2 dmol−1) that is indicative of an unfolded structure. This is in contrast to a previous report of the structure of the isolated domain 4 by CD, which shows the protein having a substantial amount of α-helix.20 We have found in the course of our studies that the presence of certain detergents such as Triton X-100 (above the critical micelle concentration) induces helix formation in domain 4, which may explain the apparent helical CD structure observed in the other report (unpublished observations). Nonetheless, if we monitor the temperature dependence at pH 8 from 5 to 60°C (at ∼198–201 nm), we observe a cooperative transition with a midpoint (TM) of ∼37°C.
Figure 4.
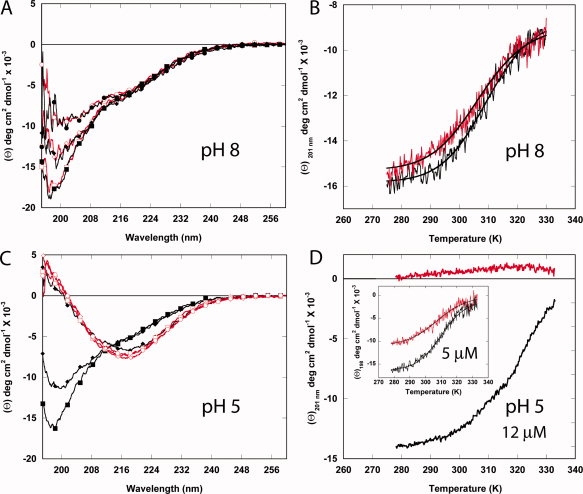
Circular dichroism analysis of domain 4 as a function of pH and temperature. Spectra as a function of temperature (A and C) and recorded at a fixed wavelength of 201 nm (B and D) at pH 8 (A and B) and pH 5 (C and D) in 5 mM BisTris/HEPES/cacodylic acid, 67 mM NaCl with 12.1 μM domain 4. In (A) and (C), spectra were recorded at 5 (▪), 33 (♦), and 60°C (•). Decreasing temperature is also shown from 60 to 5°C (red line) at 60 (○), 33 (⋄), and 5°C (□). In (B) and (D), data in black were recorded from 5 to 60°C, and data in red were recorded from 60 to 5°C. The inset for 3D (5 μM domain 4) has the first increase in temperature in black and the second increase (after rapid cooling from high temperature) is shown in red.
The temperature dependence of the CD at pH 8 was not completely reversible and was dependent on the concentration of protein and salt. At salt concentrations above 50 mM NaCl and lower protein concentrations (c < 15 μM), we observed greater reversibility, suggesting that the protein was prone to aggregation at high temperatures. At pH 5 (and 12.1 μM), the temperature dependence showed that the protein aggregated just above the TM into a more β-sheet structure that is reminiscent of amyloid fibers. Lowering the protein concentration to 5 μM and increasing the salt content (∼140 mM) resulted in less aggregation, but the transition was still not reversible [Fig. 4(D), inset]. Nonetheless, there is empirical evidence from systems studied by Sturtevant that proteins which show irreversibility in the second transition accurately follow thermodynamic behavior in the first heating.21–24 Therefore, we present (cautiously) the thermodynamic values for the first heating of domain 4 at pH 8 (12.1 μM) and pH 5 (5 μM), and the data are summarized in Table II.
Table II.
Thermodynamic Values at pH 8 and pH 5
| pH | ΔH°NUa (kcal/mol) | ΔS°NU (e.u.)b | TM,NU (K)c |
|---|---|---|---|
| 8d | 23.8 ± 1.6e | 76.9 ± 5.1 | 309.7 |
| 5 | 23.7 ± 1.6 | 76.5 ± 5.1 | 310.2 |
NU = native to unfolded, temperature ramp from 5 to 60°C.
e.u. expressed in cal mol−1 K−1.
Tm is calculated according to Eq. (9).
