

Abstract
A great need exists for prediction of antibody response for the generation of antibodies toward protein targets. Earlier studies have suggested that prediction methods based on hydrophilicity propensity scale, in which the degree of exposure of the amino acid in an aqueous solvent is calculated, has limited value. Here, we show a comparative analysis based on 12,634 affinity-purified antibodies generated in a standardized manner against human recombinant protein fragments. The antibody response (yield) was measured and compared to theoretical predictions based on a large number (544) of published propensity scales. The results show that some of the scales have predictive power, although the overall Pearson correlation coefficient is relatively low (0.2) even for the best performing amino acid indices. Based on the current data set, a new propensity scale was calculated with a Pearson correlation coefficient of 0.25. The values correlated in some extent to earlier scales, including large penalty for hydrophobic and cysteine residues and high positive contribution from acidic residues, but with relatively low positive contribution from basic residues. The fraction of immunogens generating low antibody responses was reduced from 30% to around 10% if immunogens with a high propensity score (>0.48) were selected as compared to immunogens with lower scores (<0.29). The study demonstrates that a propensity scale might be useful for prediction of antibody response generated by immunization of recombinant protein fragments. The data set presented here can be used for further studies to design new prediction tools for the generation of antibodies to specific protein targets.
Keywords: antibody response, immunogenicity, immunization, prediction
Introduction
The adaptive immune system is induced by lymphocytes and can be classified into humoral immunity, mediated by antibodies, and cellular immunity, mediated by T lymphocytes.1 Antibodies bind to pathogens, such as viruses or bacteria, and the binding triggers various immunological responses, such as phagocytosis and destruction by scavenger cells, e.g., macrophages. Thus, the humoral response is crucial in the host defense toward most pathogens and this makes efforts to predict efficient immunogens for generation of specific antibodies important, both in efforts to develop vaccines and to generate specific antibodies for use in research and therapy.
An efficient antibody response is reliant both on antigen recognition by the antibody presenting B-cells (B-cell epitope) and the delivery of a proliferation inducing T-cell signal to the B-cell (T-cell epitope). The antigen thus has to possess regions with both T- and B-cell epitopes for the effective generation of antibodies. Both antigen fusions, fusing the antigen with T-cell stimulatory partner proteins,2,3 and bioinformatic predictions1 have been used to stimulate and predict the T-cell response of an immunization. Whereas prediction of T-cell epitopes has been a field with great improvement in later years, prediction of B-cell epitopes has shown to be a more challenging task.1
B-cell epitopes can be classified into continuous and discontinuous epitopes.4 A continuous epitope, also called linear or sequential epitope, is a short sequence of amino acids that is recognized by the antibody, while a discontinuous epitope, also called conformational epitope, is composed of amino acids that are not adjacent in the protein primary sequence, but are brought together by the protein folding. Early work has suggested that most epitopes are conformational,5,6 based on antibodies mainly generated toward native protein structures. In contrast, antibodies generated to synthetic peptides will recognize linear epitopes.5 As peptides sometimes, due to their limited size, do not resemble their native folded protein, complete or nearly complete antigens are preferably used for applications where native recognition is desired, e.g., for vaccine development.7 Similarly, recent results from our group suggest that a large fraction of the antibodies within a polyclonal pool generated to partly nonfolded recombinant protein fragments (around 100 residues) are directed mainly toward linear epitopes and the corresponding fraction recognizing conformational epitopes is relatively is low.8 The relative amounts of linear and conformational epitopes recognized by an antibody are thus dependent on the choice of antigen used. It is in this context important that these differences in the behavior of immunogens should be taken into account when a particular antibody is generated, since some applications involve studying the protein target in its native form, such as live cell sorting (FACS) or serum analysis, while other applications target the protein in a completely or partially denaturated state, such as Western blot analysis, immunohistochemistry or immunofluorescence. For research applications, it might therefore, in many cases be beneficial to use synthetic peptides or larger partially unfolded protein fragments as immunogens to generate antibodies predominately with linear epitopes suitable across many analysis platforms, in which the protein may be denaturated at variable degrees. A need therefore exists to predict antibody response based on linear sequences to facilitate the selection of synthetic peptides and/or recombinant protein fragments starting with the complete sequence of the target protein.
