

Abstract
Methamphetamine (METH) is a major drug threat in the United States and worldwide. Monoclonal antibody (mAb) therapy for treating METH abuse is showing exciting promise and the understanding of how mAb structure relates to function will be essential for future development of these important therapies. We have determined crystal structures of a high affinity anti-(+)-METH therapeutic single chain antibody fragment (scFv6H4, KD= 10 nM) derived from one of our candidate mAb in complex with METH and the (+) stereoisomer of another abused drug, 3,4-methylenedioxymethamphetamine (MDMA), known by the street name “ecstasy.” The crystal structures revealed that scFv6H4 binds to METH and MDMA in a deep pocket that almost completely encases the drugs mostly through aromatic interactions. In addition, the cationic nitrogen of METH and MDMA forms a salt bridge with the carboxylate group of a glutamic acid residue and a hydrogen bond with a histidine side chain. Interestingly, there are two water molecules in the binding pocket and one of them is positioned for a C—H⋯O interaction with the aromatic ring of METH. These first crystal structures of a high affinity therapeutic antibody fragment against METH and MDMA (resolution = 1.9 Å, and 2.4 Å, respectively) provide a structural basis for designing the next generation of higher affinity antibodies and also for carrying out rational humanization.
Keywords: methamphetamine, antimethamphetamine antibody, therapeutic antibody, crystal structure, scFv
Introduction
The National Drug Intelligence Center reports (+)-methamphetamine (METH) is the second major drug threat to the United States, only behind cocaine.1 Current pharmacological therapies for the treatment of the adverse health effects of METH-like stimulants relieve some organ-based symptoms, but specific medications designed to treat direct medical complications of METH abuse are nonexistent. Most importantly, there are no FDA-approved medications that reduce or treat METH related addiction.
Antidrug immunotherapy, both active and passive, is an exciting and promising potential treatment for drug abuse. Active immunotherapy involves injection of a patient with a drug-like hapten conjugated to an antigenic carrier protein. This approach is showing promising results in early clinical trials for the treatment of nicotine and cocaine addiction.2,3 Passive immunization for drug abuse involves intravenous delivery of a bioengineered antidrug monoclonal antibody (mAbs) or a fragment such as Fab or single chain variable fragment (scFv) (Fig. 1) to attain immediate short term (minutes to hours) or long-lasting (weeks to months) protection. Both therapies provide antibodies that act as pharmacokinetic antagonists4 serving to sequester the drug in the vascular compartment and prevent the drug from entering various critical sites of action like the brain.5
Figure 1.
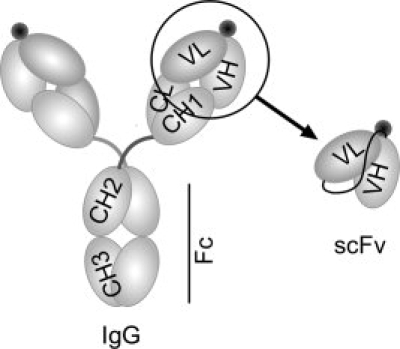
The structural representation of an intact IgG and the scFv antibody. To convert the anti-METH IgG6H4 binding region to anti-METH scFv6H4, the variable heavy (VH) and variable light (VL) regions were amplified and joined by PCR. The antigen binding sites are represented by black circles near the VH and VL domains.
The key initial step for creating both passive and active immunotherapy for METH is the design of a suitable METH-like hapten. Critical improvements have come through progressive knowledge-based design of haptens and the use of more effective antigenic carrier proteins for stimulating an immune response.6 Generating high-specificity antibodies against METH is challenging due to the presence of pharmacologically active metabolites and the potential presence of some over the counter medications that could compete for binding and reduce efficacy. An additional challenge arises from the small size of the drug molecule. Antibody interactions are mediated primarily by the six complementarity determining regions (CDRs) and antigen binding generally involves direct interactions of 15–20 amino acids7 for larger antigens like proteins or peptides. The drugs in this report likely interact with fewer residues because of their smaller surface area and paucity of charged atoms in the molecules. METH surface area is calculated as 206 Å2 and it has only one charged atom available for hydrogen bonding at physiological pH (∼7.4). Therefore, an anti-METH mAb has only a limited number of potential interaction sites for optimizing specificity and affinity. These limitations make the generation of therapeutic antibodies very challenging, but with careful hapten design mAbs were generated8 with high specificity and affinity. However, even though antibodies with these characteristics were generated, we do not yet understand the structural basis of antibody activity resulting from the hapten selection process.
Traditionally, most of the structural studies on antigen binding have been carried out using Fab fragments; however, we have chosen a recombinantly produced scFv for this study. The scFv format is only half the size of the Fab fragment and the smallest antibody fragment that retains the antigen binding properties of the parent IgG. ScFvs are generated by introducing a polypeptide linker between the variable light chain domain and the variable heavy chain domain. A key advantage of the recombinant nature of the scFvs is that it allows tailoring of the binding site for increased affinity and also humanizing the molecule rationally. Moreover, in therapeutic applications, when a short duration of action and greater extravascular penetration are needed, scFvs are better suited as they have a much shorter half life ranging from minutes to few hours.9,10 Therefore, scFvs can be of great value in overdose cases when rapid removal of the drug from the body is necessary.11
We crystallized a scFv derived from one of our highest affinity antibodies, mAb6H4 (KD = 10 nM).6 This mAb was generated using a hapten called METH-P6 [Fig. 2(A)]. ScFv6H4 was produced from the parent IgG without any loss of affinity or specificity.10 In this report, we present the crystal structures of this high affinity scFv in complex with METH and MDMA.