The concentrations of domain 4 for measurements at pH 8 were 12.1 μM, 67 mM NaCl; for pH 5, the data are values recorded at 5 μM, 140 mM NaCl.
Errors determined from the fit to Eq. (8).
GST-pulldown assay to monitor domain 4-receptor complex formation at pH 8 and 5
To determine whether the isolated domain 4 could form a complex with the VWA-domain of CMG2, we monitored complex formation using a GST-pulldown assay, in a manner similar to that carried out by Bradley and coworkers using GST-domain 41 but monitored by Coomassie blue staining (Fig. 5). In the presence of CMG2, GST-domain 4 forms a stable complex with CMG2 at pH 8, consistent with previous observations. However, at pH 5 we observe a loss in complex formation indicating release of the receptor. In this assay, the glutathione sepharose resin to which the GST moiety is bound is washed twice with the appropriate pH buffer (see experimental procedure), and thus the decrease in intensity of CMG2 (pH 5) may be a result of partial unfolding of domain 4 (Fig. 3), an increase in the off-rate at low pH or a combination of both.
Figure 5.

GST pull-down assay as a function of pH. GST or GST-domain 4 was incubated in the presence or absence of the VWA domain of CMG2 at pH 8, and then concentrated buffer (0.5 M) was added to either maintain the pH at 8 or change the pH to 5. The proteins were then bound to glutathione sepharose (GE-Healthcare) and washed twice with buffer at pH 8 or 5. Proteins remaining bound to the glutathione sepharose resin after washing at pH 8 or 5 were separated by SDS-PAGE and analyzed by Coomassie blue staining. The (*) indicates some breakdown of the GST-domain 4 (note that the band for free GST is present). Lane 1 is the vWA domain of CMG2 shown as a reference. The next four are GST (lanes 2 and 3), or GST-domain 4 proteins (lanes 4 and 5). Lanes 6–10 have CMG2 added to either GST (lanes 6 and 7) or GST-domain 4 (lanes 8 and 10).
Analytical gel filtration analysis of complex formation at pH 8 and 5
In separate experiments with isolated domain 4, we studied binding to CMG2 using gel filtration and NMR. Figure 6 shows the elution profile of CMG2, domain 4 and the complex at pH 8 and 5 by gel filtration. In these experiments, the proteins (ratio of 1:2, CMG2:domain 4) were allowed to equilibrate in phosphate-buffered saline (PBS; pH 7.4) supplemented with MgCl2 overnight on ice, followed by changing the pH either to 8 or 5, and allowing to equilibrate for an additional ∼8 h. The proteins were then injected onto a Superose-12 column (GE-Healthcare) equilibrated in pH 8 or 5 buffer containing 20 mM each of Bis-Tris/HEPES/cacodylic acid, along with 2.0 mM MgCl2 at 4°C. Although the elution times vary from pH 8 to 5, domain 4 maintains a complex with CMG2 at either pH. We note that application of chymotrypsinogen A as a standard also results in a similar elution time shift (77 min to 81 min at pH 8 and 5, respectively, data not shown), suggesting that the column properties may change slightly at low pH.
Figure 6.

Analytical gel filtration of domain 4-CMG2 complexes. A Superose 12 column (GE-Healthcare) was equilibrated at 4°C in 20 mM each of Bis-Tris/Hepes/cacodylic acid, and 2 mM MgCl2 at pH 8 (upper panel) or pH 5 (bottom panel). Samples (100 μL) of CMG2 (red peak), domain 4 (blue peak), and the complex (green peak) were loaded at a flow rate of 0.2 ml/min. The sum of domain 4 and CMG2 individual peaks is shown as a dotted line. Inset in the lower panel shows an SDS-PAGE analysis after silver staining of the peaks pertaining to the complex of CMG2 and domain 4 (A), CMG2 alone (B), and domain 4 alone (C) (pH 5), as well as the complex of CMG2 and domain 4 at pH 8 in the upper panel.
