

Abstract
Proper visualization of scientific data is important for understanding spatial relationships. Particularly in the field of structural biology, where researchers seek to gain an understanding of the structure and function of biological macromolecules, it is important to have access to visualization programs which are fast, flexible, and customizable. We present KiNG, a Java program for visualizing scientific data, with a focus on macromolecular visualization. KiNG uses the kinemage graphics format, which is tuned for macromolecular structures, but is also ideal for many other kinds of spatially embedded information. KiNG is written in cross-platform, open-source Java code, and can be extended by end users through simple or elaborate “plug-in” modules. Here, we present three such applications of KiNG to problems in structural biology (protein backbone rebuilding), bioinformatics of high-dimensional data (e.g., protein sidechain chi angles), and classroom education (molecular illustration). KiNG is a mature platform for rapidly creating and capitalizing on scientific visualizations. As a research tool, it is invaluable as a test bed for new methods of visualizing scientific data and information. It is also a powerful presentation tool, whether for structure browsing, teaching, direct 3D display on the web, or as a method for creating pictures and videos for publications. KiNG is freely available for download at http://kinemage.biochem.duke.edu.
Keywords: interactive molecular graphics, kinemage graphics, backrub motion, all-atom contact dots, Molikin, Mage, Java
Introduction
In the field of structural biology, where researchers seek to gain an understanding of the structure and function of biological macromolecules, it is important to have access to visualization programs, which are fast, flexible, and customizable. Good visualizations give researchers a more intuitive feel for how molecules behave. However, it is also important for such programs to be able to show other types of information simultaneously with the structural representation, such as 3D annotations of errors in the models or important interaction sites.
KiNG (Kinemage, Next Generation) is a Java-based, modular, easily extensible scientific visualization tool used primarily for macromolecular visualization. Like other general purpose molecular viewers such as PyMOL, SwissPdbViewer, Chimera, RasMol, and JMol,1–5 KiNG allows for real-time, interactive 3D rotation, translation, cropping, and zooming, which are critical for proper sensation of depth and for perceiving spatial relationships accurately. However, KiNG separates itself from these other programs in a number of significant ways, specifically in its molecule-agnostic kinemage graphics format, the quality of its color palette and depth cueing, and in the tools and features it offers.
KiNG builds on the three decades of successful molecular graphics experience embodied in the original protein ribbon drawings6 and in the first kinemage graphics program, Mage.7 Mage was designed as a tool for molecular illustration (with the selectivity and artistry that connotes) for journal articles and classroom settings. However, we found that Mage quickly became an integral part of the lab's research program due to its tremendous flexibility. Reimplementing the core kinemage functionality from the ground up in KiNG has enabled a similar-looking but modern user interface, a cleaner internal data structure, and greater maintainability and extensibility. Not surprisingly, KiNG has also become a central component of the lab's work.
However, instead of superseding Mage, KiNG and Mage currently coexist as separate development paths. In fact, having two different kinemage viewers in active development has encouraged improvements in both programs. For example, the high-dimensional visualization capabilities (see “Results and Discussion”) were actually, at first, independently added to both KiNG and Mage for different purposes. Then, by collaborating and combining the strengths of each separate implementation, we were able to develop functionality that was ultimately more general-purpose and more powerful.
KiNG and Mage's flexibility comes from separating the molecular details (e.g., PDB file) from the graphical representation (kinemage).7 This strategy allows traditional macromolecular structural data (models, ribbons, electron density, NMR data) to coexist naturally with secondary “annotations” of that data (e.g., helix axes, local validation outliers, interface contact dots); it also permits wholly nonmolecular visualizations using the same tool.
The kinemage format is extensively documented and numerous examples are available at http://kinemage.biochem.duke.edu (see also “Materials and Methods”). Kinemage format is plain text, designed to be both easy to edit by hand and also easily generated by programs.
When kinemage format alone is not flexible enough, KiNG itself can be modified. Although the graphics engine could be reused in a totally new program, more often new capabilities are added with plug-in modules that are written in Java and dynamically loaded at run time. The protein rebuilding and molecular illustration capabilities in this article were developed as plug-ins, while the facilities for high-dimensional analysis required modification of the core program. New modules can be written quickly and easily to extend functionality, giving KiNG very open-ended potential.
