

Abstract
Heme oxygenase (HO) is the chief regulatory enzyme in the oxidative degradation of heme to biliverdin. In the process of heme degradation, this NADPH and cytochrome P450 reductase (CPR)-dependent oxidation of heme also releases free iron and carbon monoxide. Much of the recent research involving heme oxygenase is done using a 30-kDa soluble form of the enzyme, which lacks the membrane binding region (C-terminal 23 amino acids). The goal of this study was to express and purify a full-length human HO-1 (hHO-1) protein; however, due to the lability of the full-length form, a rapid purification procedure was required. This was accomplished by use of a GST-tagged hHO-1 construct. Although the procedure permitted the generation of a full-length HO-1, this form was contaminated with a 30-kDa degradation product that could not be eliminated. Therefore, we attempted to remove a putative secondary thrombin cleavage site by a conservative mutation of amino acid 254, which replaces lysine with arginine. This mutation allowed the expression and purification of a full length hHO-1 protein. Unlike wild-type HO-1, the K254R mutant could be purified to a single 32-kDa protein capable of degrading heme at the same rate as the wild-type enzyme. The K254R full-length form had a specific activity of ~200–225 nmol bilirubin hr−1nmol−1 HO-1 as compared to ~140–150 nmol bilirubin hr−1nmol−1 for the WT form, which contains the 30-kDa contaminant. This is a 2–3-fold increase from the previously reported soluble 30-kDa HO-1, suggesting that the C-terminal 23 amino acids are essential for maximal catalytic activity. Because the membrane spanning domain is present, the full-length hHO-1 has the potential to incorporate into phospholipid membranes, which can be reconstituted at known concentrations, in combination with other ER-resident enzymes.
Heme oxygenase catalyzes the breakdown of heme to biliverdin, iron, and carbon monoxide (CO) (1). The physiological significance of these metabolites, most notably CO and biliverdin, is a reason for the escalating interest in HO research within the last 15 years (2). HO regulates iron homeostasis in mammals by recycling the iron released from the porphyrin ring. Biliverdin reductase, a soluble enzyme found in the cytosol, converts biliverdin to bilirubin. Bilirubin is a potent antioxidant that is conjugated to glucuronic acid in order to be excreted (3). Obstruction of bilirubin elimination has been implicated in common conditions such as neonatal jaundice, which can lead to neurological damage (4). CO, a third product of heme oxygenase, has been shown to have signaling properties that parallel those of nitric oxide (5–7). CO is believed to elicit its effects via the guanylate cyclase pathway, common to NO.
Two distinct isoforms of heme oxygenase have been well characterized, while at least one other is believed to exist. The inducible form, heme oxygenase-1 (HO-1), is a 32.8-kDa membrane-bound enzyme found at highest concentrations in the liver and spleen. This 288-residue protein is induced by a variety of stimuli, including porphyrins, heavy metals, and certain disease states (8–10). The constitutive form, heme oxygenase-2 (HO-2), is a 316-residue membrane-bound protein found at highest levels in the brain and testis (11). Although both HO-1 and HO-2 metabolize heme through identical pathways, the enzymes only share a 45% sequence identity (12). A C-terminal lipophilic domain secures HO-1 and HO-2 in the endoplasmic reticulum membrane (13, 14). HO-1 and HO-2 participate in heme breakdown and the production of bile pigments, but the primary role of HO-2 is believed to be CO production in the brain (6, 15).
Full-length forms of HO-1 have been isolated from various sources, including rat, pig, and bovine (8, 9, 16–18). Due to the extreme lability and proteolytic susceptibility of the enzyme, storage and multiple freeze-thaw cycles have been reported to lead to the generation of smaller contaminant bands and a decrease in HO-1 enzyme activity (16, 18, 19). Previous studies involving HO-1 from rat liver report of the extensive proteolytic degradation that the enzyme undergoes following efforts to solubilize and purify the full-length form. In the prior attempts to purify rat HO-1, the 32.8kD protein is degraded into smaller products, specifically 30-kDa and 27–28-kDa products (20). Wilks, et al. successfully expressed and purified truncated versions of both human and rat HO-1, which are used as the standard form in many recent characterization studies (21, 22).
In order to obtain a stable, full-length hHO-1 protein, we have developed a rapid solublization and purification using an N-terminal glutathione S-transferase (GST) tag. Removal of the GST-tag generates hHO-1 in its 32.8-kDa native state. The GST-tag is readily removed by thrombin treatment; however, this treatment leads to generation of a 30-kDa degradation product, attributable to an internal thrombin cleavage site. Here we report on a conservative single-site mutation eliminating the internal cleavage site, resulting in a homogeneous full-length hHO-1 protein. Characterization of these enzymes indicates that the C-terminal 23 amino acids play an important role in the interaction between HO-1 and CPR, enhancing the conversion of hemin to biliverdin.
EXPERIMENTAL PROCEDURES
Chemicals
Glucose, PBS tablets, EDTA, phenylmethanesulfonyl fluoride (PMSF), lysozyme, dithiothreitol (DTT), hemin, n-Octyl-β-D-glucopyranoside (octyl glucoside), and NADPH were purchased from Sigma. One Shot© DH5α cells, carbenicillin and IPTG came from Invitrogen. The biliverdin used in biliverdin reductase quantification was purchased from Frontier Scientific. Glutathione Sepharose© 4B and thrombin were obtained from Amersham Biosciences. Terrific broth (TB) was purchased from US Biological. Complete© Protease Inhibitor tablets were purchased from Roche. Triton X-100 and Sodium N-lauroylsarcosinate (Sarkosyl) were obtained from Acros and Fluka, respectively. All PCR primers were purchased from Integrated DNA Technologies. The restriction enzymes, BamH1 and Sal1, were acquired from New England Biolabs. BCA protein assay kit was purchased from Pierce.