Figure 2.
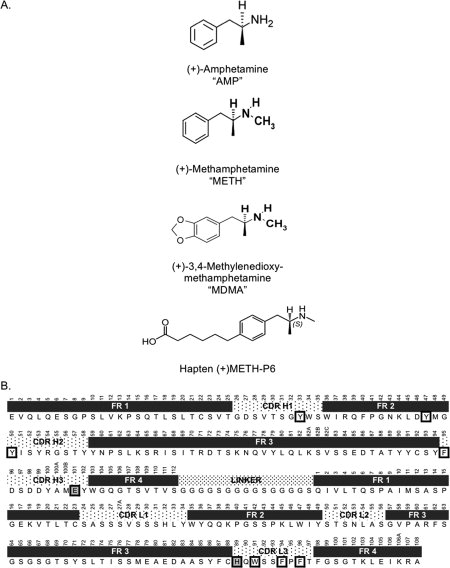
Chemical structures of stimulants and the amino acid sequence of scFv6H4. (A) Chemical structure of related stimulants (+)-amphetamine, (+)-methamphetamine, (+)-3,4-methylenedioxymethamphetamine, and hapten (+)-METH-P6, that was used to generate the parent mAb6H4. (B) The amino acid sequence of scFv6H4 with Kabat numbering.12 The framework and CDR loop regions are indicated above the sequence. Residues forming the aromatic barel around METH and MDMA are boxed in bold. Residues that form salt bridge or hydrogen bond are boxed in bold and shaded.
Results
Crystal structures
The crystal structures of ScFv6H4:METH and ScFv6H4:MDMA complexes were determined using 1.9 Å diffraction data (Rfactor = 21.0%) and 2.4 Å diffraction data (Rfactor = 21.4%), respectively. The electron density maps clearly revealed the positions of the ligands. The conformations of both ligands are similar in that they are fully extended in the binding pocket. Figure 3 depicts the electron densities of the ligands and five surrounding residues. The overall conformations of METH and MDMA are very similar to those observed in the small molecule crystal structures of METH-HCl and MDMA-HCl, respectively,13,14 with minor differences in the positions of the methyl groups attached to the cationic nitrogen atoms, indicating that the binding sites accommodate the native conformations of METH and MDMA.
Figure 3.
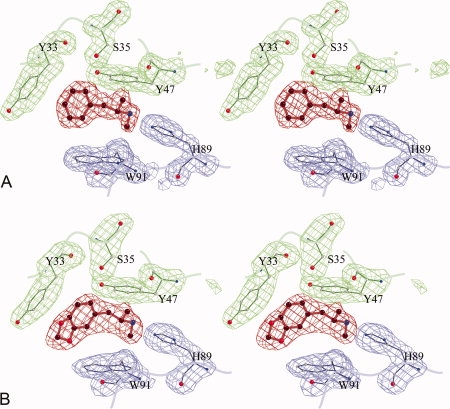
The omit maps displaying the electron densities of the ligands and surrounding amino acids. The ligands are shown in red, the heavy chain residues in green, and the light chain in blue. (A) METH and surrounding five residues. The map is computed at 1.9 Å resolution and the contours are drawn at 2.7σ level (B) MDMA and surrounding residues. The map is computed at 2.4 Å and the contours are drawn at 2.7σ level.
Molecular geometry
In the crystal lattice, scFv6H4 molecules exist solely as monomers, even though, in solution, the size exclusion elution profiles indicated the presence of dimeric and trimeric forms in addition to the monomeric form.10 The scFv6H4 is comprised of a variable light chain domain (VL) and a variable heavy chain (VH) domain, both possessing immunoglobulin fold [Fig. 4(A)]. Each domain contains two antiparallel β-pleated sheets, four strands in the first sheet and five in the other. The binding site is formed by six CDR loops; three contributed by the light chain and three by the heavy chain. These loops correspond to the six hypervariable sequence regions which are labeled as L1, L2, and L3, and H1, H2, and H3 [Figs. 2(B) and 4(A)].
Figure 4.
scFv6H4 METH binding. (A) A ribbon representation of scFv6H4:METH complex. The scFv6H4 consists of variable light chain domain (VL) (blue) and a variable heavy chain domain (VH) (green). Each domain is made up of two antiparallel pleated β-sheets. One sheet comprises four β-strands (marked as 1, 2, 4, and 5) and the other five strands (marked as 3c, 3b, 3, 6, and 7). The antigen binding site is formed by three CDR loops from the light chain (marked as L1, L2, and L3) and three CDR loops from the heavy chain (marked as H1, H2, and H3). The METH ligand is shown in red. The heavy and the light chains are joined together through a 15 amino acid peptide linker. Nine of these linker residues were not clearly visible in the electron density map of the METH complex and they are indicated by a dashed line in this figure. In the MDMA complex, only five residues are missing. The disordered residues are more than 20 Å away from the binding site. (B) A surface (solvent exclusion) representation of scFv6H4 bound to METH illustrating the deep binding pocket. The representation has been colored with the same colors used in (A). METH is shown as a ball and stick figure colored in salmon. The protein was clipped in the plane of the paper to show the interior of the binding pocket. Representation was rendered using PyMol (Delano Scientific, LLC).
