

Abstract
Wolfram syndrome 1 (WFS1, OMIM 222300), a rare genetic disorder characterized by optic nerve atrophy, deafness, diabetes insipidus and diabetes mellitus, is caused by mutations of WFS1, encoding WFS1/wolframin. Non-syndromic WFS1 variants are associated with the risk of diabetes mellitus due to altered function of wolframin in pancreatic islet cells, expanding the importance of wolframin. This study extends a previous report for the monkey retina, using immunohistochemistry to localize wolframin on cryostat and paraffin sections of human retina. In addition, the human retinal pigment epithelial (RPE) cell line termed ARPE-19 and retinas from both pigmented and albino mice were studied to assess wolframin localization. In the human retina, wolframin was expressed in retinal ganglion cells, optic axons and the proximal optic nerve. Wolframin expression in the human retinal pigment epithelium (RPE) was confirmed with intense cytoplasmic labeling in ARPE-19 cells. Strong labeling of the RPE was also found in the albino mouse retina. Cryostat sections of the mouse retina showed a more extended pattern of wolframin labeling, including the inner nuclear layer (INL) and photoreceptor inner segments, confirming the recent report of Kawano et al. (J. Comp. Neurol. 2008: 510, 1-23). Absence of these cells in the human specimens despite the use of human-specific antibodies to wolframin may be related to delayed fixation. Loss of wolframin function in RGCs and the unmyelinated portion of retinal axons could explain optic nerve atrophy in Wolfram Syndrome 1.
1. Introduction
Wolfram syndrome (WFS1, OMIM 222300) is a rare genetic disorder defined by the combination of diabetes insipidus, diabetes mellitus, optic nerve atrophy, and deafness, also called by the acronym DIDMOAD (Wolfram and Wagener, 1938; Barrett et al., 1997). Optic nerve atrophy is required for the diagnosis of Wolfram syndrome (Barrett et al., 1997). The causative gene, WFS1, was cloned in 1998 (Inoue et al., 1998; Strom et al., 1998) and the encoded protein was named WFS1 (Inoue et al., 1998) or wolframin (Strom et al., 1998). Wolfram syndrome was initially associated with mitochondrial dysfunction (reviewed by Barrett, 2000). Subsequently, wolframin was characterized as an integral, endoglycosidase H-sensitive membrane glycoprotein localized to the endoplasmic reticulum (ER) (Takeda et al., 2001; Hofmann et al., 2003; Osman et al., 2003). Wolframin plays a role in regulating calcium fluxes of the ER (Osman et al., 2003; Takei et al., 2006). Functional studies have focused on the role of wolframin in islet cells in relation to diabetes (Takeda et al., 2001; Osman et al., 2003; Ishihara et al., 2004,Fonseca et al., 2005; Philbrook et al., 2005; Ueda et al., 2005; Riggs et al., 2005; Yamada et al., 2006;). Wolfram syndrome is a recessive disorder; functional studies indicate a loss of function mechanism (Hofmann and Bauer, 2006), and local gene replacement therapy may be considered in the future.
Dysfunction of the ER provides a new pathway for optic nerve degeneration which is otherwise strongly associated with mitochondrial disorders (Carelli et al., 2004; Newman, 2005). To elucidate the pathophysiology of optic nerve atrophy, it is essential to know what cells express the protein. In a preliminary study of paraffin-sections of immersion-fixed Cynomolgus monkey eye cups, we found that wolframin was primarily localized in retinal ganglion cells (RGCs) and in glial cells in the proximal portion of the optic nerve (Yamamoto et al., 2006). Some cells in the inner nuclear layer were also labeled whereas faint immunoreactivity was present in the inner segment of photoreceptors. We proposed that dysfunction and/or loss of RGCs may be sufficient to cause optic nerve degeneration in Wolfram syndrome, and we pointed out that expression of the mutant protein in optic nerve glial cells could be also involved (Yamamoto et al., 2006). A recent very detailed study of wolframin examined perfusion-fixed cryostat sections of mouse retina and optic nerve (Kawano et al., 2008). Their results are consistent with ours in finding wolframin expression in RGCs, cells in the inner nuclear layer, photoreceptors, and glial cells of the optic nerve. Wolframin was also detected in amacrine cells, bipolar cells and Müller cells of the mouse retina (Kawano et al., 2008). The difference could be due to technical reasons, in that immersion-fixation and/or paraffin embedding may have masked immunoreactivity in our study on the monkey retina. However, species differences cannot be excluded. Therefore, this paper expands our analysis of wolframin expression to the human retina. Since human retinal specimens are fixed by immersion, we expect the immunoreactivity to be similar to that in the monkey study, i.e. cells with lower expression may not be detectable. Therefore, we used the same antibodies to wolframin to label sections of the mouse retina after immersion fixation and compared the results to those of Kawano et al. (2008).