Expression systems
The pGEX-4T-2 (Amersham) expression vector containing cDNA Human HO-1 (hHO-1) was generously provided by Dr. Mahin Maines (University of Rochester, New York). The full-length hHO-1 (WT) gene was cloned into the pGEX vector between sites BamH1 and Sal1. These restriction sites allowed for restriction analysis to verify gene size and plasmid incorporation following bacterial transformation. The truncated hHO-1 (sHO-1) expression system was supplied by Dr. Paul Ortiz de Montellano (University of California, San Francisco) and purified as previously reported (Wilks, 1995). Recombinant rat CPR, which possessed a K56Q mutation to decrease proteolytic susceptibility, was provided by Dr. Grover Paul Miller (University of Arkansas, Little Rock). It was expressed in C41 cells and purified with minor modifications to previously described methods (23, 24).
Expression and purification of WT pGEXhHO-1
Plasmid purification was carried out according to previously described methods with minor modifications (25–27). One Shot© DH5α cells were transformed with the hHO-1 expression plasmid and plated on an LB-agarose plate containing carbenicillin (100μg/ml). All of the TB contained carbenicillin (100μg/ml) and 2% glucose, unless otherwise stated. A 50 ml aliquot of TB was inoculated with a single colony from a fresh plate and incubated at 37°C overnight. All PBS buffer (PBS), pH 7.3 used here was supplemented with 20% glycerol, unless stated otherwise. Five milliliters of the overnight culture were used to inoculate 3L of TB, which was incubated in an orbital shaker at 37°C and 225 rpm until the OD at 600 nm = 0.5. Following the addition of 0.1 mM IPTG to relieve the inhibition of gene expression, the cells were shaken at 28°C and 150 rpm for 12–14 hours.
The three liter cultures were centrifuged at 4000 × g for 30 min. The pellets were re-homogenized in PBS buffer, pH 7.8, containing 20% glycerol, 1mM EDTA, 1mM PMSF, and Complete© protease inhibitors (20 ml/L) (RH Buffer). Lysozyme was added to a final concentration of 100 μg/ml, and cells incubated on ice for 15 minutes. Following the addition of 5mM DTT, the cells were lysed by the addition of 1% Sarkosyl from a 10% stock in RH buffer. The lysate was then briefly vortexed and sonicated in a water bath for 1 min. The bacterial cell lysate was then clarified by centrifuging at 10,000 × g for 5min. The supernatant was adjusted to 2% Triton X-100 from a 10% stock in RH buffer. HO-1 was purified using a batch-wise method. Glutathione Sepharose 4B (25 ml) was added to the supernatants, and the mixture was allowed to rock gently at 4°C for 30 minutes to maximize binding. The beads were washed 8–10 times with ice-cold PBS to free them of any residual detergent used to solublize the protein. Using thrombin cleavage buffer (20 mM Tris-HCl pH 8.4, 150 mM NaCl, 2.5 mM CaCl2), the appropriate amount of thrombin (2 U/ml glutathione Sepharose) was added to the beads in order to cleave the hHO-1 from the glutathione matrix. The thrombin cleavage occurred at room temperature for 2 hours. Subsequently, the beads were centrifuged and the supernatant removed. The beads were washed with a high salt wash buffer (PBS pH 7.3, 20% glycerol, 250 mM NaCl) to remove any remaining contaminant proteins. The full-length hHO-1 was then eluted from the beads using a modified elution buffer (PBS pH 7.3, 20% glycerol, 2% N-octyl glucoside). The eluent was cleared of detergent by dialysis against 2 L PBS, 20% glycerol, 0.1 mM EDTA, and pH 7.4, with one buffer exchange. Both GST-tagged WT and R254K hHO-1 were also purified as mentioned above, but were eluted from the beads with 50 mM Tris-HCl, 20% glycerol, 10 mM reduced glutathione, pH 7.6, prior to the thrombin treatment.
Thrombin titration of GST-WT
Purified GST-WT was incubated with increasing amounts of thrombin for 2 hours at room temperature (23–25°C). For this experiment, about 0.1 nmol GST-WT and previously determined thrombin concentrations were used. The ratio of GST-WT: thrombin closely resembled that used in the purification procedure. Following 2 hours at room temperature, the degradation was visualized via SDS-PAGE analysis.
Construction of pGEXhHO-1 R254K mutant expression plasmid and purification
The WT hHO-1 gene was used as a template to construct the pGEXhHO-1 R254K hHO-1 expression system. The single site-directed mutagenesis was performed using a Stratagene Mutagenesis kit. The 5′-sense oligonucleotide primer (5′ CCC CTG GAG ACT CCC AAA GGG AAG CCC CCA CTC -3′) encoded for a lysine (AAA), replacing an arginine (AGA) at amino acid position 254. The 3′-antisense oligonucleotide mutation primer (5′-GAG TGG GGG CTT CCC TTT GGG AGT CTC CAC GGG -3′) consisted of the same lysine to arginine mutation at position 254. The PCR settings were as follows: 95°C for 1 minute, followed by 95°C for 50 seconds, 60°C for 50 seconds, 68°C for 6 minutes, and lastly 68°C for 7 minutes. The amplification segment of the reaction continued for 18 cycles. Following PCR amplification of the mutated gene, Dpn1 digestion liberated the sample from the parental, non-mutated strand. The pGEXhHO-1 R254K was then transformed, plated, and purified using a Qiagen Plasmid Mini-Prep kit. The purified plasmid was screened by restriction digestion and the mutation was verified by sequencing analysis.
WT and R254K hHO-1 plasmids were handled separately, but in the same manner, as previously described above, throughout their expression and purification. R254K containing the GST-affinity tag was also purified as mentioned for the WT.
Hemin titration of purified hHO-1
HO-1 was quantified using a hemin titration1 based on a previously described method (16). Aliquots of a 1 mM stock of hemin were added to 1 ml HO-1 and spectral scans were taken from 700–350 nm. The absorbance at 405 nm was recorded and plotted against the hemin concentration. HO-1 concentration was determined by the break point at which the increased absorbance at 405 nm deviates from linearity. The extinction coefficient of the ferric heme-HO-1 complex was 140 mM−1 cm−1, which is consistent with published reports (16).