To facilitate the generation of antibodies toward continuous epitopes, a large number of algorithms, often based on hydrophilicity propensity in which each amino acid is assigned a value based on the degree of exposure of the amino acid in an aqueous solvent, have been published.9 In the early 1980s, Hopp and Woods10 developed the first linear epitope prediction methods and this was followed by many others, including Kyte and Doolittle11 and Zaslavsky et al.12 In 1993, Pellequer suggested that the propensity scale should also be based on turn propensity.13 However, in 2005 Blythe and Flowers published an extensive study of various linear epitope prediction methods14 and they concluded that even predictions based on the most accurate amino acids scales were only marginally better than random, suggesting that a more sophisticated approach is needed to predict linear epitopes. Recently, position-specific scoring matrices (PSSM) and machine-learning methods have been used15-17 to incorporate additional information for the prediction of linear epitopes, such as neighborhood parameters. These methods have been used to increase the accuracy of the linear epitope prediction as compared to single-parameter methods.1 Kawashima et al. published in 2008 an amino acid index database (AAindex) with 544 different indices intended for a wide range of bioinformatics research on protein sequences, including immunogenicity.18 The degree of predictability of these methods and propensity scales is yet to be determined, emphasizing the need for large sets of experimental data generated in a standardized manner to facilitate comparative studies and to further enhance bioinformatics development in the field.
Here, we report the evaluation of antibody responses using 12,634 recombinant human protein fragments, the largest test set of immunogens and their antibody response yet reported. The generation of the protein fragments and the immunization of rabbits have been performed in a standardized manner, making comparisons of antibody response (immunogenicity) possible. The antibody response was measured as the amount of antibody obtained after immunization and affinity purification using the antigen as ligand. Thus, the polyclonal antibody serum was converted to a monospecific antibody fraction,19 in which all antibodies are directed toward epitopes displayed by the antigen. In this study, Protein Epitope Signature Tags (PrESTs) have been used, comprising unique “signature” sequence of the protein targets and are therefore suitable immunogens for the generation of specific antibodies with low unwanted cross-reactivity.19 No epitope prediction method was used in the design of the protein fragments which makes this data set attractive for assessing which types of fragments give the best antibody responses.
Results
Design of human recombinant immunogens based on low sequence identity
A protein feature visualization tool PRESTIGE20 was used to design recombinant human protein fragments based on low sequence identity to other human proteins. The fact that the human genome sequence is known21,22 and that the coding parts of the genome can be predicted and assembled into a list of potential proteins23,24 has made it possible to exclude regions within a target protein with high sequence identity to other human proteins. Several methods have recently been published based on a sliding window algorithm to determine the sequence similarity of the various parts of a particular human protein to all the other protein sequences of the human proteome.20,25 A central feature of the PRESTIGE tool is the similarity graphs showing a sliding window of 50 and 10 amino acids, respectively, in which each part of the analyzed protein is compared to all the human proteins predicted from the genome sequence. Based on this tool, large numbers of antigens were designed and primers were synthesized and used for de novo cloning of cDNA from RNA extracted from various human tissues26 into an Escherichia coli expression vector as described earlier.27
Generation of monospecific antibodies
Recombinant PrESTs were produced in E.coli as fusion proteins linked to immunopotenting Albumin Binding Protein (ABP),2,3 validated by mass spectrometry and used for immunizations in rabbits using a standardized immunization protocol.19 The polyclonal antisera were purified using the immunogen as affinity ligand to generate monospecific antibodies.28 Here, 12,634 protein fragments from 7,231 human genes were used as antigen to generate antibodies corresponding to ∼35% of all the 21,000 protein-encoded genes.29 The genes were distributed on the chromosomes as shown in Figure 1(A). Typically 8 mL of serum was used for the purification and the monospecific antibodies were normally eluted in a 3 mL volume and stored in aliquots. The amount of the antibodies after elution varied considerably, with approximately two thirds of the samples in the range from 0.11 mg to 0.6 mg.
Figure 1.

The distribution of selected genes and PrESTs regarding human chromosomes and amino acid content. A: The number of genes on each of the human chromosomes was calculated. Altogether, 7231 genes were analyzed and the number of genes on each chromosome is shown (black bars), as well as the total number of genes on each chromosome (white bars) predicted by Ensembl version 50.36.29 B: The amino acid compositions were calculated for all of the protein-encoded genes (black bars) based on Ensembl version 50.36 and all the 9107 PrEST fragments used to generate antibodies (white bars).
The content of amino acids in the test set of antigens
To check for bias of certain amino acids in the design of the PrESTs compared to the amino acid distribution of the human proteome, an analysis of the amino acid content of the selected antigens was carried out. Altogether, 1,353,235 amino acid residues of antigens corresponding to the 12,634 antibodies were evaluated. The distribution of the amino acids is shown in Figure 1(B) together with a comparison of the total distribution of amino acids in the human predicted proteome according to Ensembl.29 The analysis shows that the distribution of amino acids used in the study is similar to the naturally occurring. This demonstrates that the antigen selection, based on low sequence identity, has not introduced any major bias for certain amino acids in the data set.
