

Abstract
CD4+ T cells from the TCR transgenic TxA23 mouse recognize a peptide from the H/K-ATPase α-chain. When TxA23 CD4+ thymocytes are differentiated into Th1, Th2, and Th17 lines, all three subpopulations induced autoimmune gastritis (AIG) upon transfer into nu/nu recipients. The induction of AIG by naïve T cells or by Th1, or Th2 cell lines could be prevented by co-transfer of polyclonal Foxp3+ T regulatory cells (nTreg), while Th17-induced AIG was resistant to suppression. We compared the capacity of different types of Treg to suppress Th17-mediated AIG. Co-transfer of either nTreg or polyclonal TGFβ-induced Treg (iTreg) did not prevent AIG, while co-transfer of TGFβ-induced antigen-specific TxA23 iTreg completely prevented development of disease. Antigen-specific iTreg were able to suppress Th17-mediated disease when injected 6 days after the Th17 effectors. The implications of these results for the use of Treg for the cellular biotherapy of autoimmune disease are discussed.
Keywords: Autoimmunity, T cells, Tolerance/Suppression
The concept that organ-specific autoimmune disease is a result of dysregulated autoantigen-specific Th1 responses has recently been broadened by the discovery of IL17-producing helper T cells (Th17) (1). Th17 cells are involved in the pathogenesis of autoimmune diseases, such as experimental autoimmune encephalomyelitis and collagen-induced arthritis, which previously were regarded as exclusively Th1-mediated (2). The cytokine IL-17 has pro-inflammatory effects and induces tissue damage during the course of various autoimmune diseases (3). We have developed a TCR transgenic mouse model of Autoimmune Gastritis (AIG) in which the TCR transgenic (TxA23) T cells recognize a peptide from the H/K-ATPase α-chain (4) and the transgenic mice spontaneously develop AIG early in life. When naïve TxA23 thymocytes are differentiated in culture into Th1, Th2, and Th17 lines, all three T cell subpopulations were capable of inducing AIG upon transfer into immunoincompetent BALB/c nu/nu recipients (5). When compared to in vitro differentiated transgenic Th1 and Th2 cells, Th17 induced a distinct histological pattern of autoimmune gastritis characterized by a highly destructive disease with cellular infiltrates composed primarily of eosinophils, accumulation of transgenic CD4+ T effectors in the gastric mucosa and draining LN, and high levels of serum IgE (5). Co-transfer of polyclonal Foxp3+ Treg (nTreg) markedly suppressed disease induced by naïve CD4+ T cells (6) or fully differentiated Th1 and Th2 lines (5). In contrast, disease induced by Th17 lines was highly resistant to suppression even at a high Treg to effector ratio (20:1).
The resistance of Th17-induced AIG to suppression by Treg raised the possibility that Th17 cells are intrinsically not responsive to suppression by Treg or that the inflammatory milieu generated by Th17 cells induced the production of cytokines that rendered the effector T cells resistant to suppression or neutralized the suppressive effects of the Treg (7). We (8) and others (9) have previously demonstrated that autoantigen-specific Treg are much more potent suppressors of the induction of autoimmune disease than polyclonal Treg. As it is relatively easy to generate large number of either polyclonal or antigen-specific activated TGFβ-induced Treg (iTreg) that are potent suppressors of the induction of organ-specific (8) and systemic autoimmune disease (10), we compared these iTreg populations for their ability to suppress AIG induced by fully differentiated autoantigen-specific Th17 lines. We demonstrate that neither polyclonal nTreg nor TGFβ-induced polyclonal iTreg could prevent disease induced by Th17 cells. In contrast, co-transfer of TGFβ-induced antigen-specific iTreg completely prevented tissue destruction in the gastric mucosa and reduced the numbers of transgenic effector T cells in the gastric lymph node. Furthermore, in a therapeutic approach, TxA23 transgenic iTreg suppressed Th17-mediated AIG when injected one week after transfer of the Th17 effectors. Thus, TGFβ-induced antigen-specific iTreg are capable of both preventing and treating AIG induced by fully differentiated Th17 effector cells.
