

Abstract
The structure of AF2331, a 11-kDa orphan protein of unknown function from Archaeoglobus fulgidus, was solved by Se-Met MAD to 2.4 Å resolution. The structure consists of an α + β fold formed by an unusual homodimer, where the two core β-sheets are interdigitated, containing strands alternating from both subunits. The decrease in solvent-accessible surface area upon dimerization is unusually large (3960 Å2) for a protein of its size. The percentage of the total surface area buried in the interface (41.1%) is one of the largest observed in a nonredundant set of homodimers in the PDB and is above the mean for nearly all other types of homo-oligomers. AF2331 has no sequence homologs, and no structure similar to AF2331 could be found in the PDB using the CE, TM-align, DALI, or SSM packages. The protein has been identified in Pfam 23.0 as the archetype of a new superfamily and is topologically dissimilar to all other proteins with the “3-Layer (BBA) Sandwich” fold in CATH. Therefore, we propose that AF2331 forms a novel α + β fold. AF2331 contains multiple negatively charged surface clusters and is located on the same operon as the basic protein AF2330. We hypothesize that AF2331 and AF2330 may form a charge-stabilized complex in vivo, though the role of the negatively charged surface clusters is not clear.
Keywords: new type of α + β fold, orphan protein, homo-oligomers, dimerization, Archaeoglobus fulgidus
Introduction
Archaeoglobus fulgidus is a sulfur-metabolizing organism, which grows at extremely high temperatures (between 60 and 95°C), and is evolutionarily unrelated to other sulfate reducers.1 The protein encoded by the AF2331 gene from A. fulgidus strain DSM 4304 has no functional annotation. Here, we report the crystal structure of AF2331 (PDB code 2FDO) determined by the multiwavelength anomalous dispersion (MAD) technique2,3 at 2.4 Å resolution. The crystal structure of AF2331 exhibits an unusual interdigitated interaction between subunits, forming a homodimer that represents a new type of fold.
AF2331 is not significantly similar by sequence to any other known protein, as determined by a sequence search of the May 2009 edition of the UniProt (Swiss-Prot + TrEMBL) Knowledgebase.4 The STRING server5–8 (http://string.embl.de/) was used to obtain functionally relevant protein interactions. We obtained one “neighborhood” protein–protein interaction: AF2330, which is located on the same operon in A. fulgidus, has a sequence similar to other proteins from Archaea, but all of the similar proteins are described as hypothetical.
Results and Discussion
Homodimer structure
The molecule of AF2331 folds into an α + β structure (Fig. 1), which consists of five β-strands, β1 (residues 2–7), β2 (residues 24–42), β3 (residues 34–42), β4 (residues 47–58), and β5 (residues 71–80), and three α helices, α1 (residues 7–19), α2 (residues 64–70) and α3 (residues 88–91). In the crystal structure, AF2331 forms an unusual interdigitated homodimer (Fig. 2), composed of chains A and B, where the subunits are related by two-fold noncrystallographic symmetry (NCS). In the homodimer, antiparallel β-sheets are formed, both containing strands from both chains. The first β-sheet is interdigitated, composed of strands β1, β2, and β5 from both chains, with the arrangement β1A-β2B-β5A-β5B-β2A-β1B (where “β1A” represents strand β1 of chain A, for example). The second β-sheet is composed of strands β3 and β4 from both chains with the arrangement β3B-β4B-β4A-β3A. These two antiparallel β-sheets pack together, forming a hydrophobic core. All three α-helices from each subunit are packed around the core composed of the two antiparallel β-sheets.
Figure 1.
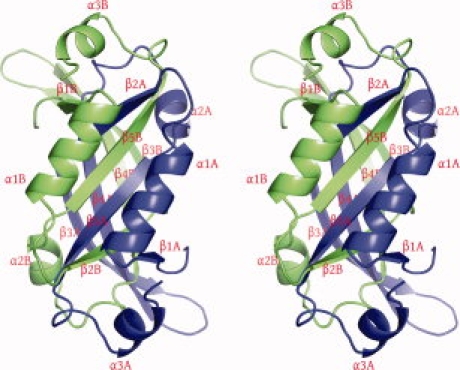
Stereo view of ribbon representation showing the distribution of structure elements in AF2331 dimer. Chains A and B are colored in blue and green, respectively. The figure was prepared with PyMOL.9
Figure 2.
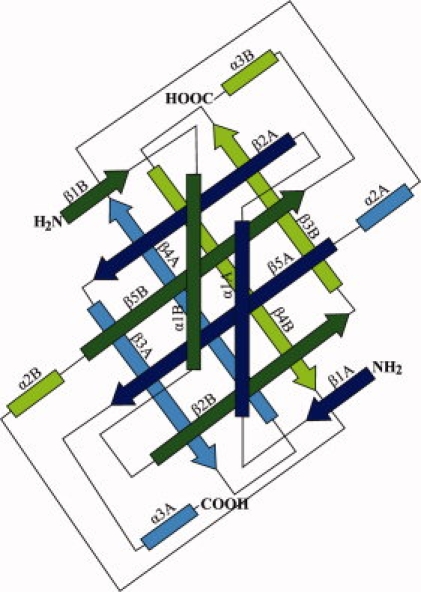
Topological diagram of AF2331, where β-strands are represented by arrows and α-helices by rectangles. The chains are colored green and blue as in Figure 1. The β strands in dark green and blue and the β strands in light green and blue correspond to the front and back β-sheets in Figure 1, respectively.
