

Abstract
Deletion analysis and alanine-scanning based on a homology-based interaction model were used to identify determinants of oligomerization in the transcriptional regulator CynR, a member of the LysR-type transcriptional regulator (LTTR) family. Deletion analysis confirmed that the putative regulatory domain of CynR was essential for driving the oligomerization of λ repressor-CynR fusion proteins. The interaction surface of a different LTTR and OxyR was mapped onto a multiple sequence alignment of the LTTR family. This mapping identified putative contacts in the CynR regulatory domain dimer interface, which were targeted for alanine-scanning mutagenesis. Oligomerization was assayed by the ability of mutant λ repressor-CynR fusions to assemble in E. coli revealing interesting similarities and differences between OxyR and CynR.
Keywords: protein-protein interactions, LysR-type transcriptional regulators, CynR, oligomerization
Introduction
The LysR-type transcriptional regulators (LTTRs) comprise a large family of proteins in which oligomerization is essential for their function. Many bacteria encode multiple LTTR paralogs in their genomes. For example, there are 47 and 123 LTTRs in Escherichia coli and P. aeruginosa, respectively.1 Based on the full-length crystal structure of CbnR from Ralstonia eutropha NH9, tetrameric LTTRs consist of a helix-turn-helix DNA binding domain connected via a coiled-coil linker to an α/β regulatory domain that resembles periplasmic binding proteins.2 The monomeric subunits form tetramers via two distinct interactions: If the subunits are labeled A, B, P, and Q the regulatory domains of subunits B and P form a dimer, as do the regulatory domains of subunits A and Q. The DNA binding domains are brought together via the α-helical linker of A and B or P and Q. Most well-studied LTTRs undergo a conformational change in response to an environmental signal.3 Because the oligomerization of the LTTRs also shows high specificity (Knapp and Hu, unpublished), these interfaces must have diverged within the constraints of a common structural framework while maintaining stability in both their activating and repressing forms.
To understand the evolution of specificity in LTTR interactions, we are analyzing the basis of regulatory domain dimerization in different members of the family. Previously, we have used the program QContacts4 and site-directed mutagenesis to identify seven potential hot spot residues in the regulatory domain interface of OxyR,5 a regulator of the oxidative stress response in E. coli.
Here, we describe studies on the oligomerization determinants of CynR, another E. coli LTTR. CynR is the transcriptional regulator of the cyn operon, which encodes genes that allow cyanate to be used as a sole source of nitrogen. The operon consists of cynT, cynS, cynX, which encode a carbonic anhydrase, a cyanase, and a protein of unknown function, respectively.6 CynR binds to DNA in vitro in the presence or absence of cyanate. CynR binding induces bending of the DNA under both conditions, but the amount of DNA bending is decreased in the presence of cyanate.7
We have used the domain structure of CbnR and analysis of the interaction surface of the OxyR regulatory domain dimer to guide deletions and site-directed mutagenesis of CynR. The results reveal interesting similarities and differences between OxyR and CynR. Unlike OxyR, a structure for the CynR regulatory domain was not available when this study was begun. During the course of this study, the structure of the regulatory domain of CynR without the inducer was deposited in the PDB (2HXR), allowing us to evaluate the performance of the homology-driven analysis of subunit interfaces.
Results
Regions sufficient for oligomerization
To genetically dissect the determinants of CynR oligomerization, we used an established genetic method, the λ repressor system, to test for oligomerization in CynR. The λcI repressor protein oligomerizes at the C-terminal domain. Fusing a protein of interest to the DNA binding domain (residues 1–102) allows for screening for oligomerization by challenging E. coli expressing a fusion protein with phage λ.8–12
Full-length CynR confers immunity to phage λ infection when fused to the DNA binding domain of λ repressor. To determine which portions of the CynR protein were necessary and sufficient for oligomerization, repressor fusions were constructed to a series of deletion mutations of CynR (Fig. 1), which were assayed for oligomerization by immunity assays. Four constructs D, E (full-length), F, and G showed immunity to phage, indicating that these constructs are able to oligomerize.
Figure 1.