Results and Discussion
KiNG is freely available to all users, multiplatform, and open source. In addition to having an extensive outside user base, KiNG has gradually become one of the most important research tools within our laboratory. Because of its modular nature, it has become a test bed for a variety of new tools and visualization techniques, enabling the discovery of many new research results. Several of these tools, techniques, and results are described here.
Conventional use
A key goal in the development of KiNG (and originally, in Mage) was to make basic navigation and user interaction as simple and intuitive as possible. To that end, in KiNG, most of the basic navigation tasks can be controlled by the mouse. Rotations, zooming, and clipping are all controlled with click and drag mouse actions. Clicking on a point displays associated information about the point in the graphics window. Clicking on two successive points displays the distance between them, an essential tool in structural biology visualization for determining interatomic distances and possible interactions. Different aspects of the kinemage display hierarchy can be turned on or off on the fly using the check boxes located along the side of the main graphics window. The main menus enable access to more advanced features of KiNG. For instance, important views can be saved and returned to instantly. More advanced measuring tools can be activated for measuring angles and dihedrals. There is a whole suite of tools for editing kinemages directly in KiNG, such as for changing colors of points or lines, deleting objects, editing coordinates, or adding additional graphical elements. Some of the more noteworthy tools and features of KiNG are described in more detail below.
Applet mode
KiNG includes an applet mode, which allows it to be run through web browsers, without users having to download KiNG separately. This functionality has been critical in our structural validation website, MolProbity,8 for display of errors within macromolecular structures. Feedback from MolProbity users has helped greatly to ensure that KiNG keeps functioning smoothly for multiple subjects and viewing styles. Also, the applet feature has allowed KiNG to be used directly on the website of the Protein Data Bank (PDB)9 repository of structures. Previously, separate kinemages had to be generated for use with KiNG on the PDB website. This led to significant maintenance difficulties, since whenever PDB files were updated, the kinemage files would also have to be updated. This issue has been resolved with the latest version of KiNG on the PDB website, as it can now use our macromolecular kinemage generation software Molikin (see below) directly to generate kinemages on the fly when a user wants to display a structure. We tested KiNG against the entire PDB (as of March 2009) and it successfully generated a valid kinemage for every file. Also, KiNG truncates input files if it determines it does not have enough memory allocated, which allows the typically memory-limited KiNG applet to still successfully generate some kind of visualization, even for the largest structures. A king in browser tutorial package on using KiNG as an applet is available for download at http://kinemage.biochem.duke.edu.
Protein backbone rebuilding
Although kinemages have been used to display everything from the flight paths of Russian MIGs to the social networks in an Antarctic research station, we use them primarily for viewing biological macromolecular structures. Also, we often want to modify the starting structure—for instance, rebuilding to make it fit the experimental data better (e.g., electron density, which can be displayed from most map file formats). Although many tools exist for rebuilding protein structures,10,11 we found KiNG to be an excellent platform for testing out some new ideas we recently developed about protein backbone.
Fitting backbone is difficult because the region of interest is generally constrained at both ends by the rest of the chain, and any local change may also disrupt perfectly good regions nearby. However, we have recently documented the “backrub” motion, a very local pattern of nondisruptive conformational change in protein backbone that frequently occurs as alternate conformations in native protein structures.12 Simulating backrub motions as a rebuilding tool has proven very successful for correcting the kinds of backbone distortion introduced by fitting a sidechain backwards into the density, which is a common mistake. Currently, KiNG is the only software package that offers backrub motions to correct these kinds of mistakes. The use of KiNG for backrub modeling, and interactive display of all-atom contacts and sidechain rotamer quality, was essential to successful improvement in accuracy for 29 high-throughput crystal structures by the SouthEast Structural Genomics Consortium.13
Figure 1 shows the backrub fitting tool in action in the KiNG graphical user interface. Although the core of KiNG knows nothing about molecules, the backrub plug-in reads PDB or mmCIF files and correlates the atom positions with the kinemage graphics. It updates the graphics as the model is being refit, and then writes a new model file at the end. Because the kinemage format acts as a graphics lingua franca for communicating to the user, KiNG can incorporate additional feedback from external programs into the rebuilding process (in nearly real time). For instance, if hydrogens have been added to a structure, our program Probe15,16 can provide extremely helpful live updates on all-atom steric contacts and collisions in the proposed model as backrub fitting is being performed. A tutorial for using the backrub fitting tool, along with Probe, is available at http://kinemage. biochem.duke.edu, under teaching/MolProbity on the navigation bar.
Figure 1.