The pattern of strands in the first β-sheet in the protein can be described as ABABAB (where A and B correspond to two different polypeptides). Such an extensive pattern of interdigitation in a β-sheet is rarely seen in the PDB. β-Sheets with a single interdigitated strand (strand pattern ABA) occur quite frequently in the PDB. β-Sheets with an ABAB pattern can be found in at least 100 protein structures. However, only three nonredundant proteins with β-sheets contain five interdigitated strands (PDB codes 1K3P, 1Q7L, and 1DGR), and only one other protein besides AF2331 contains an ABABAB β-sheet (PDB code: 2HJ1).
Dimerization interface surface area
Based on calculations by the Protein Quaternary Structure (PQS)10 server, the solvent-accessible surface area (ASA) for each monomer in the homodimer is 3960 Å2 less than the ASA of the isolated monomer. This decrease in the ASA upon dimerization is a reasonable estimate of the surface area of one monomer involved in forming the dimerization contact. We subsequently refer to the decrease in ASA upon dimerization as the “dimerization surface area.” The dimerization surface area of a monomer of AF2331 is 41.1% of the total surface area of the monomer (total ASA; see “Materials and Methods”). It should be noted that the use of interface and total ASA values for a monomer in a homo-oligomer is a technical conceit. While the solvation free energy of dimerization for AF2331 was not measured experimentally, the calculated change in solvation free energy of folding upon dimerization for AF2331 is estimated to be −74.4 kcal/mol. Given the probably very high free energy of dimerization for AF2331, it seems very unlikely that a monomer of AF2331 would be found in solution in a folded form.
To compare this dimerization surface area to other homodimer structures in the PDB, we used dimerization surface areas calculated by the PQS for a set of 4710 nonredundant homodimer structures. The dimerization surface areas are plotted as a function of molecular weight on Figure 3(A). The least-squares regression line (ΔASA = 0.055 × MW, R2 = 0.72) is very similar to the regression line calculated by Jones and Thornton11 for a much smaller hand-curated set of 32 homodimers (ΔASA = 0.06 × MW, R2 = 0.48). The dimerization surface areas as a function of total surface area (total ASA) are plotted on Figure 3(B), along with the regression line (R2 = 0.78). In both cases, the data point representing AF2331 lay far above the corresponding regression line.
Figure 3.
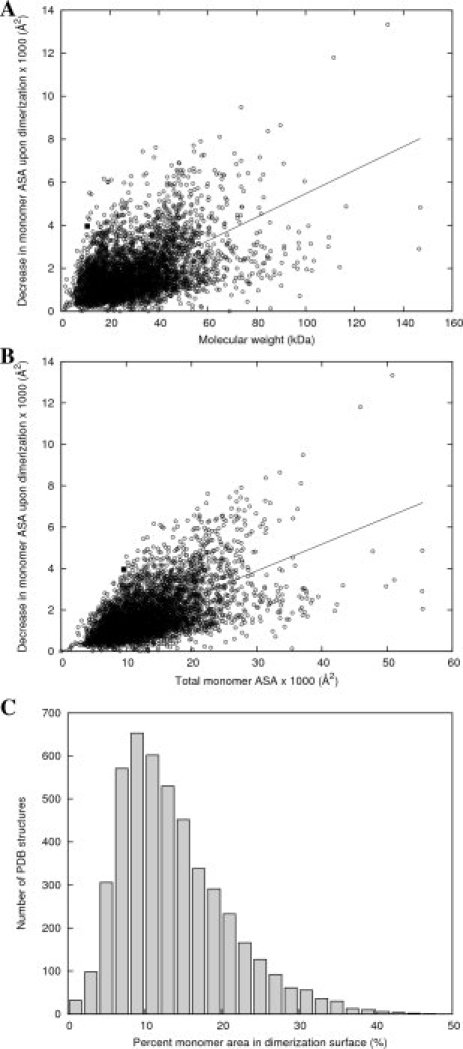
Correlation of the area of the dimerization surface as a function of molecular weight or total surface area for a nonredundant set of 4710 proteins predicted to form homodimers by the Protein Quaternary Server. (A) Area of the dimerization interface surface for each homodimer, plotted as a function of the molecular weight of one monomer. The solid line is the linear least-squares regression fit, and AF2331 is shown as a filled square. (B) Area of the dimerization interface surface for each homodimer, plotted as a function of the total ASA for one monomer. The solid line is the linear least-squares regression fit, and AF2331 is shown as a filled square. (C) Histogram of percentages of monomer ASA in the interface surface for the nonredundant set of homodimers. 41.1% of the total surface area of a monomer of AF2331 is contained within its dimerization surface.
Figure 3(C) shows a histogram of the percentage of monomer ASA in the dimerization surface for each dimer in the nonredundant set. The mean for this distribution is 13.8% (and median 12.3%) of total monomer surface area. AF2331, with a dimerization surface area of 41.1% of the total monomer ASA, is above the 99th percentile. In fact, AF2331 has the 10th highest percentage out of the 4710 structures in the set (Table I).
Table I.