Expression of wolframin in the retina raises additional issues. First, diabetes mellitus is an important element of Wolfram syndrome which can be related to the high expression of wolframin in pancreatic beta cells (Philbrook et al., 2005). Disruption of Wfs1 in mice caused progressive beta-cell loss and impaired stimulus-secretion coupling of insulin secretion (Ishihara et al., 2004). Recently, common gene variants of WFS1 (not causing Wolfram syndrome) have been linked to the risk of type 2 diabetes (Sandhu et al., 2007; Wasson and Permutt, 2008; Florez et al., 2008; Frank et al., 2008; Sparsoe et al., 2008). These WFS1 variants affect the function of wolframin in islet cells and contribute to an altered risk of diabetes (Sandhu et al., 2007). Diabetic retinopathy has been related to the effects of abnormal glucose levels on the inner retina, resulting in vascular pathology and abnormal local retinal metabolism (Frank, 2004). Recent studies have shown an early loss of RGCs (Gastinger et al., 2008), suggesting that wolframin expression in RGCs itself could also become involved. Second, reports of a pigmentary dystrophy in some patients with Wolfram syndrome (Al-Till et al., 2002) suggest that the RPE may be involved. Some expression of wolframin was noted in RPE cells of the monkey (Yamamoto et al., 2006). To analyze expression in the RPE, we examined wolframin expression in albino mouse retina and in the human RPE cell line (ARPE-19 cells), which retain differentiated characteristics in culture (Dunn et al., 1996). Finally, we screened genetic databases to determine whether any retinal disease genes have been mapped to 4p, because non-syndromic mutations of WFS1 may be associated with optic nerve degeneration or other retinal disorders.
2. Materials and Methods
2.1 Immunohistochemistry of human donor eyes
Five eyes from non-diabetic donors were made available from the Cornea Bank Amsterdam (Netherlands Institute of Neuroscience, Amsterdam, The Netherlands). After a postmortem interval of less than six hours, eyes were fixed in 4% buffered formaldehyde followed by a 24 h wash in Tris-buffered saline. After cryoprotection with sucrose (10-20-30%), 50 μm thick cryosections through the optic nerve and retina were obtained. Affinity purified rabbit anti-human wolframin N-terminal antibody (Hofmann et al., 2003) was used as primary antibody at a dilution of 1:500-1:5,000 for 24 h. No antigen retrieval protocol was used. Detection was carried out using anti-rabbit IgG (1:200; Sigma-Aldrich, St. Louis, MO, USA) and the ABC kit (1:800; VectorLabs, Burlingame, CA, USA) followed by diaminobenzidine.
Separately, a 46 year old female donor eye became available within one hour after enucleation due to severe herpes simplex keratitis (Univ. Regensburg, Germany, obtained by M.P.). The eye was fixed overnight at 4°C in 4% paraformaldehyde in PBS (pH 7.4), embedded in paraffin, and 2 μm sections prepared. Antigen retrieval was accomplished by microwaving sections in citric acid/citrate buffer (0.2 mM citric acid, 9.8 mM sodium citrate, pH 7.3) for 2 min at 800 W and further 10 min at 240 W. Immunohistochemistry was performed using the avidin-biotin technique. The wolframin antibody (1:500) was incubated using Shandon Coverplates (Thermo, Pittsburgh, PA, USA) and detected using the DakoCytomation LSAB2 System-HRP (K0672, DakoCytomation, Carpinteria, CA, USA) used for detection according to the manufacturers instructions. Sections were dehydrated and coverslipped.
2.2 Immunohistochemistry of mouse eyes
Eyes were enucleated from C57Bl/6 pigmented mice and Balb/c albino mice (n=4; 10-12 weeks old) and placed into 4% paraformaldehyde in PBS. After 30 min fixation, the cornea and lens were removed, and the eye cup was fixed for an additional few hours before it was rinsed in PBS and stored in PBS. Eyes were cryoprotected by incubation for 30 minutes each in an ascending series of sucrose in phosphate buffer (10% - 15%), kept overnight in 20% sucrose, then infiltrated 30 minutes and embedded in a 20% sucrose-OCT (2:1) mixture for 30 minutes before freezing. Cryosections (8 μm) on glass slides were rehydrated with PBS and incubated for 1 hr in blocking solution (PBS containing 4% goat serum and 0.2% Triton X-100) and stained overnight with rabbit anti-wolframin antibody (1:100 in blocking solution) followed by Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA). Sections were mounted with Vectashield hardset containing DAPI stain (VectorLabs). Sections were viewed with a Zeiss Axiovert 35 microscope equipped with a SPOT camera (RTKE, Diagnostic Instruments, Inc., Sterling Heights, MI) for image capture. The results obtained using antibody to human wolframin in the mouse retina were consistent with another study using an antibody specific to mouse wolframin in the mouse retina (Kawano et al., 2008), suggesting that sequence differences in the N-terminal region of human and mouse wolframin did not influence the interpretation.