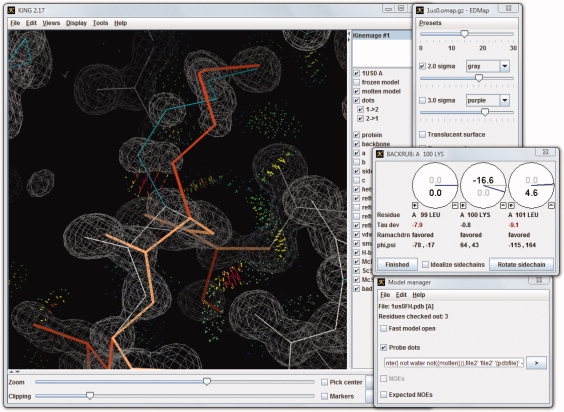
KiNG graphical user interface. An image of the KiNG graphical user interface, showing the Backrub tool being used to reconstruct an alternate conformation of a lysine residue in a protein structure (PDB: 1US0).14 Contact dots from Probe are shown (as dots and small spikes). The electron density is shown in gray wire-mesh, the original model in white and cyan, and the active model in orange. Pop-up windows for controlling aspects of the backrub motion, the electron density, and the contact dots are also visible.
High-dimensional data
For several recent projects, we have found ourselves needing to plot data points in high-dimensional spaces. One example is analyzing the conformations of protein loops, where it takes seven parameters to completely define the spatial relationship between the ends. Another is clustering the conformations of RNA, where (coincidentally) seven dihedral angles define the backbone conformation between two bases.17 For the RNA analysis, we originally used different sets of three angles to make many different 3-D plots. However, points that cluster in one plot do not necessarily cluster in the others, and correlating points from one plot to another is tedious and error-prone.
To facilitate this type of high-dimensional “info vis” use of kinemages, we added two new features both to KiNG and Mage. The first is support for high-dimensional points: rather than just XYZ, each point may have an arbitrary number of coordinates associated with it. KiNG then provides tools for quickly switching among different 3-D projections of the data, that is, three of the coordinates are mapped to XYZ and the rest are temporarily ignored. By assigning a distinctive color to an apparent cluster of points in one projection and then switching to another projection, it becomes relatively easier to disambiguate the true 7-D clusters. In the RNA case, it allowed us to resolve several distinct conformations that we had previously lumped together.
The second new high-dimensional feature in KiNG and Mage is plotting data points in parallel coordinates.18 Rather than the orthogonal arrangement of Cartesian coordinate axes, parallel coordinate displays place all of the coordinate axes side by side and parallel to one another. As such, data points become polylines that pass through the appropriate values on each axis. In theory, this allows for simultaneous viewing of an unlimited number of dimensions, although even a moderate number of data points can produce a visual clutter of criss-crossing lines that is hard to decipher.
Figure 2 shows an example of the power of using high-dimensional plotting combined with parallel coordinates, with three different representations of related rotamer data, all shown in KiNG. Recently, using a much larger data set, we have been updating our previous protein sidechain rotamer libraries.19 The conformation of the sidechain of arginine is defined by four dihedral (chi) angles. In Figure 2, only the data points with chi1 close to 180° are shown. In the traditional three-dimensional view of the data points, clusters of points are evident, but in the parallel coordinate view, the clustering of the points (as polylines) becomes even more obvious. Using KiNG's button panel control, each proposed individual cluster can be examined individually in the parallel coordinate representation, to ensure that no split in any dimension is overlooked. When transparency is enabled on the data [compare Fig. 2(D) with Fig. 2(C)], all of the major clusters can be seen even in a complex background.
Figure 2.