The 20 Homodimers with the Highest Percentage of Monomer Surface Area Involved in the Dimerization Surface in the Set of Nonredundant PDB Structures
| Rank | PDB ID | Interface surface area (Å2) | Percent total surface area (%) | Protein | Organism |
|---|---|---|---|---|---|
| 1 | 1W5U | 1490 | 47.0 | Gramicidin D | Bacillus brevis |
| 2 | 1U42 | 4583 | 45.9 | Mutant of dimerization domain of NF-kB p50 factor | Mus musculus |
| 3 | 1W7R | 1165 | 44.4 | Feglymycin | Streptomyces |
| 4 | 2RH0 | 6167 | 43.3 | NudC domain-containing protein 2 | Mus musculus |
| 5 | 1ZK9 | 5436 | 43.1 | Dimerization domain of NF-kB RelB | Mus musculus |
| 6 | 3DIE | 4290 | 42.0 | Thioredoxin W28A mutant | Staphylococcus aureus |
| 7 | 3FJ5 | 2814 | 42.0 | C-src-SH3 domain | Gallus gallus |
| 8 | 2B1Y | 5499 | 41.6 | ATU1913 | Agrobacterium tumefaciens str. C58 |
| 9 | 2I8D | 4850 | 41.2 | COG5646 | Lactobacillus casei |
| 10 | 2FDO | 3960 | 41.1 | AF2331 | Archaeoglobus fulgidus DSM 4304 |
| 11 | 1G8E | 3302 | 40.9 | Flagellar transcriptional regulator FlhD | Escherichia coli |
| 12 | 2DLB | 3270 | 40.6 | YopT | Bacillus subtilis |
| 13 | 2OPL | 6110 | 40.1 | OsmC-like protein | Geobacter sulfurreducens |
| 14 | 1WKQ | 4129 | 39.8 | Guanidine deaminase | Bacillus subtilis |
| 15 | 1MYK | 1786 | 39.5 | Arc repressor mutant PL8 | Enterobacteria phage p22 |
| 16 | 1K9U | 2542 | 39.2 | Pollen allergen Phl p 7 | Phleum pratense |
| 17 | 2OTA | 2768 | 39.1 | UPF0352 protein CPS_2611 | Colwellia psycherythraea 34h |
| 18 | 3GXZ | 4373 | 39.0 | Cyanovirin-n | Nostoc ellipsosporum |
| 19 | 2RFP | 5999 | 38.7 | Putative NTP pyrophosphohydrolase | Exiguobacterium sibiricum |
| 20 | 1WZ3 | 3570 | 38.7 | Plant ATG12 | Arabdopsis thaliana |
Among 20 homodimer structures with the highest percentages of monomer ASA in the dimerization interface, those that form interdigitated or intertwined β-sheets are overrepresented (for example, 1U42, 2RH0, 1ZK9, 3DIE, 3FJ5, 2FDO, 2DLB, and 1WZ3). The intertwined β-sheets in the dimer of the NK-κB transcription factor RelBDD (1ZK9) were confirmed in vitro by crosslinking experiments.12 By differential scanning calorimetry, the intertwined dimer of thioredoxin W28A was shown to be a kinetically trapped state for the protein rather than a thermodynamically stable one.13 However, none of these dimer structures appear to have a fold similar to that of AF2331.
Other patterns of interdigitation are observed in some of the other homodimers shown in Table I. PDB codes 1W5U and 1W7R are structures of the small natural antibiotics gramicidin D14 and feglymycin,15 respectively, which are peptides that form pores in bacterial membranes via a novel double-helical structure with β-strand-like hydrogen bonding patterns.16 Several structures in Table I have interlocking α-helices rather than β-strands, such as 1G8E, 1K9U, 2OTA, and 2RFP.
Many of these structures exhibit 3D domain swapping,17–19 including those with PDB codes 1U42, 2RH0, 3DIE, 3FJ5, 1WKQ, 1K9U, 3GXZ, and 1WZ3. In some cases, the domain swap is triggered by a mutation, such as the MLAM mutant of the dimerization domain on NF-kB p5020 or the W28A mutant of thioredoxin.13 Formation of the domain-swapped dimer of c-Src tyrosine kinase SH3 domain appears to be induced by addition of polyethylene glycol 300 to the protein solution.21
We also compared the trends in the dimerization surface area for homodimers to the oligomerization surface areas for nonredundant sets of homo-trimers, -tetramers, and so forth. The statistics for these distributions are summarized in Table II. (It should be noted that the “oligomerization surface area” was calculated identically as for the dimerization surface area, by measuring the decrease in ASA upon oligomerization. Thus, the oligomerization surface area represents the surface area of a monomer involved in binding to all of the other monomers, not just the binding area between two monomers.) The means and medians of the percentage of total surface area increase as a function of the oligomeric state of the homo-oligomers (Table II), though the distributions for some of the oligomers have too few structures for statistical significance. The mean percent monomer ASA involved in the oligomerization surface for n = 2, 3, 4, and 6 agrees with similar data measured for a smaller set of homo-oligomers by Postingl et al.22 This is to be expected, as in general a monomer must bind a greater number of other monomers in a homo-oligomer as the oligomeric state increases. The percentage of surface area involved in oligomerization for AF2331 is above the mean for every oligomeric state save for 11-mers.
Table II.