Analysis of the antibodies
A comprehensive way of validating antibodies is to use protein array technology. This allows for multiplex analysis in which the binding to the protein target antigen is compared to the binding against other independent protein targets. We used a planar microarray19 including the protein target together with 383 other arbitrarily selected human protein fragments. A validation30 was carried out to score the antibodies as supportive (pass) and nonsupportive (fail). To evaluate if the amount of antibodies obtained after the affinity purification also has bearing on the quality (specificity) of the antibody, the 12,634 antibodies were divided into five classes depending on amount obtained after affinity capture. All classes were selected to have equal number of antibodies, spanning from the lowest group with amounts less than 0.11 mg to the top group having amounts higher than 0.60 mg, respectively. The results of the protein array for all 12,634 antibodies are shown in Figure 2(A) stratified into the five classes based on antibody amount. An average success rate of 90% was obtained and interestingly the antibodies with a lower amount showed considerably lower success rate (73%) despite the fact that all antibodies were titrated to similar assay concentrations. The results show that most of the antibodies obtained after the affinity purification pass the quality assurance based on microarray, but a tendency can be observed for better quality (specificity) of antibodies with higher amounts purified. The low success rate for the antibodies with moderate amounts could be due to an immune response with a high level of residual antibodies with weak affinity to the immunogen and promiscuous behavior to other targets or the result of an immune response favoring less unique parts of the target.
Figure 2.
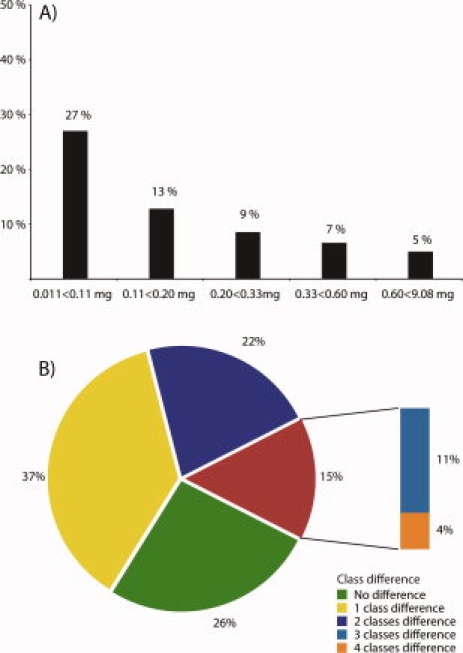
Specificity failure rate of antibodies grouped by antibody amount (A) and variation in antibody amount between re-immunizations (B). (A) The 12,634 antibodies were classified into five families based on the amounts obtained after affinity purification with the same number of antibodies in each class. Each antibody was evaluated using a standardized scoring system19,30 and the success rates (pass) and failure rates for the various classes were estimated. A supportive score (pass) was obtained if the specific binding of the antibody to the target antigen was obtained with no signal above 15% of the specific signal to all other antigens on the protein array. A nonsupportive validation score (fail) was given if the antigen has more than 40% of the signal obtained for the target protein or three antigens showing more than 15% signal as compared to the signal of the binding to the target antigen. (B) A comparison of 2456 PrESTs immunized twice in separate rabbits and their corresponding antibody amounts after purification, grouped in five equally sized groups. The majority (63%) of the immunogens gave an antibody amount within the same or closest neighboring class. 15% of the antibody fractions obtained after purification differed remarkably varying as much as three (11%) or four classes (4%).
To validate the variation between immunizations, a data set consisting of 2456 PrESTs immunized twice in two separate rabbits and their corresponding antibody amounts after purification were analyzed and compared [Fig. 2(B)]. The immunizations were grouped in the same way as aforementioned based on five equally sized classes. The results show that the majority (63%) of the immunogens gave an antibody amount within the same class or the closest neighboring class. and only 15% of the antibody fractions obtained after purification differed three classes (11%) or four classes (4%). This suggests that, although variations between immunizations exist, the majority of the antigens yield a similar immune response during repeated immunizations.
Antigen amino acid composition for the different antibody fractions
The variation in antibody amounts of the different PrESTs prompted us to investigate if a correlation could be observed between the amino acid composition of the immunogen used for immunization and the resulting antibody amount. In Figure 3, the difference of the amino acid composition relative the normalized composition [Fig. 1(B)] is shown for the two classes with the highest (white bars) and lowest (black bars) antibody amount. The results demonstrate a clear pattern with penalty for hydrophobic amino acids, such as tyrosine (Y), tryptophan (W) and phenylalanine (F) and benefit from hydrophilic residues, with glutamic acid (E) and glutamine (Q) as the most beneficial amino acids. Cysteine (C) was shown to be negatively contributing, with a high representation in antibody purifications with low antibody amount and conversely underrepresentation in purifications with high antibody amount.
Figure 3.