Determination of protein levels
Protein levels were determined using the bicinchoninic acid method supplied in a BCA protein assay kit (28). Bovine serum albumin was used as the standard.
Partial purification of rat biliverdin reductase and activity assay
Rat biliverdin reductase was partially purified from rat liver using previously described methods (29). The use of partially purified biliverdin reductase to generate bilirubin was described previously, with minor modifications (30). The assay mixture included partially purified rat liver cytosol, 5 μM biliverdin, and 100 mM KPO4 to a final volume of 1ml. Following a 2 minute pre-incubation at 37°C, 0.1 mM NADPH was added to initiate the reaction. The assay was carried out at 37°C in dark conditions. The rate of bilirubin formation was monitored by the absorbance change at 450 nm, and quantified using the extinction coefficient 53 mM−1 cm−1 (30). One unit of enzyme activity was determined to be the amount of BVR needed to form 1 nmol bilirubin hr−1.
Heme oxygenase activity assay
All of the HO-1 activity assays were done using a Spectramax M5 plate reader (Molecular Devices, Sunnyvale, CA). HO-1 activity was determined by the rate of bilirubin formation at 468 nm, using previously described conditions with minor modifications (16, 22). Reaction mixtures consisted of 0.1 μM HO-1, 15 μM hemin, 0.3 μM CPR, and an excess (50 U/ml) of partially purified BvR from rat liver cytosol. This mixture was brought to a final volume of 100 μl with 100 mM KPO4 supplemented with bovine serum albumin (12.5 mg/ml), pH 7.4. The reaction incubated at 37°C for 2 minutes prior to the addition of 0.1 mM NADPH to initiate the reaction. The increase at 468 nm corresponds to the increase in bilirubin, and was quantified using an extinction coefficient of 43.5 mM−1 cm−1. All of the reactions were measured in real time, and the increase at 468 nm remained linear for a minimum of 5 minutes in these experiments. Enzyme activity was reported in nmoles of bililirubin hr−1 nmol−1 HO-1.
CPR titration of heme oxygenase
Using a constant HO-1 concentration of 0.1 μM, activity assays were performed as described above, but CPR concentrations were varied. The eight CPR concentrations ranged from sub-saturating to saturating levels (0, 0.0125, 0.025, 0.05, 0.1, 0.2, 0.3, and 0.4 μM). The rate of bilirubin formation was measured and plotted against CPR concentration. Titration curves were constructed for the multiple HO-1 forms, including WT, R254K, and sHO1. WT and R254K hHO-1 containing the GST-affinity tags were also tested as controls. All reactions were done in a minimum of triplicates at 37°C for five minutes in the dark. Densitometry on an SDS-PAGE gel stained with Coomasie blue was used to approximate the amount of 29–30-kDa contaminant present in the WT sample.
RESULTS
Expression and purification of WT hHO-1
Under the control of an IPTG-inducible tac promoter, the expression plasmid pGEXhHO-1 encoded for a GST-tagged full-length hHO-1 gene. Terrific broth, with 2% glucose, allowed for tight control of protein expression in DH5α cells. Following induction with IPTG, expression of cells longer than 12–14 hours caused an increase in degradation of full-length hHO-1 (32.8-kDa) to a 27–28-kDa form. The induction and overnight expression resulted in a noticeably green media and pellet upon centrifugation, attributable to the accrual of bilirubin, as previously noted for the rat gene (20). As mentioned in the Methods section, 1% Sarkosyl liberates the C-terminal region from the membrane, and clarifies the sample. Very little pellet exists in the ensuing 12000 × g centrifugation, suggesting that the Sarkosyl solubilizes the entire membrane. Following the addition of 2% Triton X-100 to sequester the Sarkosyl, the GST-hHO-1 bound to the glutathione Sepharose more efficiently (27). As with standard affinity chromatography, the impurities were eluted from the sample using multiple PBS washes.
Thrombin treatment of the affinity tagged protein (~58–59-kDa) resulted not only in the formation of the 32.8-kDa full-length hHO-1, but also in multiple contaminant bands; implying non-specific proteolytic degradation. In order to demonstrate this proteolysis, purified GST-WT was subjected to a thrombin titration experiment in the absence of glutathione Sepharose. As thrombin levels were increased, the contaminant bands at 29–30-kDa and 27–28-kDa intensified, as analyzed on SDS-PAGE (Fig. 1). Because glutathione Sepharose was omitted, the GST-tag was visible at ~ 26-kDa. This data suggests that an internal thrombin cut site within the WT hHO1 amino acid sequence leads to degradation of the WT protein.
Fig. 1. Effect of thrombin treatment on GST-WT hHO-1.
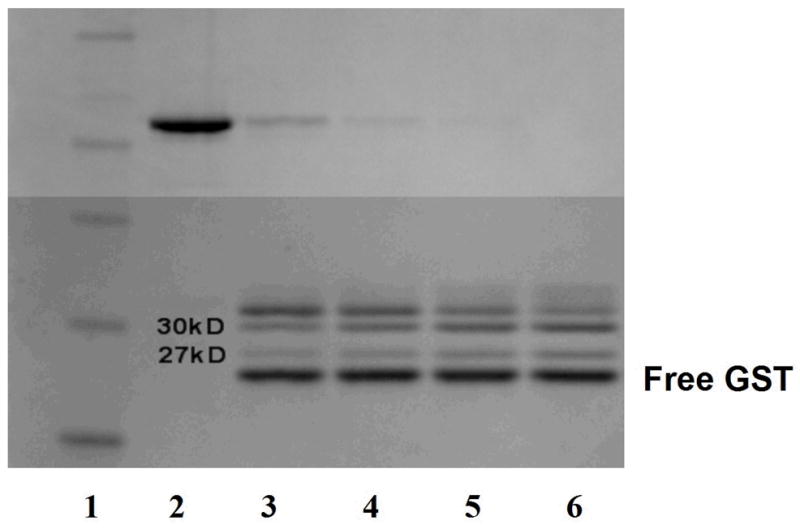
Purified GST-WT was treated for two hours at room temperature with different concentrations of thrombin ranging from 0 units (lane 2), 0.25 units (lane 3), 0.5 units (lane 4), 0.75 units (lane 5), and 1 unit (lane 6). The molecular weight standards are shown in lane 1 (bottom to top: 20, 30, 40, 50, 60, 80-kDa). As the thrombin concentration was increased, the formation of the 30-kDa contaminant was directly proportional to the degradation of the full-length hHO-1 (upper band). The 27-kDa contaminant is also labeled in lanes 3–7. The lowest band in lanes 3–7 represents the GST-tag that was removed from the HO-1.