CynR deletion mutations. The amino acid regions in CbnR are described in the top row, with the equivalent regions of CynR used to construct deletion fusions described in the second row. The regions included in the fusions are shaded, with the name of the construct on the left (A–J). The immunity of each construct against λKH54 is shown in the right hand column. I = Immunity/Oligomerization; S = Sensitivity/No oligomerization. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
cI-CynR is a higher order oligomer
To determine whether or not the four immune cI-CynR fusions were forming dimers or higher order oligomers, the clones were moved into a set of lacZ reporter strains that can distinguish between monomers and higher-order oligomers.9 In this assay (Table I), strong repression of the lacZ reporter in strain JH607 requires cooperative binding to a set of adjacent λ operators. Only the full-length CynR fusion (Table I, construct E) showed repression levels similar to that of the tetrameric control (pJH157). The constructs containing only the full regulatory domain of CynR (Table I, constructs F and G) showed repression levels similar to those of the dimeric control (pJH370). Taken together with the immunity assays, these results indicate that the full regulatory domain is sufficient for dimerization, whereas the whole protein is necessary for tetramerization. Although construct D is oligomeric in the immunity test, it represses very weakly in the lacZ reporter assay and was not studied further.
Table I.
Assay for Higher Order Oligomers
| Construct | JH607 | XZ980 |
|---|---|---|
| pZ150 | 100% ± 0% | 100% ± 0% |
| pKH101 | 34 ± 7 | 116 ± 44 |
| pJH157 (tetramer) | 4 ± 2 | 78 ± 24 |
| pJH370 (dimer) | 45 ± 21 | 45 ± 16 |
| D | 80 ± 44 | 115 ± 48 |
| E | 7 ± 5 | 69 ± 4 |
| F | 38 ± 17 | 85 ± 5 |
| G | 34 ± 10 | 81 ± 24 |
Each construct used was assayed using β-galactosidase assays (See Methods). pZ150 is the empty vector. pKH101 is the N-terminal DNA binding domain of λcI repressor. Higher order oligomerization is needed to see stronger repression of lacZ in JH607 compared to XZ980.
Prediction of the CynR interface
At the beginning of this study, only two structures of LTTR oligomerization domains were available: OxyR and CysB;13 however, because of the predicted overall similarity in the structures of the regulatory domains of homologous LTTRs, we hypothesized that the oligomerization surface from one LTTR might be similar to that of another. We chose OxyR as a structural template because it was the only LTTR for which systematic mapping of the energetic hot spots had been done.5 Using the multiple sequence alignment and the program QConAlign.pl as described in the Methods, we mapped positions in the subunit interface of OxyR onto the equivalent positions in CynR to generate predicted contact profiles of residues contact each other across the CynR dimer interface (Supporting Information Tables S1 and S2).
Of the 42 residues in the OxyR contact profile, residues at six positions in CynR were occupied in the alignment by identical amino acids: Y104, L124, E126, A221, S223, and S235. Three positions in OxyR 251, 252, and 253 aligned to gaps in the CynR sequence.
Analysis of the real CynR interface
During the course of this study, the structure of the regulatory domain of CynR without the inducer was deposited in the PDB (2HXR). The unreleased structure of the regulatory domain of CynR with sodium azide bound was graciously provided to us by Alexander Singer and Alexei Savchenko at the Midwest Centre for Structural Genomics/University of Toronto (PDB: 3HFU), allowing us to generate a real contact profile for each form of the CynR (Supporting Information Tables S3 and S4).
At the residue level, all the residues identified at the interface of the uninduced form contribute to the interface of the induced form. Only two residues, 119 and 216, were found in the interface of the induced structure that were not in the uninduced structure [Fig. 2(A)]. Examination of the contacts made by these residues showed that the uninduced structure had 59 residue-residue contacts, whereas the induced structure had 60 residue-residue contacts. Of these, 53 residue-residue contacts were the same between the two crystal forms. [Fig. 2(B)]. However, examination of the actual atomic level interactions reveal differences between the atoms in the inter residue-residue contacts. The number of nonredundant atomic interactions for the uninduced form of CynR was 245, whereas for the induced form there are 258. Of these interactions, only 159 are shared in both forms of CynR, meaning that only 65 and 62% of the uninduced and induced forms are shared, respectively. Closer examination of the I106 interacting with A226 highlights this observation. In the uninduced structure, Cβ of A226 interacts with Cδ1 of the isoleucine, whereas in the induced structure A226 Cβ interacts with I106 Cγ2. Cα of A226 makes an additional conact with I106 Cγ2 [Fig. 2(C)].