Materials and Methods
Mice and reagents
Female BALB/c Thy1.1+ and BALB/c nu/nu (4–8 wk old) were purchased from the National Cancer Institute animal facility and housed under specific pathogen-free conditions. TxA23 TCR-Tg mice have been described previously (11). All mice were maintained in our animal facility and cared for in accordance with institutional guidelines.
T cell purification and differentiation
Autoantigen-specific Th17 cell lines were generated from FACS-sorted CD4+CD8-CD25- thymocytes from TxA23 Thy1.1+/1.2+ mice as previously described (5, 8). nTreg were isolated from single cell suspensions of spleen cells from BALB/c Thy1.1+ mice as previously described (5). Optimal differentiation of Th17 cells was verified after 4-7 days of differentiation in vitro by intracellular staining for IL-17 expression. Polyclonal TGFβ-induced regulatory T cells (WT iTreg) were generated from CD4+CD25- T cells from BALB/c Thy1.1+ mice as previously described (10). Antigen-specific iTreg were generated from naïve CD4+CD8-CD25- thymocytes from TxA23 Thy1.1+ mice and were isolated and differentiated as previously described (8). On day 4 to 7 of culture, expression of FoxP3 was measured by flow cytometry and only populations with 80% to 99% FoxP3+ T cells were used for injection.
Cell transfer experiments
The TxA23 Th17 effector T cells and Treg were washed twice with PBS, and diluted into PBS such that an i.v. injection of 0.25 ml/mouse resulted in the transfer of 50×103 TxA23 Thy1.1+/Thy1.2+ Th17 cells and 1×106 Thy1.1+ Treg, respectively.
Antibodies and Flow cytometry
Cell surface staining was performed using antibodies against CD25 (PC61), Thy-1.1 and Thy-1.2 directly conjugated to fluorescein isothiocyanate (FITC) or phycoerythrin (PE) (all BD Pharmingen). Intracellular cytokine staining was performed using mAbs for intracellular stainings (anti-IL-4, anti-IL-10, anti-IL-17, anti-IFNγ, anti-TNFα) conjugated to PE or allophycocyanin (APC) (all BD Pharmingen) as described previously (5). APC-conjugated mAb to FoxP3 (FJK-16s) was purchased from eBioscience. All flow cytometry was performed on a FACScan or FACSCalibur (BD Biosciences) and analyzed using FLOWJo software (TreeStar).
Gastric pathology
6 weeks after transfer, the animals were euthanized, the stomach opened, washed, and fixed in 4% neutral buffered formalin. Fixed tissues were embedded in paraffin, stained with H&E by American Histolabs (Gaithersburg, MD) and inflammation was scored as previously published (5, 6, 8).
Statistical analysis
All group results are expressed as mean ± SD, if not stated otherwise. Student’s t-test was used for the comparison of group values and discriminatory parameters, where appropriate. For comparing group values that did not follow Gaussian distribution, Mann-Whitney’s test (2-tailed) was used. P values less than 0.05 were considered significant.
Results and Discussion
Naïve CD4+CD8-CD25- TxA23 Thymocytes were stimulated in vitro for 5 days under Th17-polarizing conditions. After restimulation with PMA/Ionomycin the majority of Th17 cells produce IL-17 (Fig. 1A, left panel). Since transfer of naïve TxA23 CD4+ T cells induces a Th1 disease with IFNγ as the main cytokine produced, we also measured IFNγ in the in vitro differentiated Th17 cells. No IFNγ was detectable by intracellular staining of different preparations of Th17 cells (Fig. 1A, right panel). The in vitro differentiated Th17 cells also did not produce Th2 cytokines (IL4, IL5, IL10 or TNFα) as measured by intracellular staining (data not shown). Stimulation of conventional CD4+CD25- T cells via the TCR in the presence of TGFβ and IL-2 induces FoxP3 expression and a regulatory phenotype (10, 12). We differentiated TGFβ-induced iTreg from naïve antigen-specific TxA23 T cells and from naïve WT CD4+CD25- T cells. On day 5 of the in vitro culture, the cells were stained for FoxP3 expression. Both populations of TGFβ-induced iTreg expressed high levels of FoxP3 comparable to freshly isolated nTreg (Fig.1B).