METH binding
Figure 4(B) depicts a surface representation of the scFv binding pocket, indicating that the METH molecule sits in a deep pocket and is encased almost completely. In fact, only 3% of the surface area of METH is solvent accessible in the structure of the complex. Both hydrophobic and hydrophilic interactions stabilize the binding. The binding is mediated mostly by four CDR loops, H1, H2, H3, and L3. CDR loops L1 and L2 do not directly participate in METH binding.
Aromatic barrel
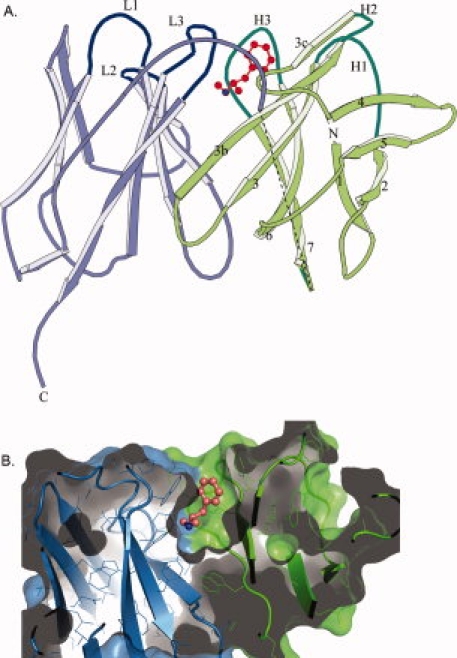
The entrance of the binding pocket is lined with seven aromatic residues; three of these are from the light chain and the other four from the heavy chain. Three residues from the L3 loop of the light chain (TrpL91, PheL94, and PheL96), one residue from H1 (TyrH33), one residue from the β-strand-3c (TyrH47), one residue from H2 (TyrH50), and one residue from H3 of the heavy chain (PheH95) form a hydrophobic barrel around the aromatic portion of METH [Fig. 5(A)]. These seven residues encase 75% of the surface area of METH in a thermodynamically favorable arrangement. It is worth noting that one of the aromatic residues, TyrH47, is located on a β-strand framework region, and TyrH50 and PheH95 are located at the beginning of the H2 and H3 CDR loops, respectively, indicating that the binding pocket runs deep into the interior of the molecule.
Figure 5.
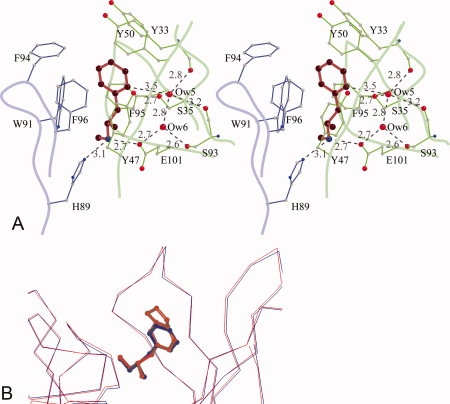
METH/MDMA binding pockets. (A) A stereoscopic view of methamphetamine binding pocket. Methamphetamine is shown in red, the light chain residues in blue, and the heavy chain residues in green. The aromatic ring of METH is surrounded by seven aromatic side chains. There is a salt bridge interaction between the cationic nitrogen and one of the carboxylate oxygen atoms of GluH101 from the heavy chain. The cationic nitrogen atom also forms a hydrogen bond with HisL89. There are two water molecules—Ow(5) and Ow(6) in the binding cavity. Ow(6) forms hydrogen bonds with Ow(5) and the side chains of GluH101 and SerH93. Ow(5), in addition, forms hydrogen bonds with the main chain carbonyl of TyrH33, SerH93, and the side chain of SerH35. Moreover, Ow(5) is positioned to form a C—H⋯O hydrogen bond with the aromatic ring of METH. The H⋯O distance is 2.6 Å and the C—H⋯O angle is 143°. (B) A comparison of METH and MDMA binding. The Cα-atoms of the scFv6H4:METH structure (blue) with scFv6H4:MDMA structure (red) are superposed (root mean square deviation of 0.23 Å for the Cα atoms). In the MDMA complex, the aromatic ring and the cationic nitrogen occupy the same locations retaining the same interactions.
Hydrophilic interactions of METH
Although the binding is mediated mostly by hydrophobic interactions, the protonated secondary amine of METH plays a crucial role in anchoring the ligand deep in the pocket. There is a salt bridge between the cationic nitrogen of METH and the carboxyl oxygen of GluH101 with a nitrogen–oxygen distance of 2.7 Å. In addition, the cationic nitrogen forms a hydrogen bond to HisL89 (3.1 Å) of the light chain [Fig. 5(A)]. The orientation of the METH molecule, with the aromatic ring near the entrance and the cationic secondary amine pointing to the bottom of the pocket is consistent with the fact that the hapten (+)METH-P6, used in generating and selecting the original IgG, has a linker between the protein and the aromatic ring of METH in the para-position [Fig. 2(A)].8
Water molecules in the binding cavity
Because METH and MDMA are predominantly hydrophobic, one would expect the binding cavities to be devoid of solvent molecules; however, in both complexes, there are two water molecules, Ow(5) and Ow(6), in the cavity separated by a distance of 2.8 Å indicating a strong hydrogen bonding interaction between them [Fig. 5(A)]. Additionally, the Ow(6) water molecule forms very strong hydrogen bonds with the side chain of SerH93 and the carboxyl oxygen atoms of GluH101. Ow(5) interacts with the side chain of SerH35 and the carbonyl atoms of SerH93 and TyrH33. Another intriguing aspect of the presence of water molecules is that the Ow(5) molecule is positioned close to the aromatic plane of METH with a C⋯O distance of 3.5 Å from the proximal ortho-carbon atom, suggesting the formation of a C—H⋯O hydrogen bond [Fig. 5(A)].