Figure 2.
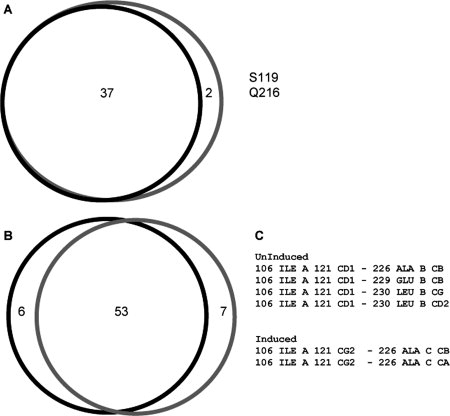
Comparison of the contact profiles generated from QContacts. Panel A shows the residues found in both contact profiles of the regulatory domains of the uninduced and induced crystal forms of CynR. B: The shared contacts between the uninduced and induced crystal forms of CynR. C: The atomic interactions of I106 contacting A226, E229, and L230 in the uninduced and induced crystal forms of CynR. Black lines, uninduced form. Gray lines, induced form.
Predicted vs. observed contact profiles
The contact profiles from the CynR crystal structures allowed us to evaluate the performance of the homology-based prediction of the dimer interface. Table II compares the residues predicted to be in contact based on the two OxyR structures (reduced and oxidized) to those observed in the two CynR structures (without and with inducer bound). Twenty-one of the 47 residues found in any of the contact profiles were shared by all four contact profiles. The predicted contacts included eight false positives (R94, V97, M110, S128, K131, A170, H172, and L228) and nine false negatives (P108, A115, P118, S119, K190, S195, E197, Q216, and L236).
Table II.
Residues from Predicted and Observed Contact Profiles for CynR
| In CynR profiles | Not found in CynR | |
|---|---|---|
| Predicted from OxyR profiles | ||
| From both OxyR profiles | S103, I106, G107, A111, Y114, L122, Q123, L124, Q125, E126, V218, I219, A221, N222, S223, S225, A226, E229, L230, R232, R233 | M110, S235 |
| From oxidized OxyR | T102, Y104, T121, V217, E220, Q247, H248 | V97, S128 |
| From reduced OxyR | I120, T234 | R94, A170, H172, L228 |
| Not predicted from OxyR profiles | ||
| In both uninduced and induced CynR | P108, A115, P118, K190, S195, E197, L236 | n/a |
| Only in induced CynR | S119, Q216 | n/a |
Figure 3 shows the contact profiles mapped onto the surface of the structure of CynR without inducer bound. The majority of the central contact surface consists of shared residues, while the false positives and false negatives lie on the edges of the dimer interface. Although similar positions are involved in the interface, the CynR monomers interact with each other at a different angle than the OxyR monomers. Comparing all residue-residue interactions found in the contact profiles of CynR and OxyR illustrates this point further. As shown in Figure 2(B), 53 residue-residue contacts are shared between both the uninduced and induced forms of CynR. Of these 53 shared interactions, only eight are shared amongst all four contact profiles of CynR and OxyR. Of these eight residue-residue contacts, one contains a hot spot in OxyR: E126 contacting S223, whereas in CynR the contact of S103 contacting E229 is of importance, as E229 is a hot spot (see below).
Figure 3.
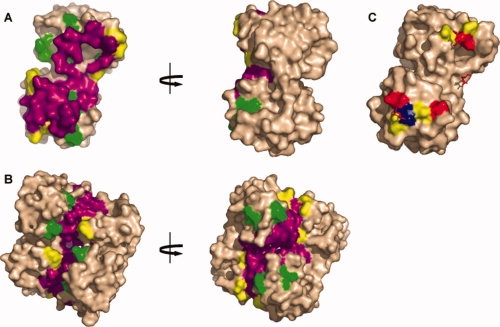
Mapped contacts of CynR comparison of the predicted and real contact profiles of CynR. The predicted and real contact profiles of CynR were compared and mapped onto the uninduced structure CynR (2HXR). In panels A and B, purple residues are detected in the predicted and real contact profiles. Yellow residues were detected in the real contact profile of CynR, whereas green was from the predicted contact profile. A: The face of one monomer of CynR uninduced. B: The dimer of CynR uninduced. In panel C, residues in contact with hot spots are highlighted. Red are hot spots, while residues in yellow are class II residues and residues in blue are class III residues. Hot spot residues from the other subunit are shown as red sticks.