The amino acid composition for the two classes of antibodies with the lowest and highest antibody concentrations after affinity purification. The amino acid composition for all antibodies was calculated based on the 20% of antibodies with lowest (below 0.11 mg) and highest amounts (above 0.60 mg). The resulting amino acid composition was compared with the average amino acid composition for all the PrESTs corresponding to the 12,634 antibodies as shown in Figure 1(B) and the difference were calculated and shown for the class of antibodies with a low amounts (white bars) and high amounts (black bars).
Correlation between prediction algorithms and antibody response
The comprehensible collection of 544 published propensity scales18 was evaluated. Theoretical grand average propensity values were calculated for each PrEST using all 544 methods and compared to the amount of antibodies obtained after affinity purification. Approximately 6.9 million data points were obtained and the theoretical values were compared with corresponding experimental antibody amount. The Pearson correlation coefficients and trend line slope values were calculated and plotted for each method (Fig. 4). Altogether eight methods, described in Table I, are high-lighted in the figure, including the methods with the highest correlation (B by Parthasarathy-Murthy, C by Vihinen, D by Wimley-White and E by Zaslavsky) and two of the earliest methods for prediction of antigenic regions10 Hopp-Woods (G) and protein hydropathy11 by Kyte-Doolittle (F). Method A by Qian-Sejnowski,31 based on scores for predicting the secondary structure of globular proteins is shown as an example of a method that is not useful for the prediction of antigenicity (Pearson correlation 0.00). The best performing propensity scales are those based on the distribution of normalized flexibility parameters (B values) (method B and C) and the hydrophobicity scale experimentally calculated by the free energies required to transfer peptides from bilayer interfaces to water (method D). Also the method (E) by Zaslavsky based on relative hydrophobicity of amino acids by partitioning in aqueous two-phase polymeric systems gives relative good (although negative) correlation. Interestingly, the propensity scales described already in 1981 by Hopp and Woods10 performs well (method G). The Pearson correlations for the best methods including (G) Hopp and Woods10 are ∼0.2 (Table I).
Figure 4.
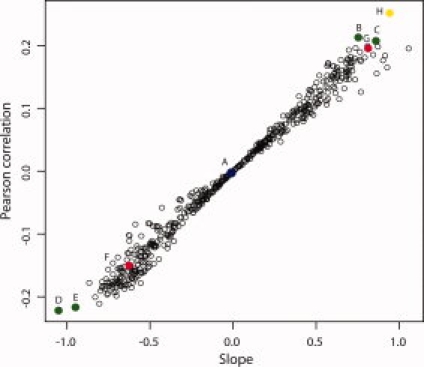
The correlation of 544 propensity scales with the 12,634 antibody amounts obtained by immunizing recombinant PrESTs. For each PrEST, the theoretical propensity value was calculated using each of the 544 propensity scale methods18 and compared with the amount of antibodies obtained. The theoretical values were compared with corresponding experimental antibody amounts and Pearson correlation coefficients and trend line slope values were calculated and plotted for each method. Eight selected methods are indicated by color and letter and are described in more detail in Table I. The top-performing methods, as measured by highest positive and negative Pearson correlation, were colored in green (B–D). Early methods for prediction of antigenic regions10 (G) and protein hydropathy11 (F) were colored in red. A least square fit of all antigen sequences to their corresponding antibody amounts yielded a new propensity scale (Supporting Information Table 1), which was used to predict antibody amounts analogously to the other methods and was plotted in yellow (H).
Table I.
Some Examples of Propensity Scales and Their Correlation to the Antibody Response of this Study
| Index | Author | Publication | Description | Slope | Pearson correlation |
|---|---|---|---|---|---|
| A | Qian and Sejnowski31 | J Mol Biol 202, 865–884 (1988) | Predicting the secondary structure of globular proteins using neural network. Weights for alpha-helix at the window position of 3. | 0.001 | 0.000 |
| B | Vihinen et al.38 | Proteins 19, 141–149 (1994) | Accuracy of protein flexibility predictions. Normalized flexibility parameters (B-values), | 0.754 | 0.213 |
| C | Parthasarathy and Murthy32 | Protein Eng 13, 9–13 (2000) | P-Values of mesophilic proteins based on the distributions of B values | 0.860 | 0.207 |
| D | Wimley and White33 | Nature Struct Biol 3, 842–848 (1996) | Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Free energies of transfer of AcWl-X-LL peptides from bilayer interface to water | −1.051 | −0.222 |
| E | Zaslavsky et al.12 | J Chromatogr 240, 21–28 (1982) | Dependence of partition coefficient on ionic strength. Measurement of relative hydrophobicity of amino acid side-chains by partition in an aqueous two-phase polymeric system: Hydrophobicity scale for nonpolar and ionogenic side-chains | −0.949 | −0.217 |
| F | Kyte and Doolittle11 | J Mol Biol 157, 105–132 (1982) | Hydropathy index. A simple method for displaying the hydropathic character of a protein | −0.626 | −0.150 |
| G | Hopp and Woods10 | Proc Natl Acad Sci USA 78, 3824–3828 (1981) | Prediction of protein antigenic determinants from amino acid sequences | 0.813 | 0.196 |
| H | This study | Antigen composition correlated with purified antibody amount | 0.943 | 0.252 |
The slope and Pearson correlation as a result of comparing the theoretical prediction using a particular propensity scale and the experimental values of antibody concentrations for the 12,634 recombinant protein fragments are shown. The references to the various propensity scales are A (Qian),31 B (Parthasarathy),32 C (Vihinen),33 D (Wimley),34 E (Zaslavsky),12 F (Kyte and Doolittle),11 and G (Hopp and Woods).10