MDMA binding
ScFv6H4 binds MDMA with an almost identical affinity as METH (KD = 17 nM, and 10 nM, respectively). Analysis of the scFv6H4:MDMA structure revealed that the antibody indeed binds to MDMA in an almost identical manner as it does to METH. Figure 5(B) depicts a superposition of the two complexes and shows that the protein conformations are the same in both structures. The root mean square deviation of superposition between the Cα carbon atoms in the two crystal structures is 0.23 Å. The METH and MDMA molecules have the same conformation as they bind to scFv6H4. Also, careful examination of the conformation of the residues forming the antigen binding site reveals that the conformations of the side chains of these residues are practically the same in both structures. Thus, the antibody is able to bind to two structurally related stimulants without any significant change in conformation.
Discussion
The therapeutic scFv6H4 binds METH and MDMA with very high affinity, but it binds their (−) isomers with much lower affinity. Similarly, scFv6H4 binds another structurally related stimulant, AMP, with much lower affinity than METH. This study provides a basis for analyzing these differences. Compared with METH, MDMA has an additional five-membered ring attached to the aromatic ring [Fig. 2(A)], yet the binding interactions with scFv6H4 are almost identical, because the extra five-membered ring moiety of MDMA is positioned at the entrance of the binding pocket in a solvent accessible manner, causing no steric hindrance (Fig. 5). The binding modes of METH and MDMA suggest strongly that the binding pocket was dictated by the linker position of the hapten at the para-carbon of the aromatic ring [Fig. 2(A)].
ScFv6H4 binding to AMP is weak
The dissociation constant of the (+) stereoisomer of AMP to the anti-METH scFv6H4 (KD = 20.7 μM) is ∼2000 times weaker than that for METH (KD = 10 nM). The AMP molecule differs from METH only in the absence of the methyl group attached to the cationic nitrogen. The aromatic ring of AMP and its cationic nitrogen could both participate in favorable interactions similar to those observed in the METH:scFv6H4 and MDMA:scFv6H4 structures. Additionally, as AMP is smaller in size than METH, it should not cause any steric hindrances when binding to scFv6H4. Therefore, a ∼2000 fold difference in affinity between AMP and METH is rather surprising.
We can envision two possible scenarios leading to lower affinity of the scFv for AMP. (i) The methyl group attached to the nitrogen remains in a hydrophobic environment and it packs tightly against the aromatic side chains of TyrL34, TrpL91, and PheH95 [Fig. 5(A)]. In the absence of the methyl group these packing interactions will be lost; however, it is difficult to imagine that the lack of close packing in just one area of the molecule would cause such a dramatic drop in binding affinity. Therefore, one possibility is that there is an overall shift in the ligand position which weakens the interactions. (ii) The other possibility is that the weaker binding is associated also with entropic effects. The creation of the void space could pave the way for the entry of a water molecule into the binding pocket weakening the binding. In one of our earlier structural studies on von Willebrand factor A1 domain, it was found that a mutation of an Ile residue to a Val residue was accompanied by an entry of a water molecule close to the position of the deleted methyl group. The presence of the water molecule caused the rearrangement of side chains in the vicinity and even caused conformational changes at a remote location resulting in a drastic change in the binding affinity.15
Stereospecificity of binding
Although scFv6H4 has an affinity of 10 nM for METH, it binds the (−) isomer of METH with an affinity of 586 nM.10 In an attempt to understand the mechanism of this stereo-specificity, we generated an in silico model for (−)-METH binding to scFv6H4 such that the interactions of the aromatic ring and hydrogen bonding interactions of the cationic nitrogen are retained. In this scenario, the methyl group attached to the chiral center of (−)-METH is too close to the Cβ atom of TrpL91, suggesting that steric hindrances weaken the binding interactions for (−)-METH (Fig. 6).
Figure 6.
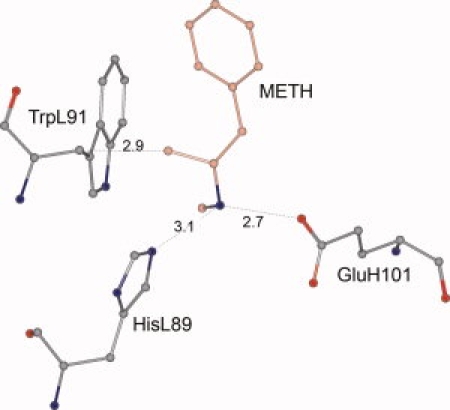
Docking of the (−) isomer of METH to scFv6H4. In this model, all the atoms other than the methyl group occupy the same location as the (+) isomer of METH in the crystal structure. The methyl group makes an unfavorable contact (2.87 Å) with the Cβ atom of TrpL91. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Aromatic pairing interactions in the binding pocket
Considering the involvement of a relatively high number of aromatic residues in the binding of METH and MDMA, we examined whether the relative orientations of the aromatic side chains favor antigen binding. It has been suggested that intramolecular aromatic–aromatic interactions contribute to the stability of globular protein structures.16,17 It was pointed out, that on an average, 60% of the aromatic side chains in proteins participate in such interactions.16 Molecular dynamics simulation calculations on model compounds indicate that one of the optimal geometries for aromatic residue pairs is a T-shaped arrangement where the edge of one ring points to the face of the other.18 In this configuration, hydrogen atoms with partial positive charges on the edge of one ring can interact favorably with the pi electrons or the partially negatively charged carbons of the other ring. In the scFv6H4 crystal structures, the aromatic rings of METH and MDMA molecules are oriented nearly perpendicular to the benzene ring of TrpL91 and similarly the PheL96 side chain is almost orthogonal to the aromatic rings of METH and MDMA, indicating that the pairing interactions play an important role in binding.