Individual residues' contributions to oligomerization
To determine the importance of individual residues in the interface to oligomerization, nonalanine or glycine residues were mutated to alanine. The mutant cynR-repressor fusions were moved to pLM1000, pAZ299 and, in some cases, pGK751. These three vectors express the repressor fusions at different levels in E. coli and have different thresholds for detection of oligomerization.5 pLM1000 requires strong oligomerization, pGK751 detects relatively weak oligomers, and pAZ299 is intermediate in sensitivity. In all three vectors, full-length, wild-type CynR forms oligomers above the assay threshold. Table III shows the immunity results for full-length CynR fusions with different mutations. The mutants can be sorted into three classes: (I) those that were not immune when expressed from any of the vectors, (II) those that were unable to oligomerize when expressed from pLM1000, but able to oligomerize in pAZ299 or pGK751 and (III) those that behaved as wild-type, being immune in all three vectors. There are four residues in Class I, 16 in Class II, and nine residues belonging to Class III.
Table III.
Oligomerization Results of CynR
| Full-length | Regulatory domain | Sub-class | |
|---|---|---|---|
| L122A | I | I | 1A |
| L228A | I | I | 1A |
| E229A | I | I | 1A |
| L124A | II | I/II | 2A |
| Q247A | II | I/II | 2A |
| Y104A | II | II | 2B |
| I120A | I/II | II | 2B |
| I121A | II | II | 2B |
| L230A | II | II | 2B |
| R233A | II | II | 2B |
| S235A | II | II | 2B |
| Y114A | II | III | 2C |
| E126A | II | III | 2C |
| Q123A | III | II | 3A |
| Q125A | III | II/III | 3A |
| H172A | III | III | 3C |
| S223A | III | III | 3C |
| E220A | I | ND | |
| R94A | II | ND | |
| L106A | II | ND | |
| M110A | II | ND | |
| K131A | II | ND | |
| V218A | II | ND | |
| R232A | II | ND | |
| H248A | II | ND | |
| S103A | III | ND | |
| S128A | III | ND | |
| I219A | III | ND | |
| S225A | III | ND | |
| T234A | III | ND |
Because residues 89–299 are sufficient to drive oligomerization, and because the tetramer contacts of the full-length protein could be masking the destabilizing effects of the alanine substitutions at the dimer interface, repressor fusions to the regulatory domain (residues 89–299) containing the mutations were constructed and assayed for the ability to confer immunity to λ (Table III). We again sorted the regulatory domain results into different classes: (I) those that were not immune when expressed from any of the vectors, (II) those that were able to confer immunity in pAZ299 or pGK751, and (III) those that behaved as wild-type. Combining the results of the full-length mutational analysis with the regulatory domain analysis allows for an overall classification of the mutant, as evident by the subclassification (Table III).
Overall, four residues were found to be required for the oligomerization of full-length CynR as assayed by λcI repressor fusions. L228 was almost completely buried in both forms of CynR. Because it is not at the surface, we conclude that the effect of L228A is likely to be on monomer stability. As the LTTRs are notoriously difficult to work with and in most cases insoluable, we cannot independently assess its role in oligomerization. The other three residues in the dimeric interface of CynR that are sensitive to mutation to alanine, L122, E220, and E229, are putative hot spots for oligomerization.
Discussion
Genomes often encode many members of paralogous gene families that encode proteins with similar structures. These proteins, which usually function in multisubunit protein complexes, must have evolved to coexist in the same cellular environment. This requires the evolution of distinct oligomerization specificities to prevent assembly of inappropriate inactive complexes. The LysR-type transcription factors provide an interesting example of this situation, as some bacteria may contain dozens to hundreds of LTTR paralogs.