1H-15N HSQC NMR of domain 4 and domain 4 bound to CMG2 at pH 8 and 5
The results using gel filtration indicate stable association with CMG2 at low pH, in contrast to the GST-pulldown assay. As a separate measure of binding as well as structure at pH 5, we measured the effect on the three-dimensional structure of domain 4 in the presence and absence of CMG2 at pH 8 and 5, using 1H-detected, 15N-edited heteronuclear single quantum coherence (HSQC) NMR. The 1H-15N-HSQC spectra of the amide NH region of an 15N-labeled domain 4, either in the presence or absence of CMG2 at pH 8 and 5 (20°C), is shown in Figure 7. First, the well-dispersed resonances in both dimensions indicate that the protein is well-structured and stable at pH 8 and 5. Also, the sequence specific resonance assignments of backbone residues and its secondary structural elements (manuscript in preparation) are in good agreement with the isolated domain 4 X-ray structure, indicating that the fold is similar in solution. In comparison of pH 8 to 5, some of the resonances do not overlay and are shifted, and the number of resonances at pH 5 increase, suggesting that at pH 5 there is some conformational heterogeneity that likely reflects partial unfolding of the protein.
Figure 7.
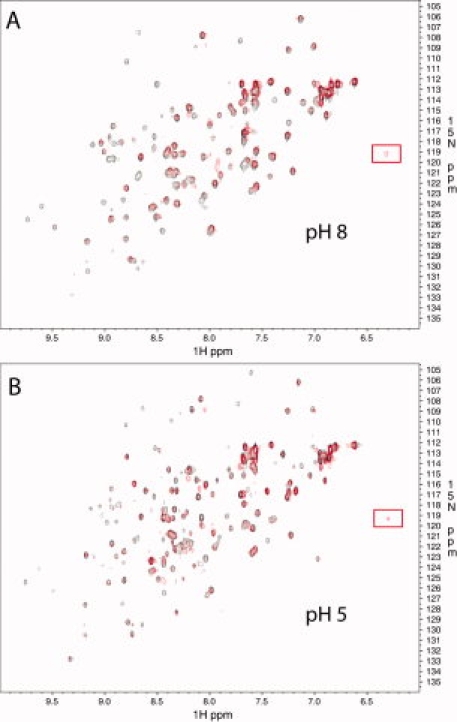
Superposition of 2D-[1H-15N] HSQC of [U-15N]-domain 4 (black) and [U-15N]-domain 4 + unlabeled CMG2 (red) at pH 8 (top panel, A) and pH 5 (bottom panel, B). The data were collected at 20°C. The resonance at ∼6.3 ppm in the presence of CMG2 is highlighted by a red box.
In the presence of CMG2, we note that although many resonances remain unchanged, several have shifted or are no longer present. In particular, new resonances are observed with a prominent peak at ∼6.3 ppm (1H), which persists at pH 5. This provides further support that domain 4 maintains structure and interactions with CMG2, albeit to a lower extent, at pH 5.
Discussion
The binding of domain 4 of PA to the cell surface receptor is a key step in anthrax pathogenesis, as it leads to endocytosis, trafficking, and finally translocation of EF and LF into the cell cytosol. At the low pH of the endosome, a major structural change occurs in which domain 2 residues form a β-barrel pore. In vitro studies have shown that at this same pH, the receptor dissociates.10,17 This implied to us that domain 4, in addition to domain 2, may also undergo a conformational change (unfolding) that results in the release the receptor.
The crystal structure of domain 4 revealed a structure with amazing adherence to the topology of domain 4 within the context of the full-length PA protein. An overlay of the structure of domain 4 (molecule A) and domain 4 within the context of PA shows that domain 4 assumes its structure irrespective of domain 2 or domain 3, and is indeed an independently folding unit.25 In this respect, we note that a search for similar structures using DALI26 revealed that domain 4 adopts a fold similar to the small, 12.7 kDa Flammulina velutipes immunomodulatory protein Fve,27 with an average root mean square deviation (RMSD) of 3.9 Å. As these authors have assigned the fold a pseudo-h-type topology, an immunoglobulin fold with characteristics of both s- and h-type folds,28 we also assign domain 4 to this topology.