Cation-π interactions
The cationic nitrogen atoms of METH and MDMA are positioned right above the center of the benzene ring of PheH95 (at about 5.1 Å), pointing to a favorable interaction of the nitrogen with the π-electrons of the Phe side chain.19 In a similar interaction with TrpL91, the nitrogen atoms are situated about 4.5 Å above the center of the pyrrol ring of the Trp residue. This type of cation-π interaction with a pyrrol ring is almost as strong as the interaction with a benzene ring20 (Dougherty, personal communication). In the crystal structure of morphine:antibody complex, the cationic amine of morphine participates in cation-π interactions with two Trp residues, but both involving the benzene rings.21
C—H⋯O hydrogen bonds in ligand binding
There are two water molecules in the binding site and they are depicted in Figure 5(A). As mentioned in the “Results” section, one of them, Ow(5) is involved in a C—H⋯O hydrogen bond with the ligand. Although the most widely recognized types of hydrogen bonds have either NH or OH groups acting as proton donors, the ability of CH groups to also act as proton donors is well appreciated.22,23 These hydrogen bonds involving CH groups are recognized as major contributors to the stabilization of protein folding and protein/protein or protein/ligand interactions, even though they are weaker than conventional hydrogen bonds like N—H⋯O and O—H⋯O (see Refs. 24 and25, and the references listed therein). Ligands bound to protein molecules often form hydrogen bonds with solvent molecules also. Analyses of the protein-ligand complexes show that ligands use C—H⋯Ow bonds more abundantly than N—H⋯Ow and O—H⋯Ow bonds.26 Ligands generally use the stronger hydrogen bonds N—H⋯Ow and O—H⋯Ow to bind to proteins,27 while keeping an amicable relationship with neighboring water molecules through weaker C—H⋯O hydrogen bonds. When METH or MDMA binds to scFv6H4, they use N—H⋯O (GluH101) and N—H⋯N (HisL89) hydrogen bonds. METH/MDMA interacts with a solvent molecule through C—H⋯Ow hydrogen bonds.
Comparison of anti-METH scFv6H4 with anticocaine and antiphencyclidine (PCP) mAb structures
We compared the binding modes of METH with cocaine28,29 and PCP30 to examine if there is any correlation between the nature of the binding site and the length of the hapten linker used for generating these antibodies. It is worth pointing out that the modes of cocaine binding by the two antibodies are entirely different despite the fact that both antibodies have equally high affinities toward cocaine. One antibody binds cocaine into a deep pocket,28 whereas the other one binds it on the surface.29 The former was generated using a longer hapten linker, whereas the latter was generated with a shorter linker. The anti-PCP mAb30 also exhibits a deep pocket and it should be pointed out that the PCP-like hapten linker is similar in length and is at the same para-position on an aromatic ring as in METH P6 [Fig. 2(A)]. Thus in light of antibody structural information, the linker length as well as the position of attachment on the drug-like molecule are vitally important aspects of hapten design.
In a previous attempt to visualize the geometry of METH binding, a homology model for scFv6H4 was constructed with a METH molecule computationally docked into the binding site.6 The modeling predicted a shallow binding pocket on the surface while crystal structure shows that METH binds in a deep pocket.
In conclusion, this analysis has provided the structural basis for understanding the binding mechanism of a therapeutic anti-METH single chain antibody, scFv6H4, with METH, MDMA, and structurally similar compounds. The scFv6H4 binds METH in a deep pocket suggesting that the length of the linker group of the METH-like hapten (P6) was sufficient to allow deep engulfment of the free METH molecule. On the basis of the crystal structure of the METH complex, we hypothesize that the diminished binding affinity of AMP is possibly due to the entry of a water molecule into the cavity. Docking studies indicate that the binding of the (−) stereo isomers of METH and MDMA will generate unfavorable contacts in the binding site, thus increasing the specificity of scFv6H4 for the (+) isomer of METH. The structures reported here represent the first crystallographic analysis of METH and MDMA interactions with antibodies. The insights gained from this study will be invaluable in designing more potent antibodies, humanizing these antibodies to reduce possible antigenicity and to tailor more effective haptens for anti-METH vaccines.