Determination of a new propensity scale based on the experimental values in this study
A propensity scale was determined using a least square fit algorithm based on the results from the 12,634 PrEST antibodies. The Pearson correlation for the new propensity scale (method H) is somewhat higher (0.25) than the previously published scales (Table I). The values for each amino acid are shown in Supporting Information Table 1 with high negative values for tryptophan (−1.9), tyrosine (−1.6), phenylalanine (−0.7) and cysteine (−0.5). Positive values were obtained for glutamic acid (2.2), aspartic acid (1.4) and glutamine (1.2). In Figure 5, the propensity scales of five of the methods from Table I have been normalized and plotted to allow comparisons between the contributions from each amino acid residue. The tendency from hydrophobic to hydrophilic residues can be seen for all methods, but individual differences for particular amino acids can be observed. Proline has a larger positive value for method B by Parthasarathy-Murthy,32 somewhat higher than other methods such a method D by Wimley-White,34 and method G by Hopp-Woods.10 The basic residue lysine (K) was found to have a relatively low positive contribution as compared to earlier propensity scales, such as methods B32 and G10, while the amino acids isoleucine and leucine showed a relatively high positive contribution in the new scale (Fig. 5).
Figure 5.

The value for each amino acid for the top performing propensity scales. The scales have been normalized from lowest (0) to highest (100) score and each residue is plotted. The methods are according to Table I, with exception for inversely correlated scales by method (D) Wimley34 (blue) and method (E) Zaslavsky12 (yellow) which have been inverted for clarification. The order of the amino acid residues is according to the propensity scale of this study (turquoise).
Prediction of antibody response for some selected propensity scales
To investigate the predictive value of the various propensity scales further, a comparison of the five classes of antibodies stratified according to amounts was made. The theoretical grand average value obtained by the eight propensity scales described in Table I was calculated for all the 12,634 antibody PrEST antigens and the antibodies were divided into five bins of equal size (∼2500 antibodies in each group) based on the obtained score. The fractions of antibodies in each class were thereafter calculated and the frequency was plotted for each method (Fig. 6). Method A by Qian-Sejnowski, based on prediction of globular proteins and with a poor Pearson correlation, showed as expected a close to random frequency of 20% in each class. In contrast, all the other seven methods showed good correlations (positive and negative correlation) between the theoretical predicted value and the antibody amounts, and the correlations were around 30% and 10% for the high and low group, respectively, as compared to a random 20% value. Based on our propensity scale, 31% of the antigens scored to be in the lowest immunogenicity group were verified experimentally as poor immunogens with amounts of antibodies less than 0.11 mg. Similarly, 33% of the PrESTs calculated to be in the highest antigenicity group were indeed verified to give antibody yields exceeding 0.60 mg. Hence by choosing antigen regions with a higher score (>0.48) over a region with a lower (0–0.29) the fraction of antibodies with highest quality and amount will increase from 8% to 33% (Fig. 6).
Figure 6.

The correlation between theoretical propensity values and experimental antibody amounts stratified into five groups of equal sizes. The theoretical value obtained by the eight propensity scales described in Table I was calculated for all the 12,634 PrESTs and the PrESTs were divided into five bins of equal size. Similarly, the antibody amounts were sorted and classified into five bins with 20% of the data points each. The fractions of antibodies in each class were thereafter calculated and the frequency was plotted for each method.
Discussion
Here, we show comparisons between 12,634 experimentally measured antibody responses of recombinant protein fragments and corresponding theoretical predictions based on a large set of propensity scales. The Pearson correlation coefficient for the propensity scales calculated from the current data set is 0.25 and ∼0.2 for the best prediction methods published previously. Although rather low, the correlation coefficient shows unambiguously that some of the antibody responses can be predicted using propensity scales. In addition, the classification into five groups of the antibody response and prediction score, respectively, showed a good correlation between experimental and predicted values, as exemplified by the fact that the fraction of antibodies with low antibody response can be decreased 50% (from 20% to 10%) by choosing PrEST sequences with high values calculated using the new propensity scale. The results somewhat contradicts the conclusion in 200514 that even predictions based on the most accurate amino acids scales are only marginally better than random, although it is important to point out that the earlier results were based on epitope prediction of a smaller experimental data set (50 proteins) generated in a nonstandardized manner. The data set used here is the largest so far published and more importantly, all antibodies have been generated with the same standard operating procedure.