Given the structure of isolated domain 4 and the similarity to the domain observed in the full-length protein, the question of how this structure might change to cause dissociation of the receptor is of great interest. Domain 4 is marginally stable and undergoes partial unfolding at pH 5 as evidenced by fluorescence, with an apparent pKa of ∼5. We cannot assess, however, at which point along the pH transition the actual unfolding of the protein occurs, because all but one of the seven tyrosines are solvent exposed, and may not reflect unfolding of the hydrophobic core. Consistent with this, the TM measured at pH 8 or 5 is virtually unchanged, suggesting that when some partial unfolding may occur at pH 5, it is unlikely to affect those elements needed to maintain global stability. Furthermore, although the NMR data shows that at pH 5 the protein may undergo some partial unfolding, the structure does not unfold to an extent that results in a complete loss in binding, as peaks associated with binding (such as that at 6.3 ppm) are maintained. The latter is also supported by gel filtration, which shows that domain 4 maintains association with the vWA domain at pH 5.
Our study suggests then that pH-induced unfolding is unlikely to be the sole mechanism responsible for receptor release, and that to achieve complete dissociation requires other structural changes to assist in the release process. It may be, for instance, that domain 4 maintains its structure in the pore state at pH 5, but is sterically prohibited from interacting with the receptor. Alternatively, if one assumes that in the context of the prepore domain–domain interactions are necessary for the stability of the protein,29,30 low pH induced unfolding of domain 2 may cause unfolding of domain 4 and lead to receptor dissociation. Clearly, further studies are required to understand the relative contributions of domain 2 and domain 4 to the stability PA as a whole, the relative contributions of each domain to binding to the cellular receptor, and structural changes in domain 4 within the context of the prepore to pore transition.31
Materials and Methods
Protein production and purification
All chemicals (buffers, salts) used were from Sigma (St. Louis, MO) or Fisher Scientific unless otherwise specified, and were >99% pure. Bis-Tris and d-glucose were from MP Biomedicals (Solon, OH). Restriction enzymes, T4-ligase were from New England Biolabs (Ipswich, MA).
The gene encoding Domain 4 was cloned using the polymerase chain reaction (PCR) (Proofstart DNA polymerase kit, Qiagen) from using the full-length PA gene as template as described previously.1 Briefly, the region 595–735 comprising domain 4 was subcloned into the NotI/BamHI restriction sites in the plasmid pGEX-4T1 (GE-HealthCare). DNA sequencing was carried out at the Protein-Nucleic acid facility at Washington University in St. Louis using BigDye Terminator premix for automated sequencing. The plasmid (pGEX-D4) was transformed into E. coli strain BL-21 and grown in either ECPM132 or, for isotopic labeling with 15N, ECPM1 without casamino acids, substituting 15N-NH4Cl for NH4Cl. Bacteria were grown in Fernbach flasks at 32°C to an optical density (OD) at 600 nm of 3.0, and subsequently induced with 1.0 mM isopropyl-β-d-galactopyranoside (IPTG; Gold Biotechnology Inc.) then allowed to grow to an OD600 of 6.0. Bacteria were harvested by centrifugation in a swinging bucket centrifuge at 4°C, and the pellet frozen at −20°C. The pellet was resuspended in ice-cold PBS pH 7.4 and subsequently lysed using a Branson sonifier. The solution was centrifuged at 20,000 × g for 30 min, supernatant removed, and filtered through a 0.22 μm filter (Millipore-ExpressPlus), and applied to a 5 mL GST-column (GE-Healthcare). The column was washed with at least five column volumes of PBS, before adding ∼400 units of thrombin (GE Healthcare) and allowed to incubate overnight at room temperature. The next day, the protein was purified