In the course of the purification, it was noted that although thrombin treatment did remove the GST-tag, it did not release the HO-1 from the beads. HO-1 was released after the thrombin treatment step by washing the beads with PBS, pH 7.3, containing 20% glycerol and 2% octyl glucoside. As a result of the ability of hHO1 to remain bound to the glutathione beads, we were able to separate the 27-kDa contaminant, but the 30-kDa form remained in the purified sample following the 2% octyl glucoside. Most of the 27-kDa product was removed in the supernatant of the initial centrifugation of the glutathione Sepharose, post-thrombin treatment. The high salt washes removed any residual 27-kDa contaminant. The 32.8-kDa and 30-kDa hHO-1 protein eluted with PBS buffer containing 2% octyl-glucoside (fig. 4, lane 2).
Fig. 4. Spectra of the ferric and the ferrous carbon monoxide complex of R254K HO-1.

The ferric hHO-1-heme complex (——) exhibited a Soret peak at 404 nm, as characterized using previously purified forms of HO-1 (16;17;21;22). Upon reduction with sodium dithionite and exposure to carbon monoxide, the Soret band shifted to 418 nm, consistent with the formation of the ferrous carbon monoxide complex (-----). The Fe2+-CO spectrum also revealed the α and β bands at 568 and 538nm, respectively. Inset SDS-PAGE analysis of WT and R254K. R254K was purified to apparent homogeneity, showing a single band at ~32–33kD (lane 3). Lane 2 shows the purified WT HO-1, which reveals a 30-kDa degradation product. Lane 1, molecular weight marker, 30-kDa.
Protein determination and hemin titration of WT hHO-1
Three liters of terrific broth yielded approximately 4.3 mg WT HO-1, as shown in Table 1. The specific activity of the protein was calculated to be 140 nmol bilirubin formed hr−1 nmol−1; which is slightly higher, but comparable to previous reports for sHO-1 (21).
Table 1.
Purification table of WT HO-1 from Ecoli DH5α. WT HO-1 was purified using a GST affinity tag. In the process of removing the tag, a 30-kDa contaminant was introduced that could not be removed from the final preparation.
| Fraction | Protein (mg) | Specific activity (nmol hr−1mg−1) | Specific activity (nmol hr−1 nmol−1) | Units | Purification (x-fold) | Specific Content (nmol mg−1) |
|---|---|---|---|---|---|---|
| Cell supernatant (crude) | 1246.5 | 85.62 | - | 106725.3 | 1 | - |
| 1.0% Sarkosyl Supernatant | 862.5 | 321.83 | - | 277578.4 | 4 | - |
| Thrombin elution | - | - | - | - | - | - |
| 2% octyl- glucoside elution | 4.26 | 4227 | 140 | 14662.9 | 40 | 28.2 |
This purification method allowed for the isolation of apo HO-1. Consequently, HO-1 concentrations were determined by heme titration as shown in Figure 2. Ferric heme-hHO-1 complex has an extinction coefficient of 140 mM−1 cm−1, whereas the extinction coefficient of hemin alone at 402 nm is approximately 87 mM−1 cm−1 (16). Consequently, as heme is added to apo hHO-1 there is a linear increase. Once the HO-1 becomes saturated, the slope decreases. As seen in figure 2, the HO-1-hemin complex is quantified by measuring the hemin concentration where the two straight lines intersect. The concentration of WT was determined to be approximately 8 μM, for a total of 120 nmoles WT. As noted in previous studies, the 30-kDa and 27-kDa forms bind heme and produce a spectrum indistinguishable from the full-length form (20, 21). The spectral properties of these forms add to the overall absorbance, hence affecting the calculation of full-length WT.
Fig. 2. Quantification of WT HO-1 by hemin titration.

In order to quantify the hHO-1, 1ul increments of a 1mM stock were added and the increase in absorbance at 405nm was recorded. The two lines intersect at 4uM, and a final concentration of 8uM WT is calculated after factoring in the 2X dilution.
Expression and purification of R254K MUT hHO1
Because of the lability of the WT form, the sequence of the hHO-1 gene was examined in an attempt to explain its sensitivity to cleavage. Sequence analysis revealed a secondary thrombin cleavage site (ProArg↓Gly) at amino acid 254. Using site directed mutagenesis, Arg at 254 was modified by a conservative mutation to a Lys.
The R254K mutation to the WT template was confirmed by DNA sequencing. The R254K expression vector varied by only one nucleotide (see Methods), allowing for the use of the GST-affinity tag in the purification procedure (Fig. 3). The R254K was expressed using conditions analogous to the WT. Overnight expression of the R254K vector provides green media and pellet, as mentioned with the WT cells. Following the two hour thrombin cleavage at room temperature, most of the 27-kDa contaminant was eluted from the glutathione Sepharose beads in the first centrifugation step. The remaining impurity was rinsed from the beads with the high salt wash buffer, whereas the full-length hHO-1 remained on the beads. Subsequently, the 2% octyl-glucoside elution process yielded a single 32.8-kDa protein, following SDS-PAGE analysis (Fig. 4 inset, lane 3). Spectral characteristics of the R254K paralleled those of the WT and of previously reported forms (Fig. 4) (16, 17, 21, 22). The Fe3+ heme-HO-1 complex exhibited the classic Soret peak at 404 nm, as characterized using previously purified forms of HO-1. Upon reduction with sodium dithionite and exposure to carbon monoxide, the Soret band shifted to 418nm, consistent with the formation of the ferrous carbon monoxide complex. The Fe2+-CO spectrum also revealed the α and β bands at 568 and 538nm, respectively.