In this study, we focused on examining the oligomerization of the CynR protein from E. coli. We first confirmed that CynR oligomerization is similar to other LTTRs and then compared the determinants of CynR oligomerization to those found in another member of the LTTR family, OxyR. Deletion analysis and in vivo cooperativity are consistent with CynR behaving like a typical LTTR. Deletion analysis confirmed that the CynR regulatory domain is sufficient for dimerization, whereas the full-length protein behaved as a higher order oligomer. This is consistent with the observation that most LTTRs are tetramers and clarifies the oligomerization state of CynR in vivo; previous reports by the Fuchs group have said that CynR is either a dimer of dimers or a dimer as determined by gel-filtration studies.7,14
Construct D in Figure 1 was made to delete the portion of the regulatory domain where the C-terminal end of the protein crosses back from the RDII subdomain to participate in the structure of the RDI subdomain. Surprisingly, this construct showed weak self-assembly in the presence of the N-terminal domains of CynR.
Using a homology model based on OxyR to guide site-directed mutagenesis, we identified three residues important that contribute to the dimerization of the regulatory domain of CynR: L122, E220, and E229. The hot spots in CynR share several of the typical characteristics of hot spots.15,16 Primarily, the hot spots make contact with each other and are surrounded by residues that are not especially important for oligomerization [Fig. 3(C)]. However, while Bogan and Thorn15 and Hu et al.16 find that the hot spots are enriched for tryptophan, tyrosine, and arginine, the CynR hot spots are two glutamic acids and the one leucine.
Just as homologous proteins reflect conserved fold topologies, with a few notable exceptions, oligomeric proteins in conserved families assemble using similar symmetry and interfaces.17–19 It is tempting to speculate that this similarity could allow inference of a code-like rule set for understanding the basis for the evolution of homomeric interactions, at least within a family of paralogous proteins. Indeed, substantial progress has been made toward predicting the specificity of coiled-coil interactions from additive predictions based on model leucine zippers.20–22
However, assembly of globular dimers appears to be very different. Comparison of the contact profiles for OxyR and CynR, the two LTTRs we have so far studied in detail, shows that while much of the same surface is used in regulatory domain dimerization, the majority of the residue-residue interactions are different reflecting a difference in rotation of the monomers along the plane of the interaction surface [Fig. 3(A,B)]. Of the seven hot spot residues from the OxyR study, three were not present in either of the contact profiles generated for CynR (Table II). Figure 4 shows the alignment of QContacts contact profiles generated using available LTTR regulatory domain crystal structures.2,13,23–26 While there are some residues that are in the contact profile of all the regulatory domain structures, very few overlap with the hot spots of CynR and OxyR.
Figure 4.

Contact residues and hot spots mapped onto the multiple sequence alignment of the LTTRs. The sequence of each protein with a crystal structure is shown.2,13,23–26 The residues in the interface, as detected by QContacts, are highlighted blue. Residues that were detected as hotspots in our screens are colored red.
Our results highlight the challenges for understanding the evolution of specificity for the assembly of LTTRs and globular proteins in general. Although naïve homology modeling predicted most of the residues involved in dimerization of the CynR regulatory domain, the detailed differences in how the interface surfaces interact would be hard to predict. Given those differences, it is perhaps not surprising that different residues on the interaction surfaces are hotspots in different LTTRs.
In this study and our previous study with OxyR, the λ repressor system has proved useful in studying the LTTR family of proteins. While we have focused on the dimeric interfaces, this system should allow for future studies on dissecting the tetrameric protein-protein interactions of the LTTRs, providing a comprehensive picture of the oligomerization of the LTTRs.
Methods
Construction of deletion mutations
Strains and plasmids used in this study are listed in Table IV. To determine the equivalent positions of CbnR in CynR, a multiple sequence alignment of COG05831 and CbnR from R. eutrophea2 was generated using CLUSTAL W (1.82). The regions used to determine the constructs are based on those described in the crystal structure article of CbnR, deletion mutations based on CysB26 and are shown in Figure 1. Oligos were designed to attach in-frame SalI and BamHI sites and ordered from IDT (Iowa). These oligos were used to amplify the appropriate fragments out of full-length CynR (pGK304) and digested with SalI and BamHI. The inserts were cleaned using a Qiagen PCR Clean-up Kit and ligated into digested pJH391 digested with SalI and BamHI generating λcI repressor fusions. Ligations were transformed into AG1688 and transformants were recovered and sequenced.