by eluting in PBS through a 5 mL benzamidine column, to afford pure domain 4. A similar protocol was used for purification of CMG238–218 (also as a GST fusion).17 Final purification of either domain 4 or CMG2 was done by applying protein to a Superdex-200 16/60 gel filtration column (GE Healthcare) equilibrated in PBS pH 7.4, 4 °C. For experiments using the GST-domain 4 fusion, purification of the fusion was carried out in a similar manner as isolated domain 4, except that rather than adding thrombin, the protein was eluted with 50 mM Tris pH 8.0 containing 10 mM reduced glutathione. GST and GST-domain 4 were then dialyzed into 20 mM Tris pH 8.0. Concentrations were determined using extinction coefficients of 11,920 M−1 cm−1 (domain 4), 13,075 M−1 cm−1 (CMG2), 47,000 M−1 cm−1 (GST), and 58,920 M−1 cm−1 (GST-D4).33
GST-pulldown assay
In this assay, two sets of four samples were made that contained GST, GST-Domain 4, GST +CMG2, and GST-Domain 4 + CMG2. One set would be used for the pH 8 study, the other for pH 5. A solution of 80 μM GST or GST-Domain 4 in 20 mM Tris pH 8 (31 μL) was combined with 43.6 μM CMG2 in PBS pH 7.4 or a similar amount of PBS (36 μL). After the addition of 1 μL of 0.1 M MgCl2, the solutions (68 μL) were allowed to incubate on ice overnight. To this, 57 μL of 0.5 M Bis-Tris/Hepes/cacodylic acid at pH 8 was added along with 1 μL of 0.1 M MgCl2 to one set of four samples, to the other set 57 μL of 0.5 M Bis-Tris/Hepes/cacodylic acid at pH 5 and 1μL of 0.1 M MgCl2. These solutions (125 μL) were allowed to incubate on ice for 1 h. Next, 50 μL of glutathione sepharose resin (GE-Healthcare) in PBS pH 7.4, 2 mM MgCl2 was added to each solution and incubated for 30 min on ice. The solutions with resin were centrifuged (Eppendorf) and the supernatant was removed. The resin was washed two times with 150 μL of 50 mM each of Bis-Tris/Hepes/cacodylic acid (pH 8 or 5), 1 mM MgCl2. The resin was resuspended in SDS sample buffer, boiled for 5 min, and 24 μL was loaded into a 4–20% SDS-PAGE gel (Bio-Rad), and run at a constant 200 V for 30 min.
Fluorescence
Excitation and emission spectra of domain 4 (1.0 μM) were recorded on a Cary Eclipse spectrofluorimeter at 20°C in 10 mM each of Bis-Tris/HEPES/cacodylic acid pH 8 or 5. The emission spectrum was recorded with an excitation wavelength of 277 nm, whereas the excitation spectrum was recorded with an emission wavelength of 305 nm. For the pH titration, two separate solutions of domain 4 (20 μM) in 10 mM Bis-Tris/HEPES/cacodylic acid and citric acid buffer solutions, incubated overnight at 4°C at pH 2 or 8, were added (50 μL) to buffers (950 μL of 10 mM Bis-Tris/HEPES/cacodylic acid and citric acid) spanning the pH range from 2 to 8. The final pH values were determined using an Orion ROSS semimicro pH probe connected to a Fisher AR20 pH meter. Emission spectra were recorded at 20°C with an excitation of 277 nm, and the peak intensity at 305 was plotted as a function of pH. Data were fit according to the Henderson-Hasselbalch equation assuming a two-state protonation equilibrium.34
Far UV CD
Circular dichroism measurements were carried out using a Jasco-J810 spectropolarimeter. Spectra of domain 4 (12.1 μM) in 5 mM each of Bis-Tris/Hepes/cacodylic acid at pH 8 or 5, 67 mM NaCl were recorded between 180 and 260 nm in a 0.5 mm thermostatted cylindrical cell at 5°C for eight accumulations at a scan speed of 10 nm min−1 and a response time of 4 s. For variable temperature experiments, the molar ellipticity was monitored at 201 nm with a response time of 16 s, and temperature adjusted from 5 to 60°C and back from 60 to 5°C using a Julabo F25/HD circulating water bath. The temperature gradient was set at 1°C min−1, and monitored continuously using an in-line thermistor placed near the CD cell. Spectra were taken at 5, 33, and 60°C for both the increase and decrease in temperature, and five accumulations were acquired with a scan speed of 20 nm min−1 and a response of 2 s.