Fig. 3. HO-1 Expression vector.
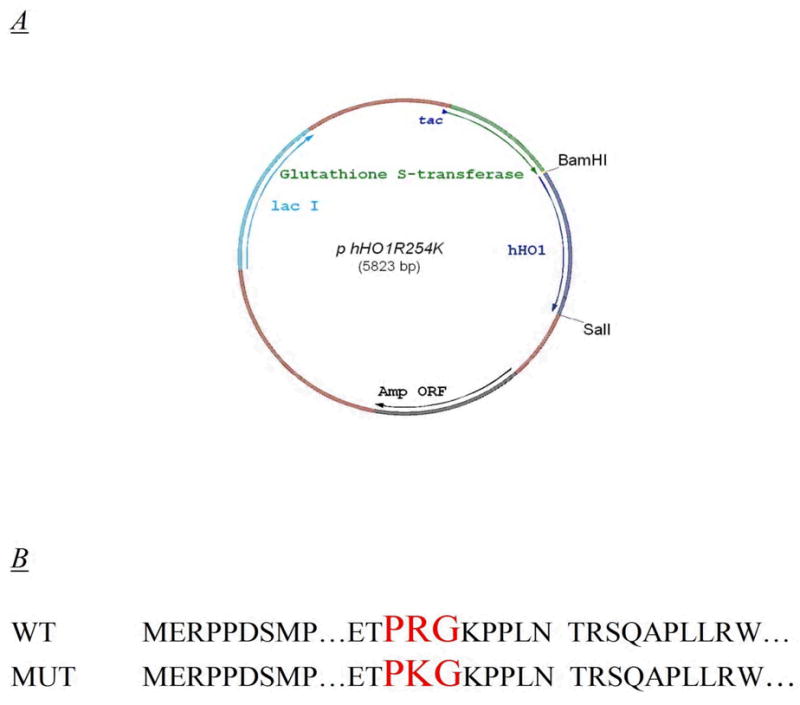
A. Expression vector pGEXhHO1R254K encoding for the R254K mutation. The 288 amino acid hHO-1 gene was ligated into a pGEX 4T-2 vector between BamHI and SalI. The R254K expression plasmid was generated by a single site mutation to the parental vector. B. DNA sequence illustrating R254K mutation. The conservative R254K mutation to the nucleotide sequence is shown here in comparison to the wild type sequence.
Protein determination and hemin titration of R254K hHO-1
From 3 liters of terrific broth, approximately 4.8 mg full-length hHO-1 (~132 nmoles) was purified. Other fractions and specific activities are listed in the purification table (Table 2). The specific activity of the full-length R254K hHO-1 was 225 nmol bilirubin hr−1 nmol−1, which is almost a 2-fold increase to that reported for the WT enzyme (Table 1) and the previously reported truncated hHO1 (21). The specific activity for the full-length R254K hHO-1 reported here is comparable to earlier forms such as rat (17, 22), bovine (18), and pig (16). The hemin titration, as described for the WT HO-1, estimated the yield of full-length hHO1 to be 12uM, for a total of 132 nmoles (data not shown). Because the conservative amino acid substitution eliminates truncation to the 30-kDa contaminant, determining the concentration of full-length HO-1 was more reliable for the R254K in comparison to the WT.
Table 2.
Purification table of R254K HO-1. The single site mutation to the WT expression vector eliminates formation of the 30-kDa hHO-1 and allows for purification of a homogenous full-length hHO-1. The specific activity of the full-length hHO-1 is 6058 nmol hr−1 mg−1, which is a 33% increase of that calculated for the WT that contains some contamination with the 30-kDa truncated species.
| Fraction | Protein (mg) | Specific activity (nmol hr−1mg−1) | Specific activity (nmol hr−1nmol−1) | Units | Purifcation (x-fold) | Specific Content (nmol mg−1) |
|---|---|---|---|---|---|---|
| Cell Supernatant (crude) | 1125 | 111.6 | - | 125550 | 1 | - |
| 1% Sarkosyl Supernatant | 903.4 | 562.8 | - | 508433 | 5 | - |
| Thrombin elution | _ | _ | - | _ | _ | - |
| 2% octyl- glucoside elution | 4.81 | 6058.2 | 200 | 29140 | 54 | 27.5 |
CPR titration of WT, R254K, and sHO1
The goal of the next study was to begin to characterize the full-length mutant hHO-1 by examining its behavior over a range of CPR concentrations, and to compare these characteristics with the partially pure WT and pure sHO-1 forms. These experiments utilized the standard HO assay at saturating CPR (3:1 CPR: HO-1). Under these conditions, the full-length protein has a specific activity twice that of the sHO1. The sHO-1 showed almost a linear increase with increasing CPR (Fig. 5). The WT form was similar to that for the sHO-1, but showed a slight increase in overall activity and appeared to saturate with excess CPR. Full-length R254K however, had a much higher activity, particularly at sub-saturating CPR, indicating an increase in affinity for its redox partner, CPR. One explanation for this increase in affinity for CPR and overall activity of the full-length enzyme can be attributed to the presence C-terminal 23 amino acids, which were deleted from the sHO-1 (21). The membrane spanning domain may play an important role in the physical interaction between HO-1 and CPR. Ideally the R254K and WT titration curves should be similar; however these results may be confounded by the presence of the 29–30-kDa contaminant in the WT enzyme not present in the R254K. Previous HO-1 studies (19, 21, 31) as well as the current study (Fig. 5) have shown that truncated or degraded forms of HO-1, specifically 27–28-kDa and 29–30-kDa, retain the ability to interact with CPR. As a result, the less active contaminant band seen in the WT sample potentially competes with the WT decreasing overall activity (Fig. 5).
Fig. 5. Effect of varying CPR levels on WT, R254K, and sHO1.