Statistics for the Distributions of Percentage of Oligomerization Surface Area as a Function of Total Surface Area
| Number of subunits (n) | Mean percentage of total ASA (%) | Median percentage of total ASA (%) | Standard deviation (%) | Number of structures |
|---|---|---|---|---|
| 2 | 13.8 | 12.3 | 7.1 | 4710 |
| 3 | 21.5 | 19.8 | 10.9 | 610 |
| 4 | 23.0 | 22.1 | 8.8 | 1515 |
| 5 | 28.8 | 29.2 | 10.5 | 55 |
| 6 | 25.5 | 24.3 | 9.1 | 485 |
| 7 | 27.5 | 28.4 | 10.9 | 19 |
| 8 | 27.2 | 26.1 | 9.6 | 198 |
| 9 | 29.4 | 33.9 | 16.7 | 4 |
| 10 | 31.2 | 29.8 | 10.8 | 47 |
| 11 | 46.5 | 48.2 | 3.4 | 3 |
| 12 | 33.2 | 32.0 | 10.2 | 103 |
| 13 | 23.9 | 23.2 | 3.8 | 3 |
| 14 | 31.7 | 30.5 | 9.6 | 14 |
| 16 | 26.8 | 25.1 | 13.4 | 14 |
| 18 | 30.9 | 30.9 | — | 1 |
| 20 | 36.9 | 36.9 | — | 1 |
| 24 | 35.3 | 36.0 | 6.1 | 37 |
| 32 | 29.4 | 29.4 | — | 1 |
| 48 | 30.4 | 30.4 | — | 1 |
| 60 | 33.7 | 42.9 | 20.1 | 6 |
The large dimerization interface area for AF2331 implies that there is a significant change of solvation free energy upon dimerization, which indicates that the protein–protein interfaces in the assembly have a strong hydrophobic character. All four β-strands that form the core β-sheets are almost exclusively composed of hydrophobic or nonpolar residues. In total, a monomer of AF2331 contains 92 residues: 45 with nonpolar sidechains, 14 with uncharged polar sidechains, and 33 residues with charged polar sidechains. In addition, AF2331 has a high aromatic amino acid content (9 Phe and 3 Tyr), which may provide additional stability due to some edge-to-face interactions between aromatic amino acids (e.g., between Phe14 and Tyr73).23
The sequence of AF2331 is dominated by acidic residues (9 Asp and 13 Glu), relative to the number of basic residues (2 Arg, 8 Lys, and 1 His), resulting in a theoretical pI of 4.3. All of the acidic amino acids are located on the surface of the AF2331 dimer (Fig. 4), where they form several negatively charged clusters. In the genome of A. fulgidus, AF2331 is found in the same operon as another protein, AF2330, which also lacks sequence homologs with known function. The sequence of AF2330 (theoretical pI = 8.4) contains an excess of basic amino acids (16 Arg, 5 Lys, and 5 His) relative to acidic residues (6 Asp and 13 Glu). We hypothesize that AF2331 and AF2330 are involved in a unique physiological function in A. fulgidus, perhaps forming a charge-stabilized complex. However, the role of the negatively charged surface of AF2331 is not clear.
Figure 4.
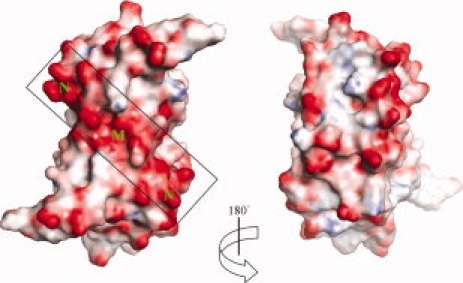
The molecular surface of the dimer shown with electrostatic potential. One of the clusters of most electronegative surface potential, labeled as M in the figure, is due to Asp 33 and Glu 52 from both of chain A and chain B. The two clusters labeled as N are due to helix α2 from each monomer. The figures were generated using PyMOL.9
Novel fold
A sequence similarity search with PSI-BLAST24 found no related sequence in the NCBI nonredundant sequence database (the highest scoring match had a BLAST E value of 2.6). Threading calculations performed by the bioinfo.pl metaserver25 also did not identify any putative homologs. This is not very surprising since most modern threading methods rely on sequence profiles or PSSMs (position-specific substitution matrices) to find matching structures. As no homologous sequences were available, these tools were unable to build a sequence profile for AF2331. These negative results from sequence-based searches suggested us that AF2331 is an orphan protein and may represent a novel protein fold.
To test this hypothesis, we used the CE program26 to compute structural alignments between AF2331 and all the proteins in PDB database, and the program failed to detect any similar structure. The longest reasonable alignments covered no more than three secondary structure elements, such as a beta-hairpin motif, which matched β2-β3 with cRMSD (coordinate-derived root mean square deviation) 1.3 Å, or a motif that matched α1-β2-β3 with cRMSD 2.6 Å. Structural similarity searches with DALI27,28 and SSM29 also did not return any significant results.
Whether or not a new protein structure contains a novel fold is difficult to determine, because there is no straightforward definition of a protein fold. Many such definitions have been proposed. A very restrictive approach was recently proposed by Zhang and Skolnick,30 where two proteins share a common fold if the TM-score distance calculated between them is higher than 0.5. Zhang and Skolnick generated a large set of random compact protein-like chains no longer that 100 amino acids. For each of these random chains they were able to find a protein structure already deposited to PDB that shares the same fold and the authors concluded that the PDB already contains all possible structural motifs. Using TM-align, we calculated structure alignments between AF2331 and all the structures in PDB. The closest similarity to AF2331 was a portion of the 1ZAX structure, where an alignment of only 21 residues had a TM-score of 0.48 and cRMSD of 1.36 Å. Thus, no significant matches to the whole fold of AF2331 were found with any of the four structure similarity comparison algorithms (CE, DALI, SSM, or TM-Align).