2.3 Immunohistochemistry of ARPE-19 cell line
ARPE-19 cells (obtained from ATCC; Dunn et al., 1996) were seeded on coverslips and cultured in 6-well plates in DMEM/F12 media containing 10% fetal calf serum for three days. Cells were fixed by 10 min exposure to 2-4% paraformaldehyde and washed with PBS. Coverslips were coated with rabbit-anti wolframin (2 h, 1:500 in PBS/0.3% Triton X-100), washed with PBS-0.3% Triton X-100, covered with Alexa 555-conjugated goat anti-rabbit IgG (1 h, 1:50 in PBS; Invitrogen), washed with 3 changes of PBS and mounted onto glass slides with Vectashield containing DAPI for nuclear counterstaining (VectorLabs). Omission of the primary antibody served as a control. Cells were viewed with a Zeiss Axiovert 35 microscope equipped with a SPOT camera. Dual label images were processed with SPOT Imaging Software and Photoshop (Adobe).
2.4 Database searches
WFS1 expression was analyzed in the Eyebrowse and EyeSAGE databases at NEIBank (Bowes Rickman et al., 2006) and in the original literature (Radeke et al., 2007). We asked whether the gene locus for WFS1 (4p16) overlaps with loci to which retinal disease genes have been mapped. This search was carried out using RetNet, RISN and NEI (Candidate Disease Loci). Since we found wolframin expressed in the RPE (see Results) and pigmentary maculopathy was described in patients with Wolfram syndrome (Al-Till et al., 2002; Dhalla et al., 2006), we screened the literature for genomic studies on age-related macular degeneration (AMD) reporting with linkage to 4p.
3. Results
3.1 Immunohistochemistry of human retinal sections
Wolframin was clearly expressed in the fiber layer and optic nerve head, but was virtually absent in the myelinated portion of the optic nerve (Fig. 1). Very few cell bodies of glial cells were labeled in the optic nerve in cryostat sections. RGCs themselves were strongly positive in their cytoplasm and proximal processes (Fig. 2). A few cells were stained in the inner nuclear layer (INL), and photoreceptor inner segments were sometimes faintly positive. In some areas, specific immunoreactivity was seen below the pigment in the basal cytoplasm of RPE cells. In paraffin sections of the rapidly fixed eye, RGCs and inner segments of photoreceptors were immunoreactive for wolframin whereas few cells were positive in the INL.
Figure 1.
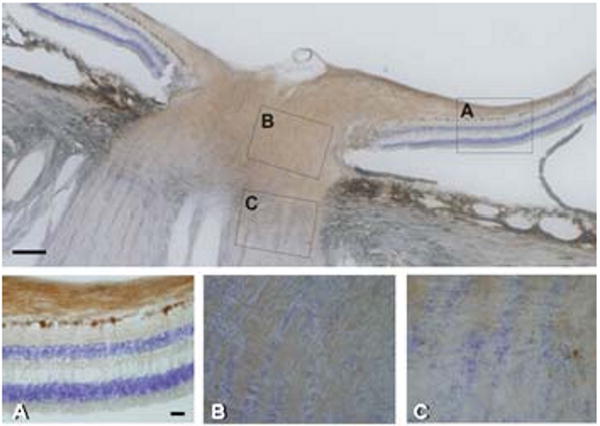
Representative overview of human retina and optic nerve immunostained for wolframin. Boxes in the upper panel designate regions of magnified images. A) Retina with optic nerve layer showing intense expression of wolframin in the nerve fiber layer and retinal ganglion cells. B) Optic nerve head with positive retinal ganglion cell axons. C) Transition to optic nerve with reduction of labeling at the interface of myelination. Magnification bar in upper panel equivalent to 100 μm; and magnification bar in panel A) equivalent to 40 μm for A) to C). Section is counterstained with cresyl violet.
Figure 2.
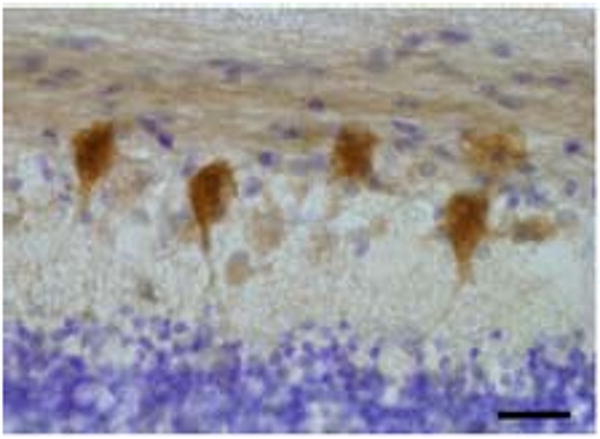
Representative high power image of human retina immunoreactive for wolframin. Retinal ganglion cells are intensely labeled for wolframin. Section is counterstained with cresyl violet. Magnification bar equivalent to 40 μm.
3.2 Immunohistochemistry of mouse retinal sections
Pigmented and albino mice showed strong wolframin labeling of neurons in the ganglion cell layer (Fig. 3a). In addition, strong label was observed over cells on the outer aspect of the INL, on scattered Müller cells and on their processes stretching across the diffusely labelled inner plexiform layer. Immunoreactivity was also observed over photoreceptor inner segments and arching over the distal end of the unstained nuclei. These results in the mouse retina are identical to those described by Kawano et al. (2008) suggesting that our antibodies to wolframin yielded the same labeling pattern. Inspection of the RPE in the pigmented mice showed occasional regions where labeling could be detected. Strong labeling of the RPE cytoplasm was found in the albino mouse (Fig. 3b). Double labeling of nuclei with DAPI was complementary to the pattern of cytoplasmic labeling of wolframin. No axonal staining was observed in the optic nerve and glial staining was very low. No labeling was present after omission of the primary antibody.