Rate of bilirubin formation was monitored in reactions containing 0.1 nmoles of HO-1 and CPR levels ranging from 0uM to 0.4uM. The homogenous full-length HO-1 (R254K) exhibited an increased activity and binding affinity for CPR, in comparison to the WT and sHO-1 enzymes. WT HO-1 had a slightly higher overall activity than the sHO-1.
CPR titration of GST-tagged full-length hHO-1
The goal of these experiments was to determine if the difference in activity of R254K HO-1 as compared to the WT enzyme was due to (1) the amino acid substitution or (2) contamination of the WT enzyme with truncated species. First, this was tested by comparing the activities of the WT and R254K enzymes without the cleavage of the GST-tag. Retention of the GST-tag eliminated the need for thrombin treatment which minimized degradation of the C-terminal amino acids. Without the contaminant, the GST-WT curve resembles that seen with the R254K (Fig. 6). These data suggest that this conservative mutation did not alter the kinetic characteristics of the full-length protein and that the decrease in WT activity may be attributable to contamination by truncated HO-1. The R254K was also purified with the C-terminal affinity tag to examine the effect of the GST on catalytic activity. As seen in Figure 6, the GST-tagged R254K curve strongly resembles the R254K, implying that the GST does not interfere with protein’s ability to degrade heme and does not appear to influence its association with CPR. These results suggest that the R to K amino acid substitution does not significantly affect the kinetic behavior of hHO-1. Future studies will be designed to characterize the effect of the GST-tag at low CPR concentrations
Fig. 6. Effect of CPR on catalytic activity of GST-tagged full-length HO-1.

In order to obtain WT HO-1 without the truncated species, WT HO-1 was purified with the GST-tag intact. The activity of the GST-WT was comparable to that seen with the R254K, suggesting that the single-site mutation was not involved in the increased activity. GST-R254K was also purified to determine if the GST-tag affected the catalytic characteristics of the full-length form.
Next, the potential for the kinetic differences between the R254K and WT proteins (Fig 5) to be due to the contamination of the WT protein with a truncated HO-1 was examined. To test this idea, sHO-1 was added to the full-length R254K HO-1 in amounts comparable to those found in the WT preparations. Densitometry of SDS-polyacrylamide gels was used to determine that 40–50% of the calculated WT sample was in fact the 29–30-kDa contaminant. With the addition of sHO1 to the full-length MUT, the activity curves declined to levels seen with the WT enzyme (Figure 7). The addition of 40% and 50% sHO1 to MUT HO1 (0.1uM final HO1) provides an overall activity that very closely resembles the WT curves. These results strongly suggest that the R to K substitution does not significantly alter the kinetics of the HO-1 protein, and that the apparent difference between the WT and R254K forms is due to contamination of WT hHO-1 with a truncated species that competes with the full-length form for CPR.
Fig. 7. Effect of the addition of sHO-1 on the catalytic characteristics of R254K.

Densitometry of SDS polyacrylamide gels was used to determine that the WT contained approximately 40–50% of a 29–30-kDa contaminant. To examine the effects of the degraded HO-1 in the WT, assays containing a combination of full-length R254K and sHO-1 were performed. A final concentration of 0.1uM HO-1 was used (60:40 and 50:50, R254K:sHO1 respectively). The addition of sHO-1 to the R254K decreased overall activity and altered the shape of the curve to more closely resemble that seen with the WT HO-1.
DISCUSSION
Full-length human HO-1 has not been well characterized due to a difficult purification procedure, lack of source tissue, and its instability. Some studies have been done using full-length HO-1 from other species (8, 9, 16–18); but most studies, including recent crystal structures and catalytic characterizations of human HO-1 have focused on the truncated forms (12, 21, 31–38). A 30-kDa soluble hHO-1, which lacks the 23 C-terminal amino acids that are thought to serve as the membrane spanning region of the enzyme, was purified and crystallized (12, 21). Rat HO-1 has also been expressed and purified in the soluble, truncated form (22), allowing for the enzyme to be studied in great detail. Although truncated rat and human HO-1 expression plasmids allow for a more rapid purification at high concentrations, its catalytic characteristics differ significantly from those of the full-length form.
With the overall goal of examining the catalytic behavior of HO-1 in the membrane, we believed that inclusion of the 23 C-terminal amino acid membrane binding domain on HO-1 was essential to more accurately address the interactions among HO-1, CPR, and the membrane. Consequently, we began purification of the full-length hHO-1 using a GST-affinity tagged construct to provide a more rapid purification (25). Using this procedure, we were able to purify GST-tagged, full-length hHO-1. The removal of the 26-kDa affinity tag was necessary to accurately study hHO-1 in protein-protein interaction experiments. However, during the thrombin cleavage procedure, there was a duration-dependent accumulation of 27 and 30-kDa contaminant bands at the expense of the 32.8-kDa form. Formation of the 30-kDa protein was attributed to the presence of a putative thrombin clip site found within the C-terminal region of the WT protein. The single-site mutation R254K eliminated this site and allowed for the isolation of a single 32.8-kDa protein. The full-length hHO-1 produced similar spectra in comparison to previously reported forms. The ferric heme-hHO-1 and ferrous carbon monoxide complexes are virtually identical to those previously reported for rat and human HO-1(17, 21, 22). Interestingly, this procedure enabled the purification of this enzyme in the apo form. The hemin titration, which was developed from a previous method in order to quantify the hHO-1, allowed for an accurate method of determining the concentration of the apo hHO-1 (16).
The activity of full-length HO-1 was characterized using a coupled assay containing biliverdin reductase to allow detection of bilirubin formation (29). When comparing the full-length form to the soluble HO-1, significant differences in kinetic characteristics were found. The most prominent characteristics were the higher activity of the full-length form and the substantially higher affinity of the full-length form for CPR. These effects could result either from anchoring and proper orientation of HO-1 in the membrane, or from a conformational difference in the full-length form that facilitates CPR-HO-1 complex formation. These differences illustrate the importance of the C-terminal region of HO-1 not only in membrane binding of the enzyme(13, 20), but also in its ability to interact with CPR as speculated by Wang et al. (35), and suggest a significant conformational difference from the soluble forms.