The geometric criteria described above employ 3D atom coordinates to detect similarity or dissimilarity between folds. Although they are very easy to use and automate, they are also known to report both false positives and false negatives. Therefore, the most respected protein classification schemes are based on manual inspection and assignment, either in a fully manual manner (Structural Classification Of Proteins or SCOP31) or a combination of manual and automated methods (Class, Architecture, Topology and Homologous superfamily classification or CATH32,33). In both cases, the boundaries of and assignments for each protein domain are determined using a combination of automated and manual procedures, which include computational techniques, empirical and statistical evidence, literature review, and expert analysis.
AF2331 belongs to a broad α + β class. The structure is composed of three layers: a β layer (sheet) of four strands, a middle β layer (sheet) with six strands, and a helical layer. According to SCOP, the protein structure has been assigned to α + β class (mainly antiparallel beta sheets—segregated alpha and beta regions). Its overall architecture could be described as a “3-Layer (BBA) Sandwich.” While many proteins of known structure adopt this architecture, the arrangement of secondary structure elements of all of them differs from that of AF2331, as indicated by the results of 3D structure comparison methods. Moreover, none of the 3-Layer (BBA) Sandwich structures have the same topology as AF2331.
The unusual topological connections between the secondary structure elements in AF2331 result from its interdigitated nature, where the two chains form a single compact globular domain. Thus, the novelty of AF2331 fold results from its quaternary rather than from tertiary structure. In contrast, this is not the case for the 2HJ1 protein, the only other protein with a six-strand interdigitated (ABABAB) β-sheet, which clearly segregates into two domains. Indeed both the SCOP and CATH classifications describe 2HJ1 as being composed of two domains of a well-known ubiquitin-like fold.
AF2331 has been annotated as a “new fold” in the SCOP database, as the founding structure of the “AF2331-like” fold, superfamily, and family. Although the structure has not been yet classified in the CATH database, AF2331 does not share topological connections with any existing fold with a 3-Layer (BBA) Sandwich architecture. Therefore, we conclude that AF2331 represents a new type of α + β fold.
Materials and Methods
Crystallization and structure solution
The AF2331 gene was cloned into a modified pET-15b plasmid, which encodes for a polyhistidine affinity tag connected through a TEV protease digestion site (MGSSHHHHHHSSGRENLYFQGH) at the N-terminus of the expressed protein. The protein was cloned, expressed, and purified by protocols developed by the Midwest Center for Structural Genomics.34 Selenomethionine (Se-Met) substituted protein was produced and crystallized, after its affinity tag was cleaved by digestion with TEV protease. Crystals suitable for X-ray diffraction experiments were obtained by hanging-drop vapor diffusion at 20°C. The drop was composed of 2 μL of a 5 mg/mL solution of Se-Met substituted AF2331 protein mixed with 2 μL of the well solution, containing 45% MPD, 0.2 M NH4 acetate, and 0.1 M Tris, at pH 8.5. The crystals were cooled in a cryogenic stream of evaporating nitrogen without additional cryoprotectant.
MAD data from Se-Met substituted protein were collected at a temperature of 100 K on beamline 19-ID35 of the Structural Biology Center at Argonne National Laboratory and processed with HKL-2000.36 Two data sets were collected at wavelengths 0.9790 and 0.9792 Å, which are the Se fluorescence peak and inflection point, respectively. The strategy of data collection was optimized to obtain the best phases.37 The peak and inflection point data sets diffracted to resolutions of 2.5 and 2.7 Å, respectively. A higher resolution data set (2.4 Å) was collected on a second isomorphous crystal and was used only for model refinement. The crystals of Se-Met substituted AF2331 protein belong to space group P212121 and contain two protein subunits per asymmetric unit. The Matthews coefficient for both crystals was 2.3 Å3/Da, corresponding to an estimated solvent content of 46%.
An initial electron density map and model was obtained with the new software package HKL-3000,38 which is coupled with the programs SHELXD and SHELXE,39,40 MLPHARE,41 DM,42,43 SOLVE/RESOLVE,44,45 CCP4,46 ARP/wARP,47 and COOT.48 The initial 2.7 Å model was extended by cycles of iterative rebuilding with RESOLVE followed by ARP/wARP (as implemented in HKL-3000). This model was extended by cycles of manual model building with COOT, followed by maximum-likelihood refinement with REFMAC49 using the 2.4 Å resolution data set. Tight main-chain and medium side-chain noncrystallographic symmetry (NCS) restraints were used during REFMAC refinement. Data collection and refinement statistics are summarized in Table III.
Table III.