Materials and Methods
Cloning of anti-METH scFv6H4
In an effort to generate a quick-acting anti-METH binding fragment with a short half-life in the body, a single chain variable fragment (scFv) was generated from an early prototype anti-METH antibody (mAb6H4).10 The scFv6H4 was constructed by joining the light and heavy chain variable domains of the parent mAb6H4 (GenBank accession nos. DQ381543 and DQ381542, respectively) with a 15 amino acid linker of (Gly4Ser)3 [Fig. 2(B)]. The genetic re-engineering of mAb6H4 IgG into scFv6H4 changed the protein from a ∼150 kDa protein with two METH binding sites to a ∼27.4 kDa protein with one METH binding site. It also converted the original IgG from a two gene product (from heavy and light chain coding sequences) into a single gene product (GenBank accession # FJ821514).
Expression of scFv6H4
The scFv6H4 was initially expressed in E. coli, but during purification it was found to form >90% insoluble inclusion bodies. To circumvent this problem, the scFv6H4 coding sequence was recloned into a Pichia pastoris yeast expression system so that the coding sequence was in-frame behind a cleavable alpha-mating factor. This allowed protein secretion into the media during methanol-induced protein expression, thereby decreasing the potential for formation of insoluble protein inclusion bodies. The scFv6H4 with an His-tag on its carboxy terminus, yielded ∼12.4 mg/L protein in 98 h of a yeast fermentation production run.10
Chromatographic purification of scFv6H4
The protein was purified to greater than 98% homogeneity in one purification step using a nickel-column. The purified scFv6H4 was a mixture of monomer (∼75%) and dimer (∼25%), with traces of a possible trimer.10 Previous studies suggest that linkers shorter than 12 amino acids can result in multimeric complexes (dimers, trimers, etc.) due to “domain swapping,” and that transition between monovalent and divalent scFv can be somewhat controlled by the linker length.31,32 Considering these findings, and with the intention of producing a predominately monomeric scFv, we used a 15 amino acid linker to connect the two chains.10
Source of METH and MDMA
METH [(+)-N-methyl-1-phenylpropan-2-amine hydrochloride] and MDMA [(+)-3,4-methylenedioxymethamphetamine] were synthesized at the Research Triangle Institute (Research Triangle Park, NC) and we obtained them from the National Institute on Drug Abuse (Bethesda, MD). The purities of the compounds were better than 99%.
Crystallization of scFv6H4 with METH and MDMA
Crystals of the scFv6H4:METH complex and the scFv6H4:MDMA complex were grown using hanging drop vapor diffusion technique. The protein stocks of 12 mg/mL were prepared in a solution of 5 mM HEPES, 150 mM sodium chloride, and 5 mM octyl β-d-glucopyranoside detergent at pH 8.3. METH or MDMA was added to the protein solution to a final concentration of 5 mM. The reservoir solution contained 1.15M sodium citrate and 0.28M imidazole-malate (at pH 8 for METH complex and pH 7.8 for MDMA complex). The hanging drops were made by combining 1 μL of the protein stock with 1 μL reservoir solution on 22 mm microscope cover slips. The crystallization tissue culture plates were kept at 14°C in an incubator.
The crystals of both complexes grew typically in about 3 weeks with dimensions of about 0.1 mm × 0.1 mm × 0.2 mm. Preliminary diffraction data were collected on an R-AXIS IV++ image plate detector system mounted on a RU-H3R Rigaku rotating anode generator. The final data sets were collected at the Stanford Synchrotron Radiation Laboratory (SSRL), Palo Alto, CA. The summary of data collection statistics of both crystals are listed in Table I.
Table I.
Data Collection and Refinement Statistics
| Crystal | scFv6H4:METH | scFv6H4:MDMA |
|---|---|---|
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 34.32, 65.25, 48.51 | 34.17, 64.81, 48.32 |
| α, β, γ | 90°, 98.72°, 90° | 90°, 98.98°, 90° |
| Resolution (Å) | 1.9 Å | 2.4 Å |
| Rmerge | 5.0% (18.8%) | 4.5% (15.1%) |
| Average I/σI | 33.2 (4.9) | 23.9 (6.8) |
| Completeness, overall (%) | 98.6 (89.8) | 99.8 (99.5) |
| Redundancy | 6.7 (4.4) | 3.75 (3.7) |
| Wavelength (Å) | 0.97946 | 0.97945 |
| Number of images | 499 | 180 |
| Oscillation range | 0.5° | 1° |
| Number of observations | 110,482 | 31,183 |
| Number of unique reflections | 16,576 | 8213 |
| Refinement statistics | ||
| Number of protein atoms | 1783 | 1799 |
| Number of ligand atoms | 11 | 14 |
| Number of water molecules | 96 | 77 |
| Rfactor/Rfreea | 21.0/23.3 (26.3/27.5) | 21.4/27.3 (34.9/37.5) |
| Rms bond lengths, angles | 0.005 Å/1.3° | 0.008 Å/1.3° |
| Ramachandran statistics | ||
| Most favored (%) | 89.0 | 89.5 |
| Additional allowed (%) | 9.5 | 8.5 |
| Generously allowed (%) | 1.0 | 1.5 |
| Disallowed | 0.5 | 0.5 |
| Luzzati error (Å) | 0.23 | 0.32 |
| Average B values | ||
| Protein (Å2) | 23.5 | 38.9 |
| Ligand (Å2) | 16.1 | 29.3 |
| Water (Å2) | 33.8 | 42.9 |
The values in the parentheses refer to the highest resolution shell: 1.99−1.90 (Å) for the METH complex and 2.48–2.40 (Å) for the MDMA complex. The diffraction data were collected at 100° K on beam line 9−1 at Stanford Synchrotron Radiation Laboratory, Palo Alto, CA. The data were reduced using the software package HKL2000.33
Seven percent of the reflections were set aside for Rfree calculations.