Figure 1.
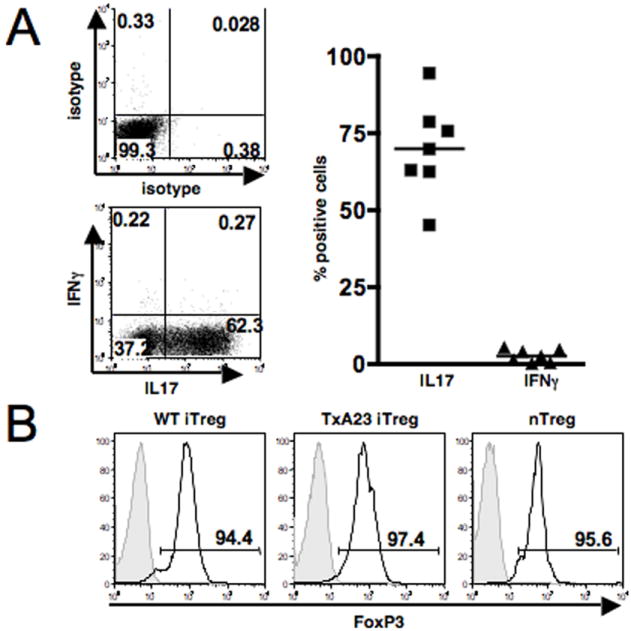
Characterization of Th17 effectors and Treg. A, CD4+CD8-CD25- TxA23 thymocytes were differentiated in vitro under Th17-polarizing conditions. On day 5, the cells were restimulated with PMA/Ionomycin and stained for intracellular cytokine expression: The left panel shows one representative experiment, the right panel shows a summary of 7 separate experiments. B, FoxP3 expression in fresh nTreg, TxA23 iTreg and polyclonal WT iTreg. nTreg were stained directly ex vivo; iTreg were induced in vitro and FoxP3 expression was determined by intracellular staining on day 5. One representative experiment of at least 10 experiments with similar results is shown.
To compare the suppressive capacity of the different types of Treg, we co-transferred Th17 effector T cells with Treg at a fixed ratio of 1:20 (50×103 Th17 effectors and 1×106 Treg). Transfer of Th17 effector cells alone induced destructive AIG (score 5-6). Six weeks after transfer, the majority of mice showed high-grade AIG as defined by severe or complete parietal cell loss with replacement by so-called ‘foamy cells’, epithelial hyperplasia and massive inflammatory infiltrates (Fig. 2A). Co-transfer of either nTreg or polyclonal iTreg did not prevent the development of destructive gastritis (Figure 2A). In contrast, co-transfer of TxA23 iTreg completely prevented development of disease, and destructive tissue pathology was not observed in any of the animals co-transferred with the antigen-specific iTreg (Fig. 2A, B). The results of a large series of co-transfers are summarized in Fig. 2B. Co-transfer of nTreg significantly reduced the percentage of animals with destructive gastritis from 85% to 66%, while only a slight reduction of the percentage of animals with destructive tissue pathology was seen when WT iTreg were co-transferred In contrast, antigen-specific iTreg completely prevented development of destructive disease (Fig. 2B). The enhanced suppressive capacity of the antigen-specific iTreg compared to the polyclonal nTreg was not secondary to their pre-activation in vitro, as in vitro pre-activated nTreg were no more efficient that freshly isolated nTreg in their capacity to prevent destructive AIG (data not shown).
Figure 2.