Another possible explanation for the diminished sHO-1 activity is an altered electron transfer from CPR. Yoshida et al. report on the inability of heme bound to a 28-kDa tryptic peptide of HO-1 (lacking hydrophobic segment) to accept the necessary reducing equivalents for the conversion of heme to biliverdin (19). The contaminant present in the WT hHO-1 affects overall activity in two ways. The 30-kDa form leads to inaccurate protein concentration of the full-length form, due to the indistinguishable heme-HO complex of the different forms. Furthermore, truncated forms of HO-1 have the potential to interact with CPR, shown by the ability of sHO1 to metabolize heme (19, 21, 22). The ability of the contaminant to bind CPR leads to a competition between the truncated and full-length HO-1, which is present at a fraction of the total HO-1. This competition and inaccurate quantification is alleviated with the MUT hHO-1, due to the absence of the truncated protein; allowing for an increased rate of bilirubin formation, particularly at sub-saturating levels of CPR.
One inconsistency seen with HO-1 characterization pertains to the variable specific activities reported throughout the literature (8, 16–18, 21, 22, 31). Although some of these numbers differ, they may represent an accurate measurement of a particular HO-1 sample. As described above, there have been various reports citing the ability of HO-1 to degrade during the purification process (16, 18, 19). Sensitivity to temperature, time, freeze-thaw, and proteolysis make for a difficult purification of homogenous full-length protein. Contaminated or degraded forms of HO-1 affect the overall activity, and may be the source of the unpredictability encountered when calculating the specific activity of the various forms of HO-1. Following the initial purification and SDS-PAGE analysis, we see a single band present. After multiple freeze-thaw cycles or exposure to 4°C for an extended period of time, we begin to see the formation of a 27–28-kDA contaminant band. Liu et al. reported on the spontaneous “aging” of HO in Nesseria meningitides in which a portion of the C-terminal region is cleaved (39). The “aging” hypothesis offers one explanation of the spontaneous degradation seen here with hHO-1 and other previously reported forms (16, 17). Although degraded products remain troublesome throughout the purification procedure, recent reports suggest that these contaminants may participate in nuclear localization and down stream signaling (40). Lin et al. describe the ability of the 27-kDa form to translocate to the cell nucleus and activate various transcription factors during oxidative stress. These recent findings suggest that the spontaneous degradation of hHO-1 may occur naturally; hence, playing an important physiological role in intracellular signaling and protection from oxidative stress.
We have purified a full-length, stable form of HO-1 by site-directed mutagenesis. This full-length R254K HO-1 has a higher affinity for CPR than does the truncated 30-kDa species, suggestive of an important role for the C-terminal region in the CPR-HO-1 interaction. We also observe that the R254K HO-1 has similar kinetic characteristics as does the full-length WT HO-1, with the differences in kinetic behavior being due to contamination of WT enzyme by truncated species. Further studies will be necessary to more completely characterize the behavior of full-length HO-1, particularly with respect to substrate binding, the effect of phospholipid, and potential to interact with other ER-resident proteins.
Acknowledgments
These studies were supported by a US Public Health Service Research Grant from the National Institute of Enviromental Health Sciences ES004344 (WLB)
We are grateful to Dr. Mahin Maines and Dr. Paul Ortiz de Montellano for supplying the full-length and truncated hHO-1 expression plasmids, respectively. The rat CPR-K56Q expression plasmid was kindly provided by Dr. Grover P. Miller. We would also like to thank Dr. Jawed Alam for his critical review of this manuscript.
ABBREVIATIONS USED:
- HO
Heme oxygenase
- HO-1
Heme oxygenase-1
- hHO-1
Human HO-1
- NADPH
Nicotinamide adenine dinucleotide phosphate, reduced form
- CPR
NADPH cytochrome P450reductase
- GST
Glutathione-s-transferase
- WT
Recombinant, wild type hHO-1
- R254K
R254K mutant hHO-1
- sHO-1
truncated, 30-kDa hHO-1
- GST-hHO-1
GST-tagged hHO-1
- CO
Carbon monoxide
- PBS
Phosphate buffered saline, pH 7.3
- PMSF
Phenylmethanesulfonyl fluoride
- DTT
Dithiothreitol
- TB
Terrific broth
- LB
Luria broth
- Sarkosyl
Sodium N-lauroylsarcosinate
- BCA
Bicinchoninic acid
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- ER
Endoplasmic reticulum
Footnotes
Warren J. Huber III and Wayne L. Backes, Quantitation of Heme Oxygenase-1: Heme Titration Increases Yield of Purified Protein, Analytical Biochemistry (submitted).