Crystallographic Data Collection, Phasing, and Refinement Statistics
| Crystal |
|||
|---|---|---|---|
| Crystal number 1a,b |
Crystal number 2a,c |
||
| Data collection | |||
| Peak | Inflection | ||
| Number of crystals used | 1 | 1 | |
| Wavelength (Å) | 0.979 | 0.989 | 0.979 |
| Resolution range (Å) | 50–2.5 | 50–2.7 | 20–2.4 |
| Highest resolution shell (Å) | 2.59–2.5 | 2.80–2.7 | 2.46–2.4 |
| I/σI | 21.8 (2.0) | 17.7 (1.4) | 33.9 (3.9) |
| Rmerge | 0.122 (0.464) | 0.119 (0.596) | 0.11 (0.42) |
| Observed reflections | 47,656 | 36,433 | 58,746 |
| Unique reflections | 6833 (547) | 5326 (397) | 7822 (702) |
| Completeness (%) | 97.1 (81.5) | 96.2 (75.9) | 97.8 (91.2) |
| Redundancy | 7.0 (3.5) | 6.8 (3.0) | 7.5 (5.9) |
| Phasing | |||
| FOM (MLPHARE) | |||
| Centric | 0.20 | ||
| Acentric | 0.30 | ||
| Phasing power (MLPHARE) | |||
| Centric | 0.38 | ||
| Acentric | 0.51 | ||
| FOM (DM) | 0.80 | ||
| Refinement | |||
| Resolution range (Å) | 20.0–2.4 (2.46–2.4) | ||
| Rfree test set size (%) | 4.7 | ||
| Rcryst/Rfree (%) | 20.1/25.3 (25.1/33.2) | ||
| Mean B-factor (Wilson) (Å2) | 46.7 | ||
| Mean B-factor (overall) (Å2) | 54.5 | ||
| Number of residues | 186 | ||
| Number of protein atoms | 1444 | ||
| Number of solvent atoms | 27 | ||
| RMSD from ideal geometry | |||
| Bond lengths (Å) | 0.021 | ||
| Bond angles (°) | 1.74 | ||
| Ramachandran plot | |||
| Res. in favored regions (%) | 97.8 | ||
| Res. in allowed regions (%) | 2.2 | ||
Data for the highest resolution shells are in parentheses.
Rmerge values were calculated with Bijvoet pairs merged.
Space group: P212121.
Cell dimensions: a = 45.5 Å, b = 48.9 Å, c = 86.3 Å.
Cell dimensions: a = 45.2 Å, b = 48.9 Å, c = 86.7 Å.
The final model contains two polypeptide monomers (residues 1–92 in both chain A and chain B) and 27 water molecules. Both of the chains include one extra residue at the amino terminus of the protein. The Ramachandran diagram shows that 97.7% of all residues are in the most favored regions and 100% of all residues are in the allowed regions, as determined by MOLPROBITY.50
Analysis of oligomeric state
To create a set of nonredundant homo-oligomeric structures, a list of putative oligomeric states and oligomer interface solvent-accessible surface areas (ASA) of X-ray structures in the PDB was downloaded from the Protein Quaternary Server (PQS) at http://pqs.ebi.ac.uk/.10 The set of 4107 nonredundant homodimer structures was obtained by passing the list of sequences to the CD-HIT program, which clusters sequences with greater than 90% identity and removes the duplicates.51 The “dimerization interface area” provided by PQS is defined as the difference of the total ASA of an isolated monomer minus the ASA of the homodimer divided by two. These values provide a reasonable estimate of the oligomerization surface area of a homo-oligomer, in other words, the area of the surface of a monomer in contact with other monomers in the homo-oligomer. The “percentage of the monomer ASA in the interface surface” is defined as the dimerization interface area divided by the total ASA for an isolated monomer. For each cluster, the structure with the highest percent of monomer ASA in the interface surface was chosen.
We noted that the PQS data set contained a number of false positives. For example, structure 1A5I, a monomeric structure that contains a tripeptide ligand, was incorrectly identified as a homodimer by sequence alignment of the protein and the peptide ligand. To filter out the false positives, we obtained information from the PQS data site about the length of the sequence alignment for each structure in the PQS data set. We filtered the PQS data by excluding homodimers where the two chains differed in length by 10% or the length of the sequence match between dimers was less than 50% of the longer sequence.
To generate the histograms for homo-oligomers with the number of subunits n ≥ 3, an identical procedure was followed, save that the removal of false positives by sequence alignment length was omitted. (A brief analysis of the higher-order homo-oligomers suggested that there were far fewer false positives as compared to the homodimers.) The oligomerization interface area was calculated in an analogous way to the dimerization interface area, by taking the total ASA for an isolated monomer minus the total ASA for the whole oligomer divided by the number of chains. The correlation of the oligomerization interface area as a function of monomer molecular weight [Fig. 3(A)] or total monomer ASA [Fig. 3(B)] was determined by linear least-squares regression, with the y-intercept constrained to 0.
Structure comparison methods
We used four different programs to calculate structural alignments: CE,26 TM-Align,30 DALI,27,28 and SSM.29 The BioShell package52 was used to automate calculations, and the CE tool was used for accurate structure-to-structure alignment and comparison. When given two protein models, CE detects highly similar hexapeptides and then tries to extend an alignment by means of a combinatorial extension algorithm. The method is accurate but relatively slow (comparison of AF2331 with the whole PDB took about 4 days). TM-Align is based on a novel distance score between two protein chains referred to below as a TM-score. The TM-score is normalized so that its value does not depend on polypeptide chain length. The similarity of two unrelated, randomly generated structures is usually close to 0.17. A TM-score value above 0.5 denotes that two chains share the same fold. Servers implementing the DALI and SSM algorithms were used with the default parameters.
Bioinformatic search for interdigitated β-strands
We define an interdigitated β-strand by three criteria: (1) the strand belongs to a β-sheet and is flanked two other β-strands by a network of hydrogen bonds; (2) the strand belongs to a different chain than the two flanking strands; and (3) the two flanking strands belong to the same chain. Thus, interdigitated β-strands form a specific pattern of chain IDs within the β-sheet, for example, ABA. Our analysis may be extended to longer patterns, for example, ABAB or even ABABAB. We used the DSSP program53 to calculate the hydrogen bond network in a PDB structure. DSSP allows for bifurcated H-bonds; thus, the main chain residue may participate in up to four bonds. In this work, we chose the two strongest bonds for a residue, one in which the residue acts as a proton donor and one where it acts as an acceptor. Graph representations of the H-bond networks in the β-sheets were generated using the BioShell package, treating the residues as nodes and the bonds as edges. These graphs were then searched via standard graph algorithms for the longest paths matching the specified patterns.