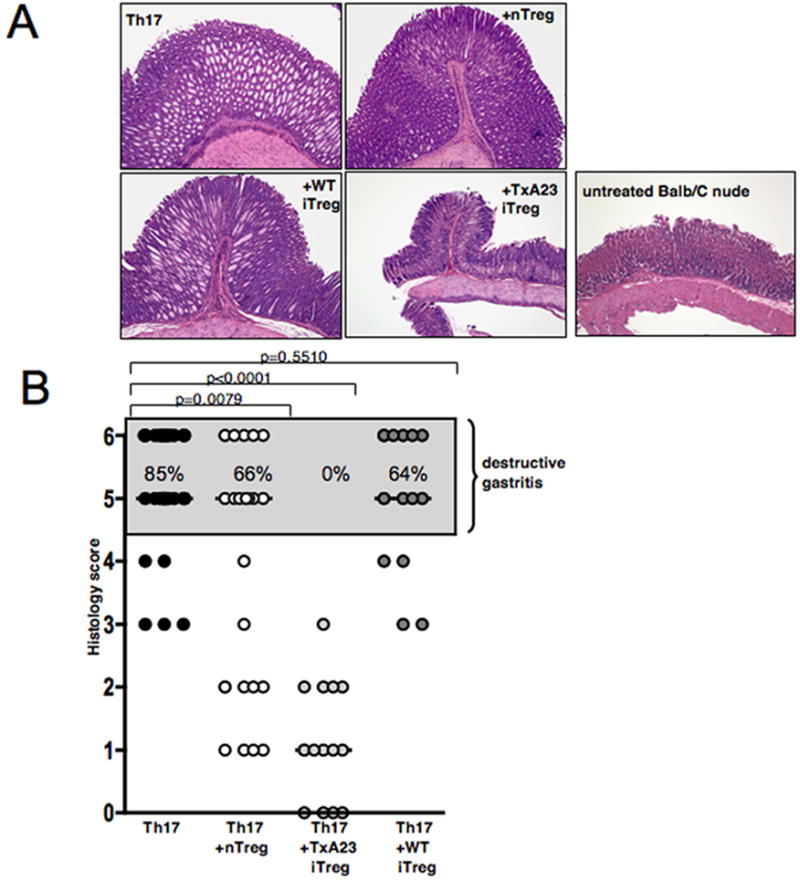
Antigen-specific iTreg suppress Th17-mediated tissue destruction in the stomach. A, Gastric pathology 6 weeks after transfer of TxA23 Th17 cells either alone or together with either nTreg, TxA23 iTreg or polyclonal WT iTreg. H&E staining, original magnification 5X. One representative experiment is shown. B, Summary of the results of 5 independent experiments; Mann Whitney test, bars, median gastritis score.
One of the proposed mechanisms for therapeutic effect of antigen-specific iTreg in the AIG model is the downregulation of costimulatory molecules on APC resulting in inhibition of the expansion of the effector T cells in the draining lymph node (8). The H/K ATPase is expressed in DC in the gastric lymph node (13), and it is likely that the initial activation of the effector cells in AIG occurs in the node with subsequent migration to the gastric mucosa. Following transfer of the Th17 effector cells, marked enhancement of the cellularity of the gastric lymph node was observed (Fig. 3A). Co-transfer of TxA23 iTreg significantly reduced the cellularity in the gastric lymph node (Fig. 3A), while neither nTreg nor WT iTreg prevented the increase in cellularity. As the Th17 effector T cells were labelled with a congenic marker, we also calculated the absolute number of Th17 effector cells. Both the percentage of Th17 effectors (Fig. 3B) and their absolute numbers (Fig. 3C) were significantly reduced only in animals that received the TxA23 iTreg, but not the nTreg or the polyclonal iTreg. The higher percentages of antigen-specific iTreg in the gLN (Fig. 3B) could either be due to their preferential homing or increased expansion in comparison to the other Treg populations. CFSE-labeling of all 3 Treg populations revealed a higher CFSE-dilution in the gLN of the antigen-specifc iTreg compared to WT iTreg or nTreg. All 3 populations displayed similar CFSE-profiles in the inguinal LN (data not shown). This result indicates that preferential expansion of the antigen-specific iTreg after antigen encounter in the draining node likely accounts for their higher numbers in the gLN and their higher suppressive capacity towards Th17 effector T cells as compared to WT iTreg or nTreg.