Figure 3.

Immunofluorescence labeling of wolframin in the retina of the albino mouse. A) Overview of cross section of the retina with different layers as indicated. B) Intense expression of wolframin expression in RPE. INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; IS, layer of inner segments; RGC, retinal ganglion cell layer; and RPE, retinal pigment epithelium. Magnification bar in A) equivalent to 50 μm; and in B) to 25 μm.
3.3 Expression in ARPE-19 cells
ARPE-19 cells exhibited intense cytoplasmic immunoreactivity of wolframin. The flattened RPE cells had a clearly discernable reticular pattern of labeling which was primarily concentrated in the peri-nuclear region (Fig. 4a). Double labeling with the nuclear dye, DAPI, indicated that wolframin was mostly absent from the nucleus (Fig. 4b). No cytoplasmic labeling was seen after omission of the primary antibodies (Fig. 4c).
Figure 4.
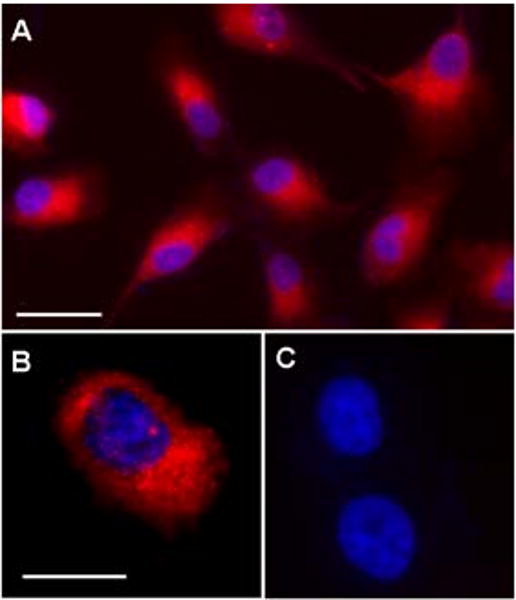
Double immunofluorescence labeling for wolframin (red) and nuclear counter stain with DAPI (blue) in ARPE-19 cells illustrating intense labeling of the RPE cytoplasm. A) Overview. B) Detailed view of double labeled cell. C) Omission control with nuclear staining by DAPI only. Magnification bar shown in A) equivalent to 40 μm; and bar in B) to 25 μm for B) and C).
3.4 Database searches
Expression of WFS1 in RPE was further confirmed using databases. RPE/choroid-derived clone CA396099 is aligned with WFS1 in the Eyebrowse data base (NEIBank). EyeSAGE lists mRNA signals in RPE (Gene Id 7466).Radeke et al. (2007) reported an exhaustive gene expression profiling of RPE/choroid of human macula and extra-macular retina, with WFS1 mRNA in the lower range of relative expression. Since specific dominant mutations of WFS1 can cause deafness in isolation (Cryns et al., 2002), novel mutations of WFS1 could also cause isolated optic nerve atrophy or other retinal disorders. The gene locus for WFS1 maps to chromosome 4p16 at 6.3 Mb (Human Genome Browser map; UCSC). None of the linkage sites for optic nerve atrophy (OPA) have been mapped to this locus. Some Wolfram syndrome patients have a pigmentary maculopathy (Al-Till et al., 2002; Dhalla et al., 2006), suggesting that wolframin dysfunction can lead to macular pathology which is supported by the expression of wolframin in the RPE found in the present study. A survey of linkage studies for AMD revealed one study with linkage to 4p16 at marker D4S3360 (Iyengar et al., 2004).
4. Discussion
Wolfram syndrome is a rare monogenic disorder associated with optic nerve atrophy. In addition, common variants of wolframin may play a role in diabetes. The present study used immunohistochemistry with an affinity-purified antibody to the N-terminal region of human wolframin (Hofmann et al., 2003) to analyze the distribution of wolframin in the human retina. To estimate the effects of fixation and tissue processing on labeling of various retinal cells, we also studied well-fixed mouse retina with our antibodies and compared the results to a previous study using mouse-specific antibodies to wolframin (Kawano et al., 2008).