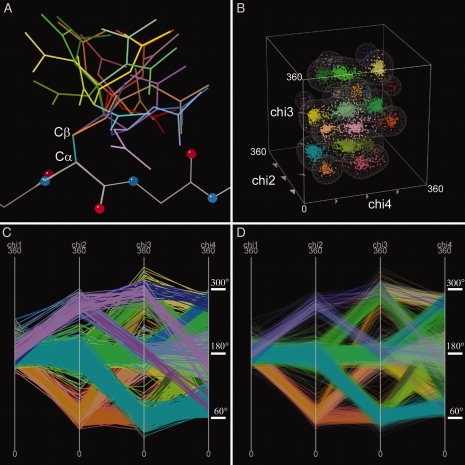
Information visualization capabilities of KiNG. Figures illustrating different types of representations in KiNG of the same data: all 21 rotamers of Arg with chi1 trans, from an updated version (unpublished) of our protein sidechain rotamer library.19 Two custom green colors were defined for this figure (see “Materials and Methods”). This figure is also available in interactive kinemage format, in Supporting Information. Panel A shows stick representations of different arginine rotamers with their backbone atoms superimposed. Backbone is colored in white, with the different sidechain rotamers in different colors. Balls are used to indicate nitrogen and oxygen backbone atoms. Text labels were added using Adobe Photoshop. Panel B shows a 3D plot of the chi2 through chi4 dihedral angles of the arginine rotamers shown in A, with identical coloring. The data points are represented by balls. Contours were drawn using kin3Dcont.20 Axis text labels were added using Adobe Photoshop. Panels C and D show the parallel coordinate representation of chi1 through chi4 of the arginine rotamers, where each polyline spanning the four axes represents one data point. Panel D shows the same data as Panel C, but with transparency increased. The tick marks and degree indicators were added using Adobe Photoshop.
The projections and parallel coordinates have complementary strengths and are most effective when used together. The 3-D projections are better for initial selection, coloring, and clustering of data, but the parallel coordinates display provides an important check that putative clusters really stay together in all dimensions. KiNG switches between the two modes with a single keystroke, and markers and point colors are shared between the two, making it easy to move back and forth while working. These capabilities of KiNG were central to the collaborative work that produced the RNA Ontology Consortium's nomenclature and conformer library for RNA backbone.21
Molecular illustration
In our teaching, we ask students to choose a protein or protein family and use kinemages to illustrate its important structural or functional features.22 Creating kinemages leads to a deeper understanding than merely examining a 3-D display, just as writing about a subject requires more knowledge than reading. Of course, making kinemage models from macromolecular coordinates is also a basic requirement for our research. Thus, any tool for converting PDB or mmCIF files to kinemages must be both easy to use and very flexible—properties often at odds with each other.
Our solution, called Molikin (see “Materials and Methods”), solves this problem by offering two modes of operation: a “quick kin” mode for rapidly generating commonly used types of visualizations of macromolecular structures, and an advanced mode that provides more control of the visualizations which are produced, down to a residue level of control if desired. Molikin is included as a plugin with the KiNG download, accessible under the “Import” option in the File menu. This allows users to read PDB files directly into KiNG and view a kinemage display almost instantly.
As mentioned previously, the kinemage format itself provides a great deal of flexibility. In addition to all the built-in kinemage editing and drawing tools in KiNG, the hand-editable nature of the format allows users to easily write enhancements to kinemages. These features mean that KiNG and the kinemage format are particularly well-suited for students creating educational projects in our classes; they can start their project with only a few decisions and gradually refine their ideas over time without starting over. Also, researchers can use KiNG and kinemages to quickly prototype new types of visualizations. For example, we have various modules and programs that produce kinemages of electron density for crystallography, NOE (Nuclear Overhauser Effect) or RDC (Residual Dipolar Coupling) restraints for NMR,23 and all-atom steric contacts.15 These kinemages are all combined seamlessly in KiNG, and so there is no need to reimplement these algorithms in one monolithic molecular graphics program.
KiNG is able to export high-resolution 2D views in a number of widely supported picture formats, including JPEG, PNG, and PDF, as well as by direct screen capture such as the ribbon diagram in Figure 3. It can also export files for use with the rendering software Raster3d25 or POV-Ray26 for generating higher quality images. It also has a movie-maker function, which makes it easy for users to create movies for use in presentations. Naturally, KiNG itself can be used during a presentation, for a live demonstration to enable even better 3D perception by the audience.
Figure 3.
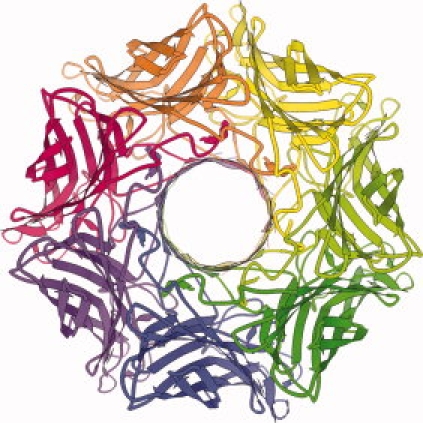
An example of ribbon graphics in KiNG. Screen-captured 2D image of a ribbon diagram of staphylococcal alpha-hemolysin, a heptameric transmembrane pore (PDB: 7AHL),24 viewed down the pore. This figure is also available in interactive kinemage format, in Supporting Information.