Table IV.
Strains, Primers and Plasmids
| pGK304 | P7107-cI-amber-CynR |
| pGK343 | P7107-cI-amber-att-CynR-att |
| pJH391 | PlacUV5-cI vector |
| pLM1000 | P7107-cI-amber-Gateway® cassette |
| pGK751 | PlacUV5-cI-amber-Gateway® cassette |
| pAZ299 | P7107-cI-Gateway® cassette |
| AG1688 | araD139 Δ(ara, leu)7697, ΔlacX74, galU, galK, hsdR, strA F′128 lacIqlacZ::Tn5 |
| JH787 | AG1688 φsu80 |
| JH607 | AG1688 λ112OsPs |
| XZ980 | AG1688 λXZ970 |
| Mach1-T1R | F− ΔlacX74 hsdR ) Δrec1398 endA1 tonA ) Δrec1398 endA1 tonA |
| CynR RD-GW-f | 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTATCTGACGCGAGGATCGCTG-3′ |
| CynR RD GW-r | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACCGTGATTCATTTCCGCCAA-3′ |
Immunity assay
Cross-streak assays27 were used to test the ability of the λcI fusions to oligomerize. In general, cells were struck out for single colonies and incubated for 16 h at 37°C. Individual colonies were challenged against lines of phage λKH54, λvir, and control phage λi21c on tryptone agar plates and incubated for 7 h at 37°C. Those that were able to grow across the λKH54 line were called immune, and those that were not able to grow were sensitive. All colonies died at the control λ21c phage. For the deletion mutation cross-streaks, stains were simply called immune or sensitive based on their phenotype to λKH54. For other described cross-streaks, strains immune to λKH54 were given a score of 1, whereas sensitive phenotypes received a score of 0. Each strain was tested three times with three individual colonies. Immune colonies were given a score of 1, with sensitive colonies given a score of 0. The average of all experiments was calculated to give an immunity score.
Dimerization and tetramerization test
To distinguish between cI repressor fusions of dimer and tetramers, the clones were moved into JH607 and XZ980 via M-13 transduction. JH607 and XZ980 are strains that are used to distinguish cooperative oligomerization and have been previously described.9 Briefly, JH607 contains the construct λ112OsPs, which contains a synthetic promoter that drives expression of cat and lacZ. A weak lambda operator overlaps the promoter, and a strong operator is upstream of the weaker promoter. Strong repression is only detected in higher order oligomerization states. XZ980 contains the same weak promoter. The strong upstream operator was replaced with a λ434 operator so that repression reflects only binding to the weak operator. β-galactosidase assays were done according to Miller.28
Generation of contact profiles
The program QContacts was utilized to generate a contact profile of the two forms of OxyR utilizing the available crystal structures of the regulatory domain.4 The combined contact profiles generated from the OxyR study were used as a template to generate predicted CynR contact profiles. The multiple sequence alignment described earlier was used. A PERL script called QConalign.pl was used to parse the residues at equivalent positions in OxyR out of CynR using the multiple sequence alignment generating a predicted contact profile for CynR.
Alanine-scanning mutagenesis
Using the contact profiles as a guide, the identified nonalanine and glycine residues were mutated to alanine. PrimerX (http://www.bioinformatics.org/primerx) was used to generate the primer pairs, and primers were ordered from IDT (Iowa). Pfu Turbo (Stratagene) or Pfx Platinum (Invitrogen) was used for the polymerase. pGK343 was the DNA template. Reactions were treated with 2U of DpnI for 2–6 h and transformed into Mach1-T1R, Top1-T1R (Invitrogen), or XL1-Blue supercompetent (Stratagene) cells. Transformants were streak purified, cultured, and the plasmid recovered and sequenced.