In this study, we have used the amount of obtained affinity purified polyclonal antibody as an indicator of antibody response, although the true specificity toward the native protein target or cross-reactivity of the antibody toward other proteins is unknown. It is reassuring that the protein array experiments show a tendency for better specificity for antibodies with higher antibody amounts and similar results have been obtained also for the functionality of the antibodies in immunohistochemistry, Western blot analysis and immunofluorescence analysis using confocal microscopy (data not shown). These results therefore suggest that antibody amounts is a relevant indicator for specific antibody response, although more specialized validation must be done to investigate the final quality of each antibody. A comparison of repeated immunizations with the same immunogen showed that the majority (63%) of the antibodies purified stayed within the same or the closest neighboring class in both immunizations.
To explain the process of antibody-antigen recognition a detailed view of the paratope-epitope complex is required. Focused studies of paratope surfaces have previously revealed prevalence for tyrosine (Y) and tryptophan (W) in the antigen combining site, both possessing a mixture of hydrophilic, hydrophobic and aromatic character, suitable for interactions with a variety of targets.7,35,36 A complication with the present data set for analogous comparisons of epitope surfaces is that relatively large recombinant protein fragments have been used as immunogens. The average size of the PrESTs in this study was 107 amino acids and the immunogens will therefore contain several independent epitopes and the relative contribution of these for the antibody response cannot be directly calculated. Recently, we have mapped the epitopes of eight PrESTs using a bacterial surface display method and the results demonstrated that each PrEST gave rise to an “oligoclonal” response of one to five epitopes spanning together less than half of the available antigen sequence.37 The recombinant protein fragment thus contains regions, which are highly immunogenic, while other regions are epitope “silent” and yields no specific antibodies. The values used here are therefore average antibody responses of protein fragments, most likely harboring several distinct epitopes, and firm predictions of specific linear epitopes cannot be made from this data set. A systematic mapping of the specific epitopes would make the data set even more valuable, but large-scale epitope mapping methods are lacking, making such efforts technically and economically difficult.
A new propensity scale was calculated based on the results of the 12,634 immunizations. This scale might be useful as a design tool when selecting recombinant protein fragments as immunogens. For example we show that by selecting a protein region with a higher score (>0.48) over a region with a lower (0–0.29) the fraction of experimentally obtained antibodies with highest quality and amount will increase from 8% to 33% (Fig. 6). The new propensity scale, listed in Supporting Information Table 1, displays several interesting differences from previously published propensity scales, such as the relatively low positive contribution of basic amino acids (arginine and lysine) as compared to some of the other propensity scales. In general, the values for many of the different amino acid residues are similar across the various methods, as exemplified by the negative contribution of hydrophobic residues and cysteines (Fig. 5). The negative contribution of cysteine is striking and it can be speculated that this might be due to the formation of local disulfide-bridges, which might, in some cases, interfere with antibody-antigen interaction. In this context, it is also possible that some of the cysteines react to various reactive sulfide-groups during the immunization to hide the epitope from the immune system.
In conclusion, the results presented here show that many of the previously published prediction models are, to some extent, relevant for the estimation of antibody response. A new amino acid propensity scale was calculated to fit the large data set of polyclonal antibody responses. The question arises if this propensity scale also may be used to design antigens for the generation of conformational epitopes or to predict antibody response using other types of antigens, such as synthetic peptides. To facilitate such bioinformatics studies, the data set corresponding to more than 10,000 antigen immunizations are available for download (www.proteinatlas.org/publ.php). This data may constitute a valuable resource for further studies to define the epitope space of the human proteome and to support the generation of new bioinformatics tools enhancing the generation of antibodies to be used for research, diagnostics or therapy.
Materials and Methods
Antibody generation and validation
9107 PrEST regions to be used as antigens were selected based on low sequence similarity25 followed by cDNA synthesis, cloning and recombinant protein production in E.coli as previously described.28 Mass spectrometry validated PrESTs were subsequently used as immunogens in New Zeeland rabbits and the antisera obtained were purified using a Äktaxpress system (GE Health Care AB) using the PrEST as ligand.19 Concentrations were determined automatically using online absorbance measurements. The antibodies were tested for specificity using protein arrays with 384 PrEST proteins, as previously described.19,30
Data analysis
Amino acid sequences for the 9107 PrEST antigens corresponding to 12,634 monospecific antibodies were obtained from the production database. Antibody amounts measured were normalized based on volume raw serum purified. Grand average scores for the whole PrEST sequences were calculated based on 544 different propensity scales18 implemented in-house using Java programming language. All prediction scores were scaled between zero and one without changing the distribution patterns. The statistic environment R38 was used for the calculation of Pearson correlations and linear slope coefficients. Matlab™ (MathWorks, Natick, MA) was used to solve an over determined equation system using least square fit of the 12,634 antibody amount values to their corresponding antigen amino acids frequencies. The resulted propensity scale was evaluated in the same way as the other methods and plotted together with all methods using the statistic environment R. Eight chosen methods were investigated in more detail by separately grouping their prediction scores into five uniformly sized bins, resembling 20% of the data points each and plotting these against antibody yield (also grouped in five uniform bins).