Conclusion
KiNG is a powerful and flexible scientific visualization software package, which has evolved into more than just a visualizer; it is an important research tool, which, due to its existing capabilities and ability to be extended, will continue to shed new light on macromolecular structures.
Materials and Methods
Kinemage format
A kinemage is a plain-text file containing both metadata (title, author, data source, and potentially extensive explanatory text) and a set of hierarchically grouped display lists of primitives such as lines, dots, balls, triangles, and labels; this structure is closely mirrored by a hierarchy of objects within KiNG's source code. Briefly, kinemage files contain two types of lines: keyword lines and point lines. Keyword lines begin with keywords, which start with the “@” symbol and define all metadata info (e.g., @text, @dimensions) and the groups, lists, and other kinemage properties (e.g., @kinemage, @group, @master). Point lines, in their simplest form, contain numbers, separated by whitespace. KiNG will keep track of the number of coordinates it has encountered, making each set of three coordinates a new point (or n coordinates when the list property “dimension = n” is used). This feature allows users to enter data into KiNG and see what it looks like very quickly. Coordinates can also be preceded by point identifier information in curly braces ({}), which is displayed in the graphics window when the point is left-clicked on. All the keywords as well as the different types of points have a wide variety of options, which are described in the “format-kinemage.pdf” document available at http://kinemage.biochem.duke.edu.
In addition to all the documented kinemage features supported by KiNG, when KiNG encounters a keyword it does not recognize, it will enter a note in the Error log (accessed through the Help menu). This mainly occurs in some of the older hand-created teaching kinemages, which contain lesser-known features that are only recognized in Mage. Conversely, Mage does not support some of the newer kinemage features introduced in KiNG.
Internal palette
Like Mage, KiNG's internal color palette consists of a set of 25 named colors. The colors are depth-cued appropriately, depending on whether they are being viewed on a black or white background. KiNG includes an “Internal palatte” kinemage (Help menu → Built-in kinemages), which allows users to see the standard colors, along with the depth-cueing. When the named colors do not provide enough options, KiNG supports a new kinemage keyword (@hsvcolor), which allows users to define custom hue/saturation/value colors, also which are also depth-cued appropriately.
Implementation
KiNG is implemented in Java, using features added in Java 1.5. The graphical user interface is implemented using the Java Swing and AWT libraries. The KiNG engine handles the viewing calculations, but it has the option of using the Java Bindings for OpenGL (JOGL) for improved graphics rendering. Using JOGL is not a requirement for running KiNG, however, meaning users with the basic Java installation, as well as web applet users, can still successfully use KiNG.
Molikin
Molikin is also implemented in Java, using features in Java 1.5. Molikin enjoys a highly modular design: just 5% of the code interfaces with KiNG, and 95% focuses on making kinemages. This made it easy to create a version of Molikin that stands alone from the KiNG plugin version, using a command-line interface. This allows scriptable control of Molikin, especially powerful for generating a large number of visualizations. Molikin has a number of different options for generating “raw materials”; these take the form of (ball and) stick renderings, space-filling models, and ribbon drawings. Other options include mainchain versus sidechains; protein versus nucleic acids versus water; which models, chains, and residues to include, and so forth, which are accessible in the plugin GUI shown when Molikin is accessed in KiNG.
Tools description
Almost all the built-in tools in KiNG are accessed under the Tools menu. Some are directly accessible from the Tools menu, and others from a number of sub-menus: Kin editing, Structural biology, and Specialty. Users can adjust the layout of the Tools menu, even creating new submenus or removing tools, by using the “Customize tools menu…” option. The Edit/draw/delete tool, as well as options for showing coordinates and measuring angles and dihedrals, is in the main menu. Kin editing includes other useful tools for editing kinemages, including superposition tools, a molecular-specific coloring tool, a tool for adjusting distances, angles, and dihedrals of points, and the aforementioned movie maker tool. Structural biology contains all the protein rebuilding tools, including the Backrub tool and sidechain mutation/rotation tools. Specialty mainly contains tools which are unsuited for general users, being either highly specific to our own research, or beta tested.