The main finding of our study was that the cytoplasm of RGCs was positive for wolframin in cryosections and paraffin sections of the immersion-fixed human retina. Consistent localization of wolframin in RGCs was also obtained in paraffin sections of immersion-fixed monkey retina (Yamamoto et al., 2006), in cryostat sections of perfusion-fixed mouse retina (Kawano et al., 2008) and in cryostat sections of immersion fixed mouse retina (this study). Thus, RGCs express high levels of the ER protein wolframin which corresponds to the presence of an elaborate ER in these neurons. Our observation of wolframin in the optic nerve layer and in fibers transversing the optic nerve head in the human eye is compatible with modern concepts of ER function in axons (Verkhratsky, 2005). In contrast to the human retina, little axonal labeling was observed in the mouse retina stained with antibodies to either human (this study) or mouse wolframin (Kawano et al. 2008). Glial cells were not labeled for wolframin in the human optic nerve layer or optic nerve. By contrast, astrocytes in the mouse optic nerve expressed wolframin (Kawano et al., 2008), and similar findings were made in the monkey optic nerve (Yamamoto et al., 2006). Few, if any, wolframin-positive cells were found in the INL of human retinal specimens, when cryostat sections were used. Wolframin-positive cells were previously noted in the INL of the monkey retina (Yamamoto et al., 2006). Our data in the mouse retina confirm the intense immunolabeling of Müller glial cells and neurons of the INL that were reported in the mouse by Kawano et al. (2008). Signals were also found in the inner segments of the mouse, as previously reported (Kawano et al., 2008). No labeling was found in the photoreceptor layer of the human eye in the study using cryostat sections, and only faint immunoreactivity was seen in the inner segments in the monkey study (Yamamoto et al., 2006). The antibodies were raised against the N-terminal sequence of human wolframin (Hofmann et al., 2003), and therefore positive findings in the human retina should be reliable in terms of protein detection. Absence of labeling for neurons of the INL, photoreceptors and glial cells in the human specimen as compared to the mouse retina (Kawano et al., 2008; this study) is best explained by loss of immunoreactivity due to delayed fixation of the human eyes. However, species differences in the levels of wolframin expression in different types of retinal cells cannot be excluded. The strong labeling for wolframin in axons in the fiber layer or optic nerve head of the human eye and the absence of axonal labeling in the mouse (both with the present antibody and the mouse-specific antibody in the study of Kawano et al., 2008) could reflect a true species difference, i.e the relative length of the unmyelinated axons. Paraffin embedding may have depressed axonal labeling in the previous study on the monkey retina (Yamamoto et al., 2006). Renewed inspection of these sections did indeed indicate weak axonal labeling (unpublished observations).
An open question then is how WFS1 mutations contributes to optic nerve atrophy in patients affected by Wolfram syndrome. Wolframin is abundant in human RCG cell bodies and the initial portion of the axons. Thus, RGCs could be slowly affected by wolframin dysfunction in the ER of the cell body itself, leading to deficits in protein synthesis, deficits in axonal transport, and, ultimately, optic nerve atrophy. Studies on optic nerve atrophy in the context of glaucoma models have shown that axons can show signs of degeneration while cell bodies of RGCs appear intact (Nickells, 2007). In the human retina, the axons of RGCs remain unmyelinated while they run in the optic fiber layer and proximal optic nerve. Propagation of action potentials in the unmyelinated portion presumably is associated with higher energy consumption than in the myelinated portion. The high density of mitochondria observed in the unmyelinated portion of optic axons has been related to the detrimental effects of mutations of the optic atrophy 1 - OPA1 protein which serves in mitochondrial functions (Delettre et al., 2000; Man et al., 2002). In a similar way, the ER could play an important role in the unmyelinated portion of the optic nerve, and mutations of the ER-protein wolframin could perturb important functions, e.g. regulation of axonal Ca2+ levels. If wolframin plays a role in optic nerve astrocytes (Yamamoto et al., 2006; Kawano et al., 2008), a dual problem would arise, because the astrocytes may fail to provide nutrients to optic axons when both are suffering from ER dysfunction due to wolframin mutations.
Dysfunction of wolframin could play a more general role in several disease processes of RGCs and the optic nerve. Wolframin plays a role in membrane trafficking, protein processing and regulation of calcium (Ca2+) homeostasis in the ER (Takeda et al., 2001; Osman et al., 2003). Ion channel activity of wolframin in the ER suggests a role in the regulation of intracellular Ca2+ homeostasis, possibly involving interactions with ER Ca2+ channels such as the IP3-receptor (Osman et al., 2003) and with calmodulin (Yurimoto et al., 2009). One of the proteins associated with wolframin is the Na+-K+-ATPase beta 1 subunit (Zatyka et al., 2008) which plays a key role in neuronal membranes. Functional studies have indicated a role for wolframin in ER stress and the unfolded protein response (UPR) (Yamaguchi et al., 2004; Ueda et al., 2005; Fonseca et al., 2005). ER dysfunction and the UPR have been associated with pathophysiologic mechanisms of neuronal cell death after brain ischemia (Paschen and Doutheil, 1999). ER stress was detected in mouse RGCs subjected to ischemia or excitotoxic injury (Awai et al., 2006; Shimazawa et al., 2007), and excessive ER stress induced cell death in a retinal ganglion cell line (Shimazawa et al., 2007). Thus, functional impairment of wolframin may play a role in RGCs degeneration in ischemia and glaucoma.