Acknowledgments
The authors are grateful to Per-Åke Nygren, Lisa Berglund, Linn Fagerberg, Åsa Sivertsson, and Mårten Sundberg for useful comments and advices. The entire staffs of the Human Proteome Resource (HPR) centre in Stockholm and Uppsala, Sweden and in Mumbai, India, are acknowledged for their tremendous efforts. This work was supported by the Knut and Alice Wallenberg foundation.
References
- 1.Lundegaard C, Lund O, Kesmir C, Brunak S, Nielsen M. Modeling the adaptive immune system: predictions and simulations. Bioinformatics. 2007;23:3265–3275. [Google Scholar]
- 2.Sjolander A, Stahl S, Nygren PA, Aslund L, Ahlborg N, Wahlin B, Scherf A, Berzins K, Uhlen M, Perlmann P. Immunogenicity and antigenicity in rabbits of a repeated sequence of Plasmodium falciparum antigen Pf155/RESA fused to two immunoglobulin G-binding domains of staphylococcal protein A. Infect Immun. 1990;58:854–859. doi: 10.1128/iai.58.4.854-859.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delmas A, Partidos CD. The binding of chimeric peptides to GM1 ganglioside enables induction of antibody responses after intranasal immunization. Vaccine. 1996;14:1077–1082. doi: 10.1016/0264-410x(95)00239-w. [DOI] [PubMed] [Google Scholar]
- 4.Atassi MZ. Antigenic structure of myoglobthe complete immunochemical anatomy of a protein and conclusions relating to antigenic structures of proteins. Immunochemistry. 1975;12:423–438. doi: 10.1016/0019-2791(75)90010-5. [DOI] [PubMed] [Google Scholar]
- 5.Barlow DJ, Edwards MS, Thornton JM. Continuous and discontinuous protein antigenic determinants. Nature. 1986;322:747–748. doi: 10.1038/322747a0. [DOI] [PubMed] [Google Scholar]
- 6.Van Regenmortel MHV. Mapping epitope structure and activity: from one-dimensional prediction to four-dimensional description of antigenic specificity. Methods. 1996;9:465–472. doi: 10.1006/meth.1996.0054. [DOI] [PubMed] [Google Scholar]
- 7.Chen SW, Van Regenmortel MH, Pellequer JL. Structure-activity relationships in peptide-antibody complexes: implications for epitope prediction and development of synthetic peptide vaccines. Curr Med Chem. 2009;16:953–964. doi: 10.2174/092986709787581914. [DOI] [PubMed] [Google Scholar]
- 8.Rockberg J, Schwenk JM, Uhlén M. Discovery of epitopes for targeting the human epidermal growth factor receptor 2 (HER2) with antibodies. Mol Oncol. 2009;3:238–247. doi: 10.1016/j.molonc.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levitt M. A simplified representation of protein conformations for rapid simulation of protein folding. J Mol Biol. 1976;104:59–107. doi: 10.1016/0022-2836(76)90004-8. [DOI] [PubMed] [Google Scholar]
- 10.Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci USA. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 12.Zaslavsky BY, Mestechkina NM, Miheeva LM, Rogozhin SV, Bakalkin G, Rjazhsky GG, Chetverina EV, Asmuko AA, Bespalova JD, Korobov NV, Chichenkov ON. Correlation of hydrophobic character of opioid peptides with their biological activity measured in various bioassay systems. Biochem Pharmacol. 1982;31:3757–3762. doi: 10.1016/0006-2952(82)90289-1. [DOI] [PubMed] [Google Scholar]
- 13.Pellequer JL, Westhof E, Van Regenmortel MH. Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol Lett. 1993;36:83–99. doi: 10.1016/0165-2478(93)90072-a. [DOI] [PubMed] [Google Scholar]
- 14.Blythe MJ, Flower DR. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005;14:246–248. doi: 10.1110/ps.041059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2.. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sollner J, Mayer B. Machine learning approaches for prediction of linear B-cell epitopes on proteins. J Mol Recognit. 2006;19:200–208. doi: 10.1002/jmr.771. [DOI] [PubMed] [Google Scholar]