Figure 3.
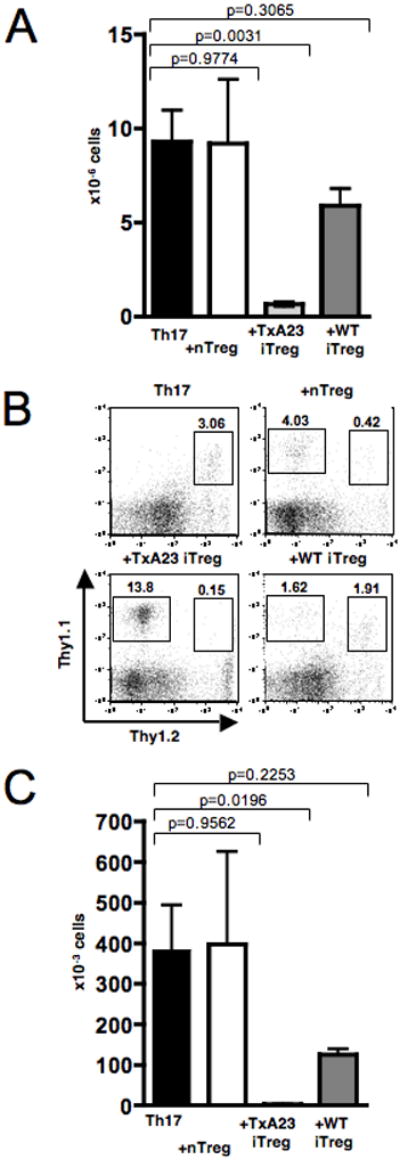
Antigen-specific TGFβ-induced iTreg reduce the cellularity and the absolute number of Th17 effector T cells in the draining lymph node. A, Total number of cells in the gLN 6 weeks after transfer of TxA23 Th17 cells alone or with nTreg, TxA23 iTreg or polyclonal iTreg. Data from 4 independent experiments were pooled for analysis, Students t-test. B, FACS analysis of the cells in the gLN 6 weeks after transfer, numbers represent percentage of cells in the lymphocyte gate. C, Absolute number of TxA23 effector T cells in the gLN 6 weeks after transfer. Data from 4 independent experiments were pooled for analysis, Students t-test.
Since the antigen-specific iTreg had a strong suppressive effect on development of AIG in co-transfer experiments, we next asked if these cells were also capable of suppressing disease in a therapeutic experimental setting. We used the same Treg:T effector ratio of 20:1 as in the previous experiments, but injected the antigen-specific iTreg 6 days after transfer of the in vitro differentiated Th17 effector cells. When analyzed 6 weeks after transfer of the Th17 effector T cells, marked inhibition of their expansion was seen in the animals that received the iTreg (Fig. 4A). All animals in the group that received Th17 effector cells alone had developed destructive AIG, whereas co-transfer of antigen-specific iTreg completely prevented development of destructive diasease (Fig. 4B). Furthermore, the co-transferred TxA23 iTreg were also readily identifiable in cell suspensions prepared from the gastric mucosa and their presence was associated with a reduced percentage of infiltrating Th17 effector T cells (Fig. 4C).
Figure 4.