In our prior study of the monkey retina (Yamamoto et al., 2006), some immunoreactivity was found in the cytoplasm of the RPE. In the present analysis of human retinal sections, the brown peroxidase-product for wolframin was difficult to distinguish from the pigment in RPE cells. However, cultured human RPE cells (ARPE-19 cells) showed strong cytoplasmic expression of wolframin which is consistent with wolframin expression in the ER (Takeda et al., 2001). Strong labeling of the RPE was found in the albino mouse (this study) and in the Wistar rat (Schmidt-Kastner, unpublished observations). These immunohistochemical findings are in line with the listing of wolframin gene expression in human RPE / choroid cells at the NEIBank database and with gene expression profiling of the human RPE/choroid (Radeke et al., 2007). Therefore, the data has established that the human RPE cells expresses wolframin. This expression may be clinically relevant, since a pigmentary maculopathy was reported in Wolfram syndrome (Al-Till et al., 2002; Dhalla et al., 2006). Abnormal RPE function could cause photoreceptor dysfunction in Wolfram syndrome. Evidence for photoreceptor dysfunction in Wolfram syndrome includes changes in dark-adaptation (Mtanda et al., 1986) and the ERG (Barrett et al., 1997). Expression of the ER protein wolframin in the RPE directs the interest towards AMD, because a role for the ER stress response in the development of AMD has been suggested (Sauer et al., 2008).He et al. (2008) showed that oxidative stress in ARPE-19 cells induced ER stress. Intriguingly, wolframin-deficient fibroblasts upregulate fibulin-3 (Philbrook et al., 2005). Fibulin-3 is also known as EFEMP1 (OMIM 126600), mutations of which cause Doyne Honeycomb Retinal Dystrophy (DHRD) that affects the macula. Accumulation of mutant fibulin-3/EFEMP1 in the ER activated the unfolded protein response of the ER in ARPE-19 RPE cells (Roybal et al., 2005) which theoretically could involve wolframin.
The recent development of optical coherence tomography (OCT) permits repeated imaging studies of the retina and optic nerve head. OCT studies should be performed in patients affected by WFS1 to define whether early changes occur in RGCs or optic nerve head. More detailed ERG studies could shed light on the involvement of photoreceptors, and the electro-oculogram may provide more data on the RPE. The present findings may have importance for possible therapeutic approaches to optic nerve degeneration in WFS1, because the recessive gene deficit could be corrected by local gene therapy. RGCs could be made to produce normal wolframin by the intraocular application of a therapeutic viral vector expressing the corrective gene. Whether optic nerve glia, photoreceptors, and RPE would have to be targeted as well remains to be determined. Finally, it is unclear how the expression of mutant wolframin in the retina itself relates to diabetic retinopathy in patients affected by Wolfram syndrome. Initial reports indicated that patients with Wolfram syndrome have less diabetic retinopathy than would be expected from their metabolic disorder, but a systematic comparison of Wolfram syndrome patients with patients afflicted by type 1 diabetes showed no significant differences (Cano et al., 2007).
Acknowledgments
Supported by FAU College of Biomedical Science and NEI 1R15EY018947-01 grants to CKD. We thank Dr. Sabine Hofmann for providing the wolframin antibody.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Till M, Jarrah NS, Ajlouni KM. Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur J Ophthalmol. 2002;12:84–88. doi: 10.1177/112067210201200202. [DOI] [PubMed] [Google Scholar]
- Awai M, Koga T, Inomata Y, Oyadomari S, Gotoh T, Mori M, Tanihara H. NMDA-induced retinal injury is mediated by an endoplasmic reticulum stress-related protein, CHOP/GADD153. J Neurochem. 2006;96:43–52. doi: 10.1111/j.1471-4159.2005.03502.x. [DOI] [PubMed] [Google Scholar]
- Barrett TG, Bundey SE, Fielder AR, Good PA. Optic atrophy in Wolfram (DIDMOAD) syndrome. Eye. 1997;11:882–888. doi: 10.1038/eye.1997.226. [DOI] [PubMed] [Google Scholar]
- Barrett TG, Scott-Brown M, Seller A, Bednarz A, Poulton K, Poulton J. The mitochondrial genome in Wolfram syndrome. J Med Genet. 2000;37:463–6. doi: 10.1136/jmg.37.6.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes Rickman C, Ebright JN, Zavodni CJ, Yu L, Wang T, Daiger SP, Wistow G, Boon K, Hauser MA. Defining the human macula transcriptome and candidate retinal disease genes using EyeSAGE. Invest Ophthalmol Vis Sci. 2006;47:2305–2316. doi: 10.1167/iovs.05-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Molines L, Valero R, Simonin G, Paquis-Flucklinger V, Vialettes B French Group of Wolfram Syndrome. Microvascular diabetes complications in Wolfram syndrome (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness [DIDMOAD]) Diabetes Care. 2007;30:2327–2330. doi: 10.2337/dc07-0380. [DOI] [PubMed] [Google Scholar]
- Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89. doi: 10.1016/j.preteyeres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Collier DA, Barrett TG, Curtis D, Macleod A, Arranz MJ, Maassen JA, Bundey S. Linkage of Wolfram syndrome to chromosome 4p16.1 and evidence for heterogeneity. Am J Hum Genet. 1996;59:855–863. [PMC free article] [PubMed] [Google Scholar]
- Cryns K, Pfister M, Pennings RJ, Bom SJ, Flothmann K, Caethoven G, Kremer H, Schatteman I, Köln KA, Tóth T, Kupka S, Blin N, Nürnberg P, Thiele H, van de Heyning PH, Reardon W, Stephens D, Cremers CW, Smith RJ, Van Camp G. Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum Genet. 2002;110:389–394. doi: 10.1007/s00439-002-0719-1. [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Dhalla MS, Desai UR, Zuckerbrod DS. Pigmentary maculopathy in a patient with Wolfram syndrome. Can J Ophthalmol. 2006;41:38–40. doi: 10.1016/S0008-4182(06)80064-5. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Florez JC, Jablonski KA, McAteer J, Sandhu MS, Wareham NJ, Barroso I, Franks PW, Altshuler D, Knowler WC. Testing of diabetes-associated WFS1 polymorphisms in the Diabetes Prevention Program. Diabetologia. 2008;51:451–457. doi: 10.1007/s00125-007-0891-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, Urano F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem. 2005;280:39609–39615. doi: 10.1074/jbc.M507426200. [DOI] [PubMed] [Google Scholar]
- Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- Franks PW, Rolandsson O, Debenham SL, Fawcett KA, Payne F, Dina C, Froguel P, Mohlke KL, Willer C, Olsson T, Wareham NJ, Hallmans G, Barroso I, Sandhu MS. Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologia. 2008;51:458–463. doi: 10.1007/s00125-007-0887-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastinger MJ, Kunselman AR, Conboy EE, Bronson SK, Barber AJ. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Invest Ophthalmol Vis Sci. 2008;49:2635–2642. doi: 10.1167/iovs.07-0683. [DOI] [PubMed] [Google Scholar]
- He S, Yaung J, Kim YH, Barron E, Ryan SJ, Hinton DR. Endoplasmic reticulum stress induced by oxidative stress in retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 2008;246:677–683. doi: 10.1007/s00417-008-0770-2. [DOI] [PubMed] [Google Scholar]
- Hofmann S, Philbrook C, Gerbitz KD, Bauer MF. Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet. 2003;12:2003–2012. doi: 10.1093/hmg/ddg214. [DOI] [PubMed] [Google Scholar]
- Hofmann S, Bauer MF. Wolfram syndrome-associated mutations lead to instability and proteasomal degradation of wolframin. FEBS Lett. 2006;580:4000–4004. doi: 10.1016/j.febslet.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Iyengar SK, Song D, Klein BEK, Klein R, Schick JH, Humphrey J, Millard C, Liptak R, Russo K, Jun G, Lee KE, Fijal B, Elston RC. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am J Hum Genet. 2004;74:20–39. doi: 10.1086/380912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, Mueckler M, Marshall H, Donis-Keller H, Crock P, Rogers D, Mikuni M, Kumashiro H, Higashi K, Sobue G, Oka Y, Permutt MA. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat Genet. 1998;20:143–148. doi: 10.1038/2441. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, Yamada T, Inoue H, Soga H, Katagiri H, Tanizawa Y, Oka Y. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet. 2004;13:1159–1170. doi: 10.1093/hmg/ddh125. [DOI] [PubMed] [Google Scholar]
- Kawano J, Tanizawa Y, Shinoda K. Wolfram syndrome 1 (Wfs1) gene expression in the normal mouse visual system. J Comp Neurol. 2008;510:1–23. doi: 10.1002/cne.21734. [DOI] [PubMed] [Google Scholar]
- Man PY, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet. 2002;39:162–169. doi: 10.1136/jmg.39.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtanda AT, Cruysberg JRM, Pinckers AJLG. Optic atrophy in Wolfram syndrome. Ophthalmic Paediatr Genet. 1986;7:159–165. doi: 10.3109/13816818609004133. [DOI] [PubMed] [Google Scholar]
- Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517–523. doi: 10.1016/j.ajo.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Nickells RW. From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Can J Ophthalmol. 2007;42:278–287. [PubMed] [Google Scholar]
- Osman AA, Saito M, Makepeace C, Permutt MA, Schlesinger P, Mueckler M. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem. 2003;278:52755–52762. doi: 10.1074/jbc.M310331200. [DOI] [PubMed] [Google Scholar]
- Paschen W, Doutheil J. Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J Cereb Blood Flow Metab. 1999;19:1–18. doi: 10.1097/00004647-199901000-00001. [DOI] [PubMed] [Google Scholar]
- Philbrook C, Fritz E, Weiher H. Epressional and functional studies of Wolframin, the gene function deficient in Wolfram syndrome, in mice and patient cells. Exp Gerontol. 2005;40:671–678. doi: 10.1016/j.exger.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Swift RG, Swift M. Linkage of the gene for Wolfram syndrome to markers on the short arm of chromosome 4. Nat Genet. 1994;8:95–97. doi: 10.1038/ng0994-95. [DOI] [PubMed] [Google Scholar]