- 17.Sweredoski MJ, Baldi P. COBEpro: a novel system for predicting continuous B-cell epitopes. Protein Eng Des Sel. 2009;22:113–120. doi: 10.1093/protein/gzn075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M. AAindex: amino acid index database, progress report 2008. Nucl Acids Res. 2008;36:D202–D205. doi: 10.1093/nar/gkm998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilsson P, Paavilainen L, Larsson K, Odling J, Sundberg M, Andersson AC, Kampf C, Persson A, Al-Khalili Szigyarto C, Ottosson J, Rimini R, Rockberg J, Runeson M, Sivertsson A, Skollermo A, Steen J, Stenvall M, Sterky F, Stromberg S, Tegel H, Tourle S, Wahlund E, Walden A, Wan J, Wernerus H, Westberg J, Wester K, Wrethagen U, Xu LL, Hober S, Ponten F. Towards a human proteome atlas: high-throughput generation of mono-specific antibodies for tissue profiling. Proteomics. 2005;5:4327–4337. doi: 10.1002/pmic.200500072. [DOI] [PubMed] [Google Scholar]
- 20.Berglund L, Bjorling E, Jonasson K, Rockberg J, Fagerberg L, Al-Khalili Szigyarto C, Sivertsson A, Uhlen M. A whole-genome bioinformatics approach to selection of antigens for systematic antibody generation. Proteomics. 2008;8:2832–2839. doi: 10.1002/pmic.200800203. [DOI] [PubMed] [Google Scholar]
- 21.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, Fitzhugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 22.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 23.Curwen V, Eyras E, Andrews TD, Clarke L, Mongin E, Searle SM, Clamp M. The Ensembl automatic gene annotation system. Genome Res. 2004;14:942–940. doi: 10.1101/gr.1858004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clamp M, Fry B, Kamal M, Xie X, Cuff J, Lin MF, Kellis M, Lindblad-Toh K, Lander ES. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci USA. 2007;104:19428–19433. doi: 10.1073/pnas.0709013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindskog M, Rockberg J, Uhlen M, Sterky F. Selection of protein epitopes for antibody production. Biotechniques. 2005;38:723–727. doi: 10.2144/05385ST02. [DOI] [PubMed] [Google Scholar]
- 26.Agaton C, Galli J, Hoiden Guthenberg I, Janzon L, Hansson M, Asplund A, Brundell E, Lindberg S, Ruthberg I, Wester K, et al. Affinity proteomics for systematic protein profiling of chromosome 21 gene products in human tissues. Mol Cell Proteomics. 2003;2:405–414. doi: 10.1074/mcp.M300022-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Larsson M, Graslund S, Yuan L, Brundell E, Uhlen M, Hoog C, Stahl S. High-throughput protein expression of cDNA products as a tool in functional genomics. J Biotechnol. 2000;80:143–157. doi: 10.1016/s0168-1656(00)00258-3. [DOI] [PubMed] [Google Scholar]
- 28.Agaton C, Falk R, Hoiden Guthenberg I, Gostring L, Uhlen M, Hober S. Selective enrichment of monospecific polyclonal antibodies for antibody-based proteomics efforts. J Chromatogr A. 2004;1043:33–40. doi: 10.1016/j.chroma.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Flicek P, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et al. Ensembl 2008. Nucl Acids Res. 2008;36:D707–D714. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjorling E, Uhlen M. Antibodypedia—a portal for sharing antibody and antigen validation data. Mol Cell Proteomics. 2008;7:2028–2037. doi: 10.1074/mcp.M800264-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Qian N, Sejnowski TJ. Predicting the secondary structure of globular proteins using neural network models. J Mol Biol. 1988;202:865–884. doi: 10.1016/0022-2836(88)90564-5. [DOI] [PubMed] [Google Scholar]
- 32.Parthasarathy S, Murthy MR. Protein thermal stability: insights from atomic displacement parameters (B values) Protein Eng. 2000;13:9–13. doi: 10.1093/protein/13.1.9. [DOI] [PubMed] [Google Scholar]
- 33.Vihinen M, Torkkila E, Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19:141–149. doi: 10.1002/prot.340190207. [DOI] [PubMed] [Google Scholar]
- 34.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 35.Collis AV, Brouwer AP, Martin AC. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J Mol Biol. 2003;325:337–354. doi: 10.1016/s0022-2836(02)01222-6. [DOI] [PubMed] [Google Scholar]
- 36.Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J Mol Biol. 2006;357:100–114. doi: 10.1016/j.jmb.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 37.Rockberg J, Lofblom J, Hjelm B, Uhlen M, Stahl S. Epitope mapping of antibodies using bacterial surface display. Nat Methods. 2008;5:1039–1045. doi: 10.1038/nmeth.1272. [DOI] [PubMed] [Google Scholar]
- 38.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]