Structure determination of scFv6H4:METH
The structure of scFv6H4:METH complex was determined by the molecular replacement technique. Using the known geometry of prototype Fab structures, a homology model was constructed and it was used as a search model. The rotation and translation searches were carried out with 4 Å diffraction data using CNS suite of programs,34 which yielded a unique solution. The initial model was examined using a 2Fo-Fc map and a Fo-Fc map (2.7 Å) and the regions of the model which did not match with the maps, particularly the loop regions, were rebuilt into the well-defined continuous electron densities of these regions. The model was further refined using 2.5 Å data and a difference electron density map was computed, which revealed the position and the conformation of the ligand METH. After two more cycles of rebuilding and refinements, a total of 96 water molecules were located using a difference electron density map. At the close of the refinements, Rfactor and Rfree were 21.0 and 23.3%, respectively, for the 1.9 Å diffraction data (Table I).
Structure determination of scFv6H4:MDMA
The crystals of the scFv6H4:MDMA complex have the same space group and nearly identical cell dimensions at the scFv6H4:METH structure. Therefore, the coordinates of the scFv6H4:METH structure (excluding the METH molecule and the solvent molecules) were used as the starting model. A difference electron density map computed after 20 cycles of rigid body refinement calculations revealed the entire MDMA molecule. A total of 77 water molecules were located using a different electron density map. The final Rfactor and Rfree were 21.4 and 27.3%, respectively, for the 2.4 Å diffraction data. The refinement statistics are listed in Table I.
Accession numbers
The atomic coordinates and structure factors for scFv6H4:METH (Research Collaboratory for Structural Bioinformatics Protein Databank = PDB# 3GKZ) and 3GKZ (Research Collaboratory for Structural Bioinformatics Protein Databank = PDB# 3GM0) have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Databank, Rutgers University, New Brunswick, NJ (http://www.rcsb.org).
Glossary
Abbreviations:
- AMP
(+)-amphetamine
- cDNA
complementary DNA
- CDR
complementarity determining region
- IgG
immunoglobulin gamma
- mAb
monoclonal antibody
- MDMA
(+)-3,4-methylenedioxymethamphetamine
- METH
(+)-methamphetamine
- scFv
single chain variable fragment
- VH
variable heavy chain
- VL
variable light chain.
References
- 1.NDIC. National drug treatment summary. Johnstown, PA: National Drug Intelligence Center; 2008. [Google Scholar]
- 2.Cornuz J, Zwahlen S, Jungi WF, Osterwalder J, Klingler K, Van MG, Bangala Y, Guessous I, Muller P, Willers J, Maurer P, Bachmann MF, Cerny T. A vaccine against nicotine for smoking cessation: A randomized controlled trial. PLoS ONE. 2008;3(6):e2547. doi: 10.1371/journal.pone.0002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martell BA, Mitchell E, Poling J, Gonsai K, Kosten TR. Vaccine pharmacotherapy for the treatment of cocaine dependence. Biol Psychiatry. 2005;58:158–164. doi: 10.1016/j.biopsych.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 4.Kosten T, Owens SM. Immunotherapy for the treatment of drug abuse. Pharmacol Ther. 2005;108:76–85. doi: 10.1016/j.pharmthera.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Gentry WB, Laurenzana EM, Williams DK, West JR, Berg RJ, Terlea T, Owens SM. Safety and efficiency of an anti-(+)-methamphetamine monoclonal antibody in the protection against cardiovascular and central nervous system effects of (+)-methamphetamine in rats. Int Immunopharmacol. 2006;6:968–977. doi: 10.1016/j.intimp.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Peterson EC, Gunnell M, Che Y, Goforth RL, Carroll FI, Henry R, Liu H, Owens SM. Using hapten design to discover therapeutic monoclonal antibodies for treating methamphetamine abuse. J Pharmacol Exp Ther. 2007;322:30–39. doi: 10.1124/jpet.106.117150. [DOI] [PubMed] [Google Scholar]
- 7.Poljak RJ. X-ray crystallographic studies of immunoglobulins. Contemp Top Mol Immunol. 1973;2:1–26. [PubMed] [Google Scholar]
- 8.Byrnes-Blake KA, Carroll FI, Abraham P, Owens SM. Generation of anti-(+)methamphetamine antibodies is not impeded by (+)methamphetamine administration during active immunization of rats. Int Immunopharmacol. 2001;1:329–338. doi: 10.1016/s1567-5769(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 9.Goel A, Colcher D, Baranowska-Kortylewicz J, Augustine S, Booth BJ, Pavlinkova G, Batra SK. Genetically engineered tetravalent single-chain Fv of the pancarcinoma monoclonal antibody CC49: improved biodistribution and potential for therapeutic application. Cancer Res. 2000;60:6964–6971. [PubMed] [Google Scholar]