- Radeke MJ, Petersen KE, Johnson LV, Anderson DH. Disease susceptibility of the human macula: differential gene transcription in the retinal pigmented epithelium/choroid. Exp Eye Res. 2007;85:366–380. doi: 10.1016/j.exer.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, Murray J, Schmidt RE, Herrera PL, Permutt MA. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48:2313–2321. doi: 10.1007/s00125-005-1947-4. [DOI] [PubMed] [Google Scholar]
- Roybal CN, Marmorstein LY, Vander Jagt DL, Abcouver SF. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest Ophthalmol Vis Sci. 2005;46:3973–3979. doi: 10.1167/iovs.05-0070. [DOI] [PubMed] [Google Scholar]
- Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, Lango H, Frayling TM, Neumann RJ, Sherva R, Blech I, Pharoah PD, Palmer CN, Kimber C, Tavendale R, Morris AD, McCarthy MI, Walker M, Hitman G, Glaser B, Permutt MA, Hattersley AT, Wareham NJ, Barroso I. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–953. doi: 10.1038/ng2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer T, Patel M, Chan CC, Tuo J. Unfolding the therapeutic potential of chemical chaperones for age-related macular degeneration. Expert Rev Ophthalmol. 2008;3:29–42. doi: 10.1586/17469899.3.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazawa M, Inokuchi Y, Ito Y, Murata H, Aihara M, Miura M, Araie M, Hara H. Involvement of ER stress in retinal cell death. Mol Vision. 2007;13:578–587. [PMC free article] [PubMed] [Google Scholar]
- Sparsoe T, Andersen G, Albrechtsen A, Jorgensen T, Borch-Johnson K, Sandbaek A, Lauritzen T, Wasson J, Permutt MA, Glaser B, Madsbad S, Pedersen O, Hansen T. Impact of polymorphisms in WFS1 on prediabetic phenotypes in a population-based sample of middle-aged people with normal and abnormal glucose regulation. Diabetologica. 2008;51:1646–1652. doi: 10.1007/s00125-008-1064-2. [DOI] [PubMed] [Google Scholar]
- Strom TM, Hortnagel K, Hofmann S, Gekeler F, Scharfe C, Rabl W, Gerbitz KD, Meitinger T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet. 1998;7:2021–2028. doi: 10.1093/hmg/7.13.2021. [DOI] [PubMed] [Google Scholar]
- Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabe Y, Shinoda K, Oka Y. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet. 2001;10:477–484. doi: 10.1093/hmg/10.5.477. [DOI] [PubMed] [Google Scholar]
- Takei D, Ishihara H, Yamaguchi S, Yamada T, Tamura A, Katagiri H, Maruyama Y, Oka Y. WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett. 2006;580:5635–40. doi: 10.1016/j.febslet.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Ueda K, Kawano J, Takeda K, Yujiri T, Tanabe K, Anno T, Akiyama M, Nozaki J, Yoshinaga T, Koizumi A, Shinoda K, Oka Y, Tanizawa Y. Endoplasmic reticulum stress induces Wfs1 gene expression in pancreatic beta-cells via transcriptional activation. Eur J Endocrinol. 2005;153:167–176. doi: 10.1530/eje.1.01945. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- Wasson J, Permutt MA. Candidate gene studies reveal that the WFS1 gene joins the expanding list of novel type 2 diabetes genes. Diabetologia. 2008;51:391–393. doi: 10.1007/s00125-007-0920-9. [DOI] [PubMed] [Google Scholar]
- Wolfram DJ, Wagener HP. Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc. 1938;13:715–718. [Google Scholar]
- Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, Tokita A, Satake C, Tashiro F, Katagiri H, Aburatani H, Miyazaki J, Oka Y. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum Mol Genet. 2006;15:1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Ishihara H, Tamura A, Yamada T, Takahashi R, Takei D, Katagiri H, Oka Y. Endoplasmic reticulum stress and N-glycosylation modulate expression of WFS1 protein. Biochem Biophys Res Commun. 2004;325:250–256. doi: 10.1016/j.bbrc.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Hofmann S, Hamasaki DI, Yamamoto H, Kreczmanski P, Schmitz C, Parel JM, Schmidt-Kastner R. Wolfram syndrome 1 (WFS1) protein expression in retinal ganglion cells and optic nerve glia of the cynomolgus monkey. Exp Eye Res. 2006;83:1303–1306. doi: 10.1016/j.exer.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Yurimoto S, Hatano N, Tsuchiya M, Kato K, Fujimoto T, Masaki T, Kobayashi R, Tokumitsu H. Identification and characterization of wolframin, the product of Wolfram Syndrome Gene (WFS1), as a novel calmodulin-binding protein. Biochemistry. 2009;48:3946–3955. doi: 10.1021/bi900260y. [DOI] [PubMed] [Google Scholar]
- Zatyka M, Ricketts C, da Silva Xavier G, Minton J, Fenton S, Hofmann-Thiel S, Rutter GA, Barrett TG. Sodium-potassium ATPase 1 subunit is a molecular partner of Wolframin, an endoplasmic reticulum protein involved in ER stress. Hum Mol Genet. 2008;17:190–200. doi: 10.1093/hmg/ddm296. [DOI] [PMC free article] [PubMed] [Google Scholar]