Inset far UV CD variable temperature
The CD of domain 4 (5 μM) in 5 mM each of Bis-Tris/Hepes/cacodylic acid at pH 5, 140 mM NaCl was recorded at 198 nm as a function of temperature with a slope of 1°C min−1, and a response of 16 s between 5 and 60°C. Because of problems with aggregation, the cooled down process from 60°C to 5°C was done rapidly by adding ice to the water bath. Then, once the temperature stabilized at 5°C, a second variable temperature study was initiated from 5 to 60°C.
Determination of thermodynamic values at pH 8 and 5 by CD
The temperature dependence on the CD201 were fit assuming that the transition approximates a simple two-state mechanism35:
| (1) |
The corresponding equilibrium constant will be:
| (2) |
where fU and fN are the fraction unfolded and fraction folded, respectively, which for a reversible transition is equal to 1 at any temperature:
| (3) |
Then, the CD (θobs) at any point along the transition will be:
| (4) |
where θN and θU correspond to the CD of the native and unfolded states, respectively.
Substituting Eq. (3) into (4) and solving for fU gives:
| (5) |
From
| (6) |
| (7) |
nonlinear least squares analysis of the entire transition curve can be represented using the equation:
| (8) |
As ΔG° = 0 at the TM,
 |
(9) |
Gel filtration
Two sets (pH 8 or 5) of three samples were made that included domain 4 (16 μM), CMG2 (8 μM), or the complex domain 4 (16 μM) and CMG2 (8 μM) in PBS with 2 mM MgCl2. All samples were incubated for 8 h on ice. After incubation, 27 μL of 1 M Bis-Tris/Hepes/cacodylic acid pH 5 or 8 was added to each of the three samples (total volume 128 μL) and were further incubated overnight on ice. Approximately 100 μL of sample was loaded onto a Superose-12 gel filtration column (GE-Healthcare) and eluted at a flow rate of 0.2 mL min−1 using an AKTA-Prime system (GE-Healthcare), which allowed continuous monitoring of the UV absorbance at 280 nm. The individual peaks were analyzed for the presence of CMG2 and domain 4 by SDS-PAGE on a 4–20% gradient gel and stained using the PlusOne Silver Staining kit (GE-Healthcare) according to the manufacturer's instructions.
HSQC NMR studies of domain 4
Two sets of two samples were set up for the 15N-HSQC. A total of 168 μL of 149 μM [U-15N]-domain 4 in PBS, 2 mM MgCl2 were added to each sample. To one sample, 260 μL of 96.3 μM unlabeled CMG2 was added and to the other 260 μL of PBS. All samples were allowed to incubate overnight on ice. The next day, 117 μL of 1 M Bis-Tris/Hepes/cacodylic acid at pH 8 or 5 was added to each sample. After addition of 5 μL D2O for a spin-lock, they were allowed to incubate overnight on ice before being analyzed by NMR. All NMR spectra were recorded at 25°C using Bruker Avance 800 MHz NMR equipped with a triple resonance TCI cryoprobe with a triple axis pulsed field gradient. Each data set was collected using 2048 × 128 complex points with 12820.51 Hz and 2580.65 Hz spectral widths in the F2 and F1 dimensions. We used gradient sensitivity enhanced 1H-15N-HSQC with WATERGATE for water suppression. Forty-eight transients with 1 s relaxation delay/recycle time were used. Data were processed and analyzed using NmrPipe and NmrDraw.36
Crystallization and data collection
Protective Antigen Domain D4 concentrated to 10 mg mL−1 in 140 mM NaCl, 5 mM Hepes pH 7.0 was screened for crystallization in Compact Jr. sitting drop vapor diffusion plates (Emerald biosystems) using 1 μL of crystallization solution and 1 μL of protein equilibrated against 100 μL of the latter. Crystals displaying a plate morphology were obtained at 4°C within 24 h from the Precipitant Synergy screen (Emerald biosystems) condition #51 (20% PEG 8000, 100 mM imidazole pH 6.5, 3% MPD). Single crystals were transferred to a solution containing 20% PEG 8000, 100 mM imidazole pH 6.6, 20% glycerol, and frozen in liquid nitrogen for data collection. Data were collected at 93 K using a Rigaku RU-H3R rotating anode generator (Cu-Kα) equipped with Osmic Blue focusing mirrors and a Rigaku Raxis IV++ image plate detector.