Conclusions
AF2331 forms a homodimer with two highly interdigitated β-sheets, and the percentage of its surface involved in the dimerization interaction is very high, even when compared to other homodimers, though this pattern of interdigitated β-sheets has been observed in other structures. The percentage of its surface involved in dimerization is above the mean for all higher-order oligomers as well. Its small size, the high percentage of hydrophobic or nonpolar residues in the interdigitated β-sheets, and the high percentage of aromatic amino acids explain why the structure of AF2331 is capable of forming a dimer with such a large interface surface area.
AF2331 is an orphan protein, which forms an α + β fold with a “3-Layer (BBA) Sandwich” architecture. By analysis of multiple sequence- and structure-based similarity algorithms, no homologs for AF2331 were found. Furthermore, in two different curated databases of protein folds, no folds similar to that of AF2331 were identified. The protein has been classified as a novel fold by the SCOP database, where it is the founding structure of a new fold, superfamily, and family. While it has not yet been formally classified by the CATH database, no other 3-Layer (BBA) Sandwich fold shares the same topology with AF2331. Thus, it is very likely that AF2331 represents a novel α + β fold.
The function of the protein is still unknown, and the physiological role for forming such a highly stable dimer is not clear. One possibility is that the largely negatively charged surface of the protein may be involved in charged-stabilized interaction with AF2330.
Acknowledgments
The authors would like to thank Andrzej Joachimiak and the members of the Structural Biology Center at the Advanced Photon Source and the Midwest Center for Structural Genomics for help and discussions. We would also like to thank Alex Wlodawer and Zbyszek Dauter for numerous discussions. The results shown in this report are derived from work performed at Argonne National Laboratory, at the Structural Biology Center of the Advanced Photon Source. Argonne is operated by University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Glossary
Abbreviations:
- ASA
(solvent-)accessible surface area
- cRMSD
coordinate-based root mean square deviation
- MAD
multiwavelength anomalous dispersion
- MPD
2-methyl-1,3-propanediol
- PDB
Protein Data Bank
- PQS
Protein Quaternary Structure Server
- Se-Met
selenomethionine
- TEV
tobacco etch virus.
References
- 1.Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, Richardson DL, Kerlavage AR, Graham DE, Kyrpides NC, Fleischmann RD, Quackenbush J, Lee NH, Sutton GG, Gill S, Kirkness EF, Dougherty BA, McKenney K, Adams MD, Loftus B, Peterson S, Reich CI, McNeil LK, Badger JH, Glodek A, Zhou L, Overbeek R, Gocayne JD, Weidman JF, McDonald L, Utterback T, Cotton MD, Spriggs T, Artiach P, Kaine BP, Sykes SM, Sadow PW, D'Andrea KP, Bowman C, Fujii C, Garland SA, Mason TM, Olsen GJ, Fraser CM, Smith HO, Woese CR, Venter JC. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 2.Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrickson WA. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science. 1991;254:51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 4.Bairoch A, Bougueleret L, Altairac S, Amendolia V, Auchincloss A, Puy GA, Axelsen K, Baratin D, Blatter MC, Boeckmann B, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, Saux VBL, deCastro E, Ciampina L, Coral D, Coudert E, Cusin I, David F, Delbard G, Dornevil D, Duek-Roggli P, Duvaud S, Estreicher A, Famiglietti L, Farriol-Mathis N, Ferro S, Feuermann M, Gasteiger E, Gateau A, Gehant S, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hulo N, Innocenti A, James J, Jain E, Jimenez S, Jungo F, Junker V, Keller G, Lachaize C, Lane-Guermonprez L, Langendijk-Genevaux P, Lara V, Le Mercier P, Lieberherr D, Lima TD, Mangold V, Martin X, Michoud K, Moinat M, Morgat A, Nicolas M, Paesano S, Pedruzzi I, Perret D, Phan I, Pilbout S, Pillet V, Poux S, Pozzato M, Redaschi N, Reynaud S, Rivoire C, Roechert B, Sapsezian C, Schneider M, Sigrist C, Sonesson K, Staehli S, Stutz A, Sundaram S, Tognolli M, Verbregue L, Veuthey AL, Vitorello C, Yip L, Zuletta LF, Apweiler R, Alam-Faruque Y, Barrell D, Bower L, Browne P, Chan WM, Daugherty L, Donate ES, Eberhardt R, Fedotov A, Foulger R, Frigerio G, Garavelli J, Golin R, Horne A, Jacobsen J, Kleen M, Kersey P, Laiho K, Legge D, Magrane M, Martin MJ, Monteiro P, O'Donovan C, Orchard S, O'Rourke J, Patient S, Pruess M, Sitnov A, Whitefield E, Wieser D, Lin Q, Rynbeek M, di Martino G, Donnelly M, van Rensburg P, Wu C, Arighi C, Arminski L, Barker W, Chen YX, Crooks D, Hu ZZ, Hua HK, Huang HZ, Kahsay R, Mazumder R, McGarvey P, Natale D, for UniProt Consortium The universal protein resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442–3444. doi: 10.1093/nar/28.18.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P. STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33:D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Kruger B, Snel B, Bork P. STRING 7—recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35:D358–D362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLano WL. Use of PYMOL as a communications tool for molecular science. Abstr Papers Am Chem Soc. 2004;228:U313–U314. [Google Scholar]