References
- 1.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244:6388–6394. [PubMed] [Google Scholar]
- 2.Alam J. Heme oxygenase-1: past, present, and future. Antioxid Redox Signal. 2002;4:559–562. doi: 10.1089/15230860260220049. [DOI] [PubMed] [Google Scholar]
- 3.Schmid R. In: Academic Press. Dolphin D, McDonagh AF, editors. New York: 1979. pp. 257–292. [Google Scholar]
- 4.Hyman CB, Keaster J, Hanson V, Harris I, Sedgwick R, Wursten H, Wright AR. CNS abnormalities after neonatal hemolytic disease or hyperbilirubinemia. A prospective study of 405 patients. Am J Dis Child. 1969;117:395–405. doi: 10.1001/archpedi.1969.02100030397002. [DOI] [PubMed] [Google Scholar]
- 5.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259:381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 6.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 7.Snyder SH, Jaffrey SR, Zakhary R. Nitric oxide and carbon monoxide: parallel roles as neural messengers. Brain Res Brain Res Rev. 1998;26:167–175. doi: 10.1016/s0165-0173(97)00032-5. [DOI] [PubMed] [Google Scholar]
- 8.Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. 1986;261:411–419. [PubMed] [Google Scholar]
- 9.Maines MD, Ibrahim NG, Kappas A. Solubilization and partial purification of heme oxygenase from rat liver. J Biol Chem. 1977;252:5900–5903. [PubMed] [Google Scholar]
- 10.Maines MD. CRC Press, Inc. Boca Raton, FL: 1992. pp. 203–266. [Google Scholar]
- 11.Trakshel GM, Kutty RK, Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem. 1986;261:11131–11137. [PubMed] [Google Scholar]
- 12.Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida T, Sato M. Posttranslational and direct integration of heme oxygenase into microsomes. Biochem Biophys Res Commun. 1989;163:1086–1092. doi: 10.1016/0006-291x(89)92332-2. [DOI] [PubMed] [Google Scholar]
- 14.Rotenberg MO, Maines MD. Characterization of a cDNA-encoding rabbit brain heme oxygenase-2 and identification of a conserved domain among mammalian heme oxygenase isozymes: possible heme-binding site? Arch Biochem Biophys. 1991;290:336–344. doi: 10.1016/0003-9861(91)90549-x. [DOI] [PubMed] [Google Scholar]
- 15.Zakhary R, Poss KD, Jaffrey SR, Ferris CD, Tonegawa S, Snyder SH. Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon monoxide. Proc Natl Acad Sci U S A. 1997;94:14848–14853. doi: 10.1073/pnas.94.26.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida T, Kikuchi G. Purification and properties of heme oxygenase from pig spleen microsomes. J Biol Chem. 1978;253:4224–4229. [PubMed] [Google Scholar]
- 17.Yoshida T, Kikuchi G. Purification and properties of heme oxygenase from rat liver microsomes. J Biol Chem. 1979;254:4487–4491. [PubMed] [Google Scholar]
- 18.Yoshinaga T, Sassa S, Kappas A. Purification and properties of bovine spleen heme oxygenase. Amino acid composition and sites of action of inhibitors of heme oxidation. J Biol Chem. 1982;257:7778–7785. [PubMed] [Google Scholar]
- 19.Yoshida T, Ishikawa K, Sato M. Degradation of heme by a soluble peptide of heme oxygenase obtained from rat liver microsomes by mild trypsinization. Eur J Biochem. 1991;199:729–733. doi: 10.1111/j.1432-1033.1991.tb16177.x. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa K, Sato M, Yoshida T. Expression of rat heme oxygenase in Escherichia coli as a catalytically active, full-length form that binds to bacterial membranes. Eur J Biochem. 1991;202:161–165. doi: 10.1111/j.1432-1033.1991.tb16357.x. [DOI] [PubMed] [Google Scholar]
- 21.Wilks A, Black SM, Miller WL, Ortiz de Montellano PR. Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase. Biochem. 1995;34:4421–4427. doi: 10.1021/bi00013a034. [DOI] [PubMed] [Google Scholar]
- 22.Wilks A, Ortiz de Montellano PR. Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species. J Biol Chem. 1993;268:22357–22362. [PubMed] [Google Scholar]
- 23.Yasukochi Y, Masters BS. Some properties of a detergent-solubilized NADPH-cytochrome c(cytochrome P-450) reductase purified by biospecific affinity chromatography. J Biol Chem. 1976;251:5337–5344. [PubMed] [Google Scholar]
- 24.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 25.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 26.Frankel S, Sohn R, Leinwand L. The use of sarkosyl in generating soluble protein after bacterial expression. Proc Natl Acad Sci U S A. 1991;88:1192–1196. doi: 10.1073/pnas.88.4.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 28.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 29.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutty RK, Maines MD. Purification and characterization of biliverdin reductase from rat liver. J Biol Chem. 1981;256:3956–3962. [PubMed] [Google Scholar]
- 31.Wilks A, Medzihradszky KF, Ortiz de Montellano PR. Heme oxygenase active-site residues identified by heme-protein cross-linking during reduction of CBrCl3. Biochem. 1998;37:2889–2896. doi: 10.1021/bi972720x. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Ortiz de Montellano PR. Reaction intermediates and single turnover rate constants for the oxidation of heme by human heme oxygenase-1. J Biol Chem. 2000;275:5297–5307. doi: 10.1074/jbc.275.8.5297. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Moenne-Loccoz P, Loehr TM, Ortiz de Montellano PR. Heme oxygenase-1, intermediates in verdoheme formation and the requirement for reduction equivalents. J Biol Chem. 1997;272:6909–6917. doi: 10.1074/jbc.272.11.6909. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Lu S, Moenne-Loccoz P, Ortiz de Montellano PR. Interaction of nitric oxide with human heme oxygenase-1. J Biol Chem. 2003;278:2341–2347. doi: 10.1074/jbc.M211131200. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, de Montellano PRO. The Binding Sites on Human Heme Oxygenase-1 for Cytochrome P450 Reductase and Biliverdin Reductase. J Biol Chem. 2003;278:20069. doi: 10.1074/jbc.M300989200. [DOI] [PubMed] [Google Scholar]
- 36.Lad L, Schuller DJ, Shimizu H, Friedman J, Li H, Ortiz de Montellano PR, Poulos TL. Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J Biol Chem. 2003;278:7834–7843. doi: 10.1074/jbc.M211450200. [DOI] [PubMed] [Google Scholar]
- 37.Lad L, Friedman J, Li H, Bhaskar B, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1 in a complex with biliverdin. Biochem. 2004;43:3793–3801. doi: 10.1021/bi035451l. [DOI] [PubMed] [Google Scholar]
- 38.Lightning LK, Huang H, Moenne-Loccoz P, Loehr TM, Schuller DJ, Poulos TL, de Montellano PR. Disruption of an active site hydrogen bond converts human heme oxygenase-1 into a peroxidase. J Bioly Chem. 2001;276:10612–10619. doi: 10.1074/jbc.M010349200. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Ma LH, Zhang X, Yoshida T, Satterlee JD, La Mar GN. Characterization of the spontaneous “aging” of the heme oxygenase from the pathological bacterium Neisseria meningitidis via cleavage of the C-terminus in contact with the substrate. Implications for functional studies and the crystal structure. Biochem. 2006;45:3875–3886. doi: 10.1021/bi0523097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, Dennery PA. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem. 2007 doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]