Structure solution and refinement
Intensities were integrated and scaled using the HKL200037 package. The program POINTLESS38 was used to confirm that the Laue class 2/m and space group C2 were correct. Structure solution was carried out by molecular replacement with MOLREP39 using domain 4 (residues 595–735) of the full length PA structure (PDB: 1ACC) as the search model. Rotation and translation searches for two molecules in the asymmetric unit yielded a clear solution with a correlation coefficient of 0.583. Structure refinement and manual model building were performed with REFMAC40 and COOT,41 respectively. Structure validation was conducted using MOLPROBITY42 and figures were prepared using the RIBBONS43 software package. Coordinates and structure factors have been deposited to the Protein Databank with the accession code 3INO.
Acknowledgments
The authors would like to thank Dr. David VanderVelde for assistance with the 1H-15N-HSQC experiments. They would also like to thank Maheshinie Rajapaksha, D. Shyamali Wimalasena, and Duncan Murphy for assistance in cloning and purification of domain 4. This work supported by an NIH COBRE-PSF award to the University of Kansas.
References
- 1.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 2.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 3.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, Davis GE. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci. 2001;114:2755–2573. doi: 10.1242/jcs.114.15.2755. [DOI] [PubMed] [Google Scholar]
- 5.Young JA, Collier RJ. Anthrax toxreceptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 6.Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004;166:645–651. doi: 10.1083/jcb.200312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 2005;309:777–781. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krantz BA, Finkelstein A, Collier RJ. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol. 2006;355:968–979. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 9.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rainey GJ, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, Young JA. Receptor-specific requirements for anthrax toxin delivery into cells. Proc Natl Acad Sci USA. 2005;102:13278–13283. doi: 10.1073/pnas.0505865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacy DB, Wigelsworth DJ, Scobie HM, Young JA, Collier RJ. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc Natl Acad Sci USA. 2004;101:6367–6372. doi: 10.1073/pnas.0401506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradley KA, Mogridge J, Jonah G, Rainey A, Batty S, Young JA. Binding of anthrax toxin to its receptor is similar to alpha integrin-ligand interactions. J Biol Chem. 2003;278:49342–49347. doi: 10.1074/jbc.M307900200. [DOI] [PubMed] [Google Scholar]
- 13.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430:905–908. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 14.Scobie HM, Marlett JM, Rainey GJ, Lacy DB, Collier RJ, Young JA. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin pore formation. PLoS ONE. 2007;2:e329. doi: 10.1371/journal.pone.0000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nassi S, Collier RJ, Finkelstein A. PA63 channel of anthrax toxan extended b-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 16.Benson EL, Huynh PD, Finkelstein A, Collier RJ. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 1998;37:3941–3948. doi: 10.1021/bi972657b. [DOI] [PubMed] [Google Scholar]
- 17.Rajapaksha M, Eichler JF, Hajduch J, Anderson DE, Kirk KL, Bann JG. Monitoring anthrax toxin receptor dissociation from the protective antigen by NMR. Protein Sci. 2009;18:17–23. doi: 10.1002/pro.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Vernier G, Wigelsworth DJ, Collier RJ. Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. J Biol Chem. 2007;282:1059–1065. doi: 10.1074/jbc.M609869200. [DOI] [PubMed] [Google Scholar]