Generation of λcI repressor fusions with different expression levels
For each mutant, a Gateway Entry Clone29 was generated. pGK343 is a λcI repressor fusion of full-length CynR with Gateway attachment sites flanking the full-length CynR. The mutated cynR gene was moved into pDONR201 via the back reaction to generate the entry clone, transformed into Mach1-T1R cells, and selected on LB plates containing kanamycin. Candidates were screened for the loss of ampicillin resistance by streak purification on LB kanamycin and LB ampicillin plates. The entry clones were used to move the gene into several destination vectors: pLM1000,11 pAZ299, and pGK751 via the LR reaction. The reactions were transformed into Mach1-T1R cells. Candidates were screened for loss of growth on LB kanamycin, LB chlorenphenicol, and for the ability to grow on LB ampicillin. All constructs were subjected to restriction mapping to ensure that the appropriate clone was generated. Depending on the fusion, the plasmid was transformed into either JH78730 or AG1688.31
In pAZ299 and pLM100, cI transcription is under the control of a weak, constitutive promoter, P7107.11 In pGK751, cI is transcribed from Placuv5. Both pGK751 and pLM1000 contain an amber mutation at codon 103 that is suppressed in JH787. Use of these three fusions allows for tiered levels of protein expression.
Generation of λcI regulatory domain fusions
Entry clones were generated of the mutated regulatory domains. Residues 89–299 of the regulatory domain of the full-length CynR mutants were amplified with the CynR RD GW-f and CynR RD GW-r or attB2 in-frame attB sites. Using the PCR product, entry clones were generated in pDONR201. To generate λcI regulatory domain fusions, most of the mutated genes (as entry clones) were subsequently moved to pLM1000, pAZ299, and pGK751.
Acknowledgments
The authors thank Jerry Tsai for help with bioinformatics, Alexander Singer and Alexei Savchenko of the Midwest Centre for Structural Genomics for providing the coordinates of the azide-bound CynR structure, and Deborah Siegele for helpful discussion and comments on the manuscript.
References
- 1.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucl Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J Mol Biol. 2003;328:555–566. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]
- 3.Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 4.Fischer TB, Holmes JB, Miller IR, Parsons JR, Tung L, Hu JC, Tsai J. Assessing methods for identifying pair-wise atomic contacts across binding interfaces. J Struct Biol. 2006;153:103–112. doi: 10.1016/j.jsb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Knapp GS, Tsai JW, Hu JC. The oligomerization of OxyR in Escherichia coli. Protein Sci. 2009;18:101–107. doi: 10.1002/pro.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sung YC, Fuchs JA. Characterization of the cyn operon in Escherichia coli K12. J Biol Chem. 1988;263:14769–14775. [PubMed] [Google Scholar]
- 7.Lamblin AF, Fuchs JA. Functional analysis of the Escherichia coli K-12 cyn operon transcriptional regulation. J Bacteriol. 1994;176:6613–6622. doi: 10.1128/jb.176.21.6613-6622.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng X, Zhu H, Lashuel HA, Hu JC. Oligomerization properties of GCN4 leucine zipper e and g position mutants. Protein Sci. 1997;6:2218–2226. doi: 10.1002/pro.5560061016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng X, Hu JC. Detection of tetramerization domains in vivo by cooperative DNA binding to tandem lambda operator sites. Gene. 1997;185:245–249. doi: 10.1016/s0378-1119(96)00652-x. [DOI] [PubMed] [Google Scholar]
- 10.Marino-Ramirez L, Campbell L, Hu JC. Screening peptide/protein libraries fused to the lambda repressor DNA-binding domain in E. coli cells. Methods Mol Biol. 2003;205:235–250. doi: 10.1385/1-59259-301-1:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marino-Ramirez L, Minor JL, Reading N, Hu JC. Identification and mapping of self-assembling protein domains encoded by the Escherichia coli K-12 genome by use of lambda repressor fusions. J Bacteriol. 2004;186:1311–1319. doi: 10.1128/JB.186.5.1311-1319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu JC, O'shea EK, Kim PS, Sauer RT. Sequence requirements for coiled-coils: analysis with lambda repressor-GCN4 leucine zipper fusions. Science. 1990;250:1400–1403. doi: 10.1126/science.2147779. [DOI] [PubMed] [Google Scholar]