- 10.Peterson EC, Laurenzana EM, Atchley WT, Hendrickson HP, Owens SM. Development and preclinical testing of a high-affinity single-chain antibody against (+)-methamphetamine. J Pharmacol Exp Ther. 2008;325:124–133. doi: 10.1124/jpet.107.134395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shelver WL, Keyler DE, Lin G, Murtaugh MP, Flickinger MC, Ross CA, Pentel PR. Effects of recombinant drug-specific single chain antibody Fv fragment on [3H]-desipramine distribution in rats. Biochem Pharmacol. 1996;51:531–537. doi: 10.1016/0006-2952(95)02233-3. [DOI] [PubMed] [Google Scholar]
- 12.Johnson G, Wu TT. Kabat Database and its applications: future directions. Nucl Acids Res. 2001;29:205–206. doi: 10.1093/nar/29.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hakey P, Quellette J, Zubieta J, Korter T. Redetermination of (+)-methamphetamine hydrochloride at 90 K. Acta Crystallogr. 2008;E64:940. doi: 10.1107/S1600536808011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morimoto BH, Lovel S, Kahr B. Ecstasy: 3,4-methylenedioxymethamphetamine (MDMA) Acta Crystallogr. 1998;C54:229–231. doi: 10.1107/s0108270197012390. [DOI] [PubMed] [Google Scholar]
- 15.Celikel R, Ruggeri ZM, Varughese KI. von Willebrand factor conformation and adhesive function is modulated by an internalized water molecule. Nat Struct Biol. 2000;7:881–884. doi: 10.1038/79639. [DOI] [PubMed] [Google Scholar]
- 16.Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 17.Burley SK, Petsko GA. Weakly polar interactions in proteins. Adv Protein Chem. 1988;39:125–189. doi: 10.1016/s0065-3233(08)60376-9. [DOI] [PubMed] [Google Scholar]
- 18.Chelli R, Gervasio FL, Procacci P, Schettino V. Stacking and T-shape competition in aromatic-aromatic amino acid interactions. J Am Chem Soc. 2002;124:6133–6143. doi: 10.1021/ja0121639. [DOI] [PubMed] [Google Scholar]
- 19.Burley SK, Petsko GA. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 20.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci USA. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pozharski E, Wilson MA, Hewagama A, Shanafelt AB, Petsko G, Ringe D. Anchoring a cationic ligand: the structure of the Fab fragment of the anti-morphine antibody 9B1 and its complex with morphine. J Mol Biol. 2004;337:691–697. doi: 10.1016/j.jmb.2003.12.084. [DOI] [PubMed] [Google Scholar]
- 22.Steiner T, Saenger W. The ordered water cluster in vitamin B12 coenzyme at 15 K is stabilized by C-H⋯O hydrogen bonds. Acta Crystallogr D Biol Crystallogr. 1993;49:592–593. doi: 10.1107/S0907444993005360. [DOI] [PubMed] [Google Scholar]
- 23.Wahl MC, Sundaralingam M. C-H⋯O hydrogen bonding in biology. Trends Biochem Sci. 1997;22:97–102. doi: 10.1016/s0968-0004(97)01004-9. [DOI] [PubMed] [Google Scholar]
- 24.Jiang L, Lai L. CH⋯O hydrogen bonds at protein-protein interfaces. J Biol Chem. 2002;277:37732–37740. doi: 10.1074/jbc.M204514200. [DOI] [PubMed] [Google Scholar]
- 25.Scheiner S. Contributions of NH⋯O and CH⋯O hydrogen bonds to the stability of beta-sheets in proteins. J Phys Chem B. 2006;110:18670–18679. doi: 10.1021/jp063225q. [DOI] [PubMed] [Google Scholar]
- 26.Panigrahi SK, Desiraju GR. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins. 2007;67:128–141. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 27.Sarkhel S, Desiraju GR. N-H⋯O, O-H⋯O, and C-H⋯O hydrogen bonds in protein-ligand complexes: strong and weak interactions in molecular recognition. Proteins. 2004;54:247–259. doi: 10.1002/prot.10567. [DOI] [PubMed] [Google Scholar]
- 28.Larsen NA, Zhou B, Heine A, Wirsching P, Janda KD, Wilson IA. Crystal structure of a cocaine-binding antibody. J Mol Biol. 2001;311:9–15. doi: 10.1006/jmbi.2001.4839. [DOI] [PubMed] [Google Scholar]
- 29.Pozharski E, Moulin A, Hewagama A, Shanafelt AB, Petsko GA, Ringe D. Diversity in hapten recognition: structural study of an anti-cocaine antibody M82G2. J Mol Biol. 2005;349:570–582. doi: 10.1016/j.jmb.2005.03.080. [DOI] [PubMed] [Google Scholar]
- 30.Lim K, Owens SM, Arnold L, Sacchettini JC, Linthicum DS. Crystal structure of monoclonal 6B5 Fab complexed with phencyclidine. J Biol Chem. 1998;273:28576–28582. doi: 10.1074/jbc.273.44.28576. [DOI] [PubMed] [Google Scholar]
- 31.Atwell JL, Breheney KA, Lawrence LJ, Mccoy AJ, Kortt AA, Hudson PJ. scFv multimers of the anti-neuraminidase antibody NC10: length of the linker between VH and VL domains dictates precisely the transition between diabodies and triabodies. Protein Eng. 1999;12:597–604. doi: 10.1093/protein/12.7.597. [DOI] [PubMed] [Google Scholar]
- 32.Volkel T, Korn T, Bach M, Muller R, Kontermann RE. Optimized linker sequences for the expression of monomeric and dimeric bispecific single-chain diabodies. Protein Eng. 2001;14:815–823. doi: 10.1093/protein/14.10.815. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]