- 20.Krishnanchettiar JSS, Caffrey M. Expression and purification of the Bacillus anthracis protective antigen. Protein Expr Purif. 2002;27:325–330. doi: 10.1016/s1046-5928(02)00612-5. [DOI] [PubMed] [Google Scholar]
- 21.Edge V, Allewell NM, Sturtevant JM. High-resolution differential scanning calorimetric analysis of the subunits of Escherichia coli aspartate transcarbamoylase. Biochemistry. 1985;24:5899–5906. doi: 10.1021/bi00342a032. [DOI] [PubMed] [Google Scholar]
- 22.Edge V, Allewell NM, Sturtevant JM. Differential scanning calorimetric study of the thermal denaturation of aspartate transcarbamoylase of Escherichia coli. Biochemistry. 1988;27:8081–8087. doi: 10.1021/bi00421a017. [DOI] [PubMed] [Google Scholar]
- 23.Manly SP, Matthews KS, Sturtevant JM. Thermal denaturation of the core protein of lac repressor. Biochemistry. 1985;24:3842–3846. doi: 10.1021/bi00336a004. [DOI] [PubMed] [Google Scholar]
- 24.Hu CQ, Sturtevant JM. Thermodynamic study of yeast phosphoglycerate kinase. Biochemistry. 1987;26:178–182. doi: 10.1021/bi00375a025. [DOI] [PubMed] [Google Scholar]
- 25.Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 26.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 27.Paaventhan P, Joseph JS, Seow SV, Vaday S, Robinson H, Chua KY, Kolatkar PR. A 1.7 Å structure of Fve, a member of the new fungal immunomodulatory protein family. J Mol Biol. 2003;332:461–470. doi: 10.1016/s0022-2836(03)00923-9. [DOI] [PubMed] [Google Scholar]
- 28.Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 29.Brandts JF, Hu CQ, Lin LN, Mos MT. A simple model for proteins with interacting domains. Applications to scanning calorimetry data. Biochemistry. 1989;28:8588–8596. doi: 10.1021/bi00447a048. [DOI] [PubMed] [Google Scholar]
- 30.Bann JG, Frieden C. Folding and domain-domain interactions of the chaperone PapD measured by 19F NMR. Biochemistry. 2004;43:13775–13786. doi: 10.1021/bi048614u. [DOI] [PubMed] [Google Scholar]
- 31.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 32.Bernard A, Payton M. Fermentation and growth of Escherichia coli for optimal protein production. In: Coligan JE, Dunn BM, Plough HL, Speicher DW, Wingfield PT, editors. Current Protocols in Protein Science. New York: Wiley; 1995. pp. 1–18. Unit 5.3. [DOI] [PubMed] [Google Scholar]
- 33.Pace CN, Schmid FZ. How to determine the molar absorbance coefficient of a protein. In: Creighton TE, editor. Protein Structure: A Practical Approach. New York, NY: Oxford Press; 1997. pp. 253–259. [Google Scholar]
- 34.Barrick D, Baldwin RL. Three-state analysis of sperm whale apomyoglobin folding. Biochemistry. 1993;32:3790–3796. doi: 10.1021/bi00065a035. [DOI] [PubMed] [Google Scholar]
- 35.Pace CN, Scholtz JM. Measuring the conformational stability of a protein. In: Creighton TE, editor. Protein Structure: A Practical Approach. New York, NY: Oxford Press; 1997. pp. 299–320. [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Vol. 276. New York: Academic Press; 1997. pp. 307–326. Methods in enzymology, macromolecular crystallography, Part A. [DOI] [PubMed] [Google Scholar]
- 38.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Part 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 39.Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Part 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Part 12, Part 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Ca geometry: phi,psi and Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 43.Carson M. Ribbons. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]