- 10.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 11.Jones S, Thornton JM. Protein–protein interactions—a review of protein dimer structures. Prog Biophys Mol Biol. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 12.Huang DB, Vu D, Ghosh G. NF-kappaB RelB forms an intertwined homodimer. Structure. 2005;13:1365–1373. doi: 10.1016/j.str.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Pino A, Martinez-Rodriguez S, Wahni K, Wyns L, Loris R, Messens J. Coupling of domain swapping to kinetic stability in a thioredoxin mutant. J Mol Biol. 2009;385:1590–1599. doi: 10.1016/j.jmb.2008.11.040. [DOI] [PubMed] [Google Scholar]
- 14.Glowka ML, Olczak A, Bojarska J, Szczesio M, Duax WL, Burkhart BM, Pangborn WA, Langs DA, Wawrzak Z. Structure of gramicidin D-RbCl complex at atomic resolution from low-temperature synchrotron data: interactions of double-stranded gramicidin channel contents and cations with channel wall. Acta Crystallogr D Biol Crystallogr. 2005;61:433–441. doi: 10.1107/S0907444905000399. [DOI] [PubMed] [Google Scholar]
- 15.Bunkoczi G, Vertesy L, Sheldrick GM. The antiviral antibiotic feglymycfirst direct-methods solution of a 1000+equal-atom structure. Angew Chem Int Ed. 2005;44:1340–1342. doi: 10.1002/anie.200461933. [DOI] [PubMed] [Google Scholar]
- 16.Wallace BA. Recent advances in the high resolution structures of bacterial channels: gramicidin A. J Struct Biol. 1998;121:123–141. doi: 10.1006/jsbi.1997.3948. [DOI] [PubMed] [Google Scholar]
- 17.Schlunegger MP, Bennett MJ, Eisenberg D. Oligomer formation by 3D domain swapping: a model for protein assembly and misassembly. Adv Protein Chem. 1997;50:61–122. doi: 10.1016/s0065-3233(08)60319-8. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett MJ, Choe S, Eisenberg D. Domain swapping—entangling alliances between proteins. Proc Natl Acad Sci USA. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chirgadze DY, Demydchuk M, Becker M, Moran S, Paoli M. Snapshot of protein structure evolution reveals conservation of functional dimerization through intertwined folding. Structure. 2004;12:1489–1494. doi: 10.1016/j.str.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Camara-Artigas A, Martin-Garcia JM, Morel B, Ruiz-Sanz J, Luque I. Intertwined dimeric structure for the SH3 domain of the c-Src tyrosine kinase induced by polyethylene glycol binding. FEBS Lett. 2009;583:749–753. doi: 10.1016/j.febslet.2009.01.036. [DOI] [PubMed] [Google Scholar]
- 22.Ponstingl H, Thomas KB, Gorse D, Thornton JM. Morphological aspects of oligorneric protein structures. Prog Biophys Mol Biol. 2005;89:9–35. doi: 10.1016/j.pbiomolbio.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Burley SK, Petsko GA. Aromatic–aromatic interaction—a mechanism of protein-structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 24.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 26.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 27.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 28.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 29.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murzin AG, Brenner SE, Hubbard T, Chothia C. Scop—a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 32.Cuff AL, Sillitoe I, Lewis T, Redfern OC, Garratt R, Thornton J, Orengo CA. The CATH classification revisited—architectures reviewed and new ways to characterize structural divergence in superfamilies. Nucleic Acids Res. 2009;37:D310–D314. doi: 10.1093/nar/gkn877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R-G, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, Arrowsmith CH, Clarke S, Edwards A, Joachimiak A, Savchenko A. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Minor W, Tomchick DR, Otwinowski Z. Strategies for macromolecular synchrotron crystallography. Struct Fold Des. 2000;8:R105–R110. doi: 10.1016/s0969-2126(00)00139-8. [DOI] [PubMed] [Google Scholar]
- 38.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 39.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 40.Sheldrick GM. Macromolecular phasing with SHELXE. Z Kristallogr. 2002;217:644–650. [Google Scholar]
- 41.Otwinowski Z. In: Proceedings of the CCP4 study weekend. Isomorphous replacement and anomalous scattering. Wolf W, Evans PR, Leslie AGW, editors. Warrington, UK: Daresbury Laboratory; 1991. pp. 80–86. [Google Scholar]
- 42.Cowtan KD, Zhang KY. Density modification for macromolecular phase improvement. Prog Biophys Mol Biol. 1999;72:245–270. doi: 10.1016/s0079-6107(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 43.Cowtan KD, Main P. Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints. Acta Crystallogr D Biol Crystallogr. 1993;49:148–157. doi: 10.1107/S0907444992007698. [DOI] [PubMed] [Google Scholar]
- 44.Terwilliger TC. Automated structure solution, density modification and model building. Acta Crystallogr D Biol Crystallogr. 2002;58:1937–1940. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 46.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 50.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 51.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 52.Gront D, Kolinski A. BioShell—a package of tools for structural biology computations. Bioinformatics. 2006;22:621–622. doi: 10.1093/bioinformatics/btk037. [DOI] [PubMed] [Google Scholar]
- 53.Kabsch W, Sander C. Dictionary of protein secondary structure—pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]