Developers can create either a Plugin, which adds new menu items or dialogs, or a Tool, which can also alter the effects of mouse actions. Multiple Plugins can run at the same time, but only one Tool can be active at a time. Creating a new tool or plugin is straightforward; developers interested in developing new KiNG functionality only have to subclass either Plugin or BasicTool, add new functionality with Java methods, create a configuration file listing the new tool(s) and/or plugin(s), and package it in a Java JAR file. After adding the new JAR package to the Java classpath or the “plugins” directory in KiNG, KiNG will load the tools and plugins from the JAR file; tools and plugins are discovered and loaded dynamically when KiNG is run, and so users do not have to change any code within KiNG itself to use new tools. A more thorough explanation of how to create tools or plugins is included in the “hacking-king.pdf” document in the KiNG download, and a sample set of simple plugins (with source code) is available on the KiNG software webpage.
Acknowledgments
We thank present and former members of the Richardson lab, specifically Bryan Arendall, Jeremy Block, Jeff Headd, Daniel Keedy, Gary Kapral, Laura Murray, Jane Richardson, Lizbeth Videau, Christopher Williams, and Bob Immormino, who have provided much feedback and suggestions during the course of KiNG's development. We also thank Stuart Endo-Streeter, a long-time user of KiNG. A particularly useful plugin, NOEdisplay, was contributed by Brian Coggins.
References
- 1.DeLano WL. 2002. http://www.pymol.org) The PyMOL molecular graphics system on world wide web.
- 2.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 3.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera—a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 4.Sayle R. 1992. RasMol: a program for fast realistic rendering of molecular structures with shadows. Proceedings of the 10th Eurographics UK '92 Conference. York, UK, University of Edinburgh, Scotland: Abingdon Press.
- 5. Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/
- 6.Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 7.Richardson DC, Richardson JS. The kinemage: a tool for scientific illustration. Protein Sci. 1992;1:3–9. doi: 10.1002/pro.5560010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucl Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 11.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 12.Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Arendall WB, III, Tempel W, Richardson JS, Zhou W, Wang S, Davis IW, Liu Z-J, Rose JP, Carson WM, Luo M, Richardson DC, Wang BC. A test of enhancing model accuracy in high-throughput crystallography. J Struct Funct Gen. 2005;6:1–11. doi: 10.1007/s10969-005-3138-4. [DOI] [PubMed] [Google Scholar]
- 14.Howard EI, Sanishvili R, Cachau RE, Mitschler A, Chevrier B, Barth P, Lamour V, Van Zandt M, Sibley E, Bon C, Moras D, Schneider TR, Jochiamiak A, Podjarny A. Ultrahigh resolution drug design I: details of interactions in human aldose reductase-inhibitor complex at 0.66 Angstroms. Proteins Struct Funct Bioinform. 2004;55:792–804. doi: 10.1002/prot.20015. [DOI] [PubMed] [Google Scholar]
- 15.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogens. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 16.Word JM, Bateman RC, Jr, Presley BK, Lovell SC, Richardson DC. Exploring steric constraints on protein mutations using Mage/Probe. Protein Sci. 2000;9:2251–2259. doi: 10.1110/ps.9.11.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray LJW, Arendall WB, III, Richardson DC, Richardson JS. RNA backbone is rotameric. Proc Nat Acad Sci USA. 2003;100:13904–13909. doi: 10.1073/pnas.1835769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inselberg A. The plane with parallel coordinates. Vis Comput. 1985;1:69–91. [Google Scholar]
- 19.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 20.Word JM. 2000. All-atom small-probe contact surface analysis: an information-rich description of molecular goodness-of-fit, Dissertation, Duke University, Durham, NC.
- 21.Richardson JS, Schneider B, Murray LW, Kapral GJ, Immormino RM, Headd JJ, Richardson DC, Ham D, Hershkovits E, Williams LD, Keating KS, Pyle AM, Micallef D, Westbrook J, Berman HM, RNA Oncology Consortium RNA backbone: consensus all-angle conformers and modular string nomenclature (an RNA Ontology Consortium contribution) RNA. 2008;14:465–481. doi: 10.1261/rna.657708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson DC, Richardson JS. Teaching molecular 3-D literacy. Biochem Mol Biol Educ. 2002;30:21–26. [Google Scholar]
- 23.Block J, Zielinski D, Chen V, Davis I, Vinson EC, Brady R, Richardson J, Richardson D. KinImmerse: macromolecular VR for NMR ensembles. Source Code Biol Med. 2009;4:3. doi: 10.1186/1751-0473-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 25.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 26.Persistance of Vision (TM) Raytracer. 2004. http://www.povray.org/