- 13.Choli H, Kim S, Mukhopadhyay P, Cho S, Woo J, Storz G, Ryu S. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001;105:103–113. doi: 10.1016/s0092-8674(01)00300-2. [DOI] [PubMed] [Google Scholar]
- 14.Anderson PM, Sung YC, Fuchs JA. The cyanase operon and cyanate metabolism. FEMS Microbiol Rev. 1990;7:247–252. doi: 10.1111/j.1574-6968.1990.tb04920.x. [DOI] [PubMed] [Google Scholar]
- 15.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 16.Hu Z, Ma B, Wolfson H, Nussinov R. Conservation of polar residues as hot spots at protein interfaces. Proteins. 2000;39:331–342. [PubMed] [Google Scholar]
- 17.Levy ED, Boeri Erba E, Robinson CV, Teichmann SA. Assembly reflects evolution of protein complexes. Nature. 2008;453:1262–1265. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aloy P, Ceulemans H, Stark A, Russell RB. The relationship between sequence and interaction divergence in proteins. J Mol Biol. 2003;332:989–998. doi: 10.1016/j.jmb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Grigoryan G, Keating AE. Structure-based prediction of bZIP partnering specificity. J Mol Biol. 2006;355:1125–1142. doi: 10.1016/j.jmb.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 21.Fassler J, Landsman D, Acharya A, Moll JR, Bonovich M, Vinson C. B-ZIP proteins encoded by the Drosophila genome: evaluation of potential dimerization partners. Genome Res. 2002;12:1190–1200. doi: 10.1101/gr.67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR, Bonovich M. Classification of human B-ZIP proteins based on dimerization properties. Mol Cell Biol. 2002;22:6321–6335. doi: 10.1128/MCB.22.18.6321-6335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stec E, Witkowska-Zimny M, Hryniewicz MM, Neumann P, Wilkinson AJ, Brzozowski AM, Verma CS, Zaim J, Wysocki S, Bujacz GD. Structural basis of the sulphate starvation response in E. coli: crystal structure and mutational analysis of the cofactor-binding domain of the Cbl transcriptional regulator. J Mol Biol. 2006;364:309–322. doi: 10.1016/j.jmb.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 24.Smirnova IA, Dian C, Leonard GA, McSweeney S, Birse D, Brzezinski P. Development of a bacterial biosensor for nitrotoluenes: the crystal structure of the transcriptional regulator DntR. J Mol Biol. 2004;340:405–418. doi: 10.1016/j.jmb.2004.04.071. [DOI] [PubMed] [Google Scholar]
- 25.Ezezika OC, Haddad S, Clark TJ, Neidle EL, Momany C. Distinct effector-binding sites enable synergistic transcriptional activation by BenM, a LysR-type regulator. J Mol Biol. 2007;367:616–629. doi: 10.1016/j.jmb.2006.09.090. [DOI] [PubMed] [Google Scholar]
- 26.Lochowska A, Iwanicka-Nowicka R, Plochocka D, Hryniewicz MM. Functional dissection of the LysR-type CysB transcriptional regulator. Regions important for DNA binding, inducer response, oligomerization, and positive control. J Biol Chem. 2001;276:2098–2107. doi: 10.1074/jbc.M007192200. [DOI] [PubMed] [Google Scholar]
- 27.Demerec M, Fano U. Bacteriophage-resistant mutants in Escherichia Coli. Genetics. 1945;30:119–136. doi: 10.1093/genetics/30.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller JH. Experiments in molecular genetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.; 1972. [Google Scholar]
- 29.Walhout AJ, Temple SF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- 30.Marino-Ramirez L, Hu JC. Isolation and mapping of self-assembling protein domains encoded by the Saccharomyces cerevisiae genome using lambda repressor fusions. Yeast. 2002;19:641–650. doi: 10.1002/yea.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu JC, Newell NE, Tidor B, Sauer RT. Probing the roles of residues at the e and g positions of the GCN4 leucine zipper by combinatorial mutagenesis. Protein Sci. 1993;2:1072–1084. doi: 10.1002/pro.5560020701. [DOI] [PMC free article] [PubMed] [Google Scholar]