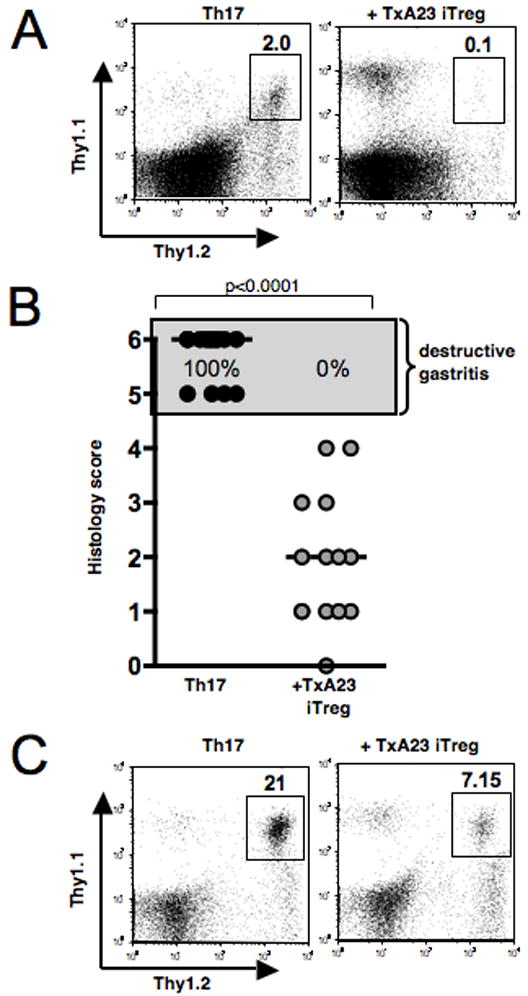
Antigen-specific TGFβ-induced iTreg prevent Th17-mediated AIG when injected 6 days after disease induction. A, TxA23 iTreg were transferred 6 days after transfer of TxA23 Th17 effector cells. FACS staining of the cells in the gLN 6 weeks after transfer of the Th17 effector cells. Numbers represent percentage of Thy1.1+/Thy1.2+ TxA23 T cells in the lymphocyte gate. B, Gastric pathology 6 weeks after transfer of TxA23 Th17 effector cells and TxA23 iTreg 6 days later. One representative experiment of 2 experiments with similar results is shown. Mann-Whitney test, bars, median gastritis score. C, FACS staining of the cells flushed out of the gastric mucosa 6 weeks after transfer of TxA23 Th17 effector cells alone or TxA23 Th17 effector cells and TxA23 iTreg 6 days later. Numbers represent the percentage of Thy1.1+/Thy1.2+ TxA23 T cells in the lymphocyte gate.
Th17 cells play an important role in the pathogenesis of many autoimmune diseases, and some studies have suggested that Th17 cells may be resistant to suppression by Treg as they produce or induce the production of cytokines such as IL6 and TNFα, that dampen Treg function. For example, even though high numbers of Treg could be detected in the CNS of animal with MOG35-55-induced EAE, they could not prevent the function of the pathogenic T effector cells and T effector cells from the acutely inflamed CNS were resistant to suppression by Treg in vitro (7). Similarly, in our AIG model, disease induced by Th17 effectors was highly aggressive and resistant to suppression by polyclonal nTreg. It was therefore of interest to compare the capacity of different Foxp3+ Treg subpopulations to suppress the induction of AIG by fully differentiated Th17 cells. Consistent with our previous results, both polyclonal nTreg and polyclonal iTreg were ineffective in preventing the induction of AIG. In contrast, antigen-specific iTreg were potent inhibitors of disease induction and even exerted considerable therapeutic effects when administered 6 days after transfer of the Th17 at a time point when the inflammatory response in the target organ had already been initiated. Taken together, these results demonstrate that Th17-induced AIG is susceptible to suppression, but only by antigen-specific Treg. As Th17 cells may play an important role in many human autoimmune diseases, these findings have important implications regarding the use of Treg for cellular biotherapy.
Acknowledgments
The authors thank C. Henry and T. Moyer for cell sorting and the staff of the animal facilities for their work maintaining the mice.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
Disclosures The authors do not have any financial, commercial or other conflict of interest.
References
- 1.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 2.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 3.Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suri-Payer E, Kehn PJ, Cheever AW, Shevach EM. Pathogenesis of post-thymectomy autoimmune gastritis. Identification of anti-H/K adenosine triphosphatase-reactive T cells. J Immunol. 1996;157:1799–1805. [PubMed] [Google Scholar]
- 5.Stummvoll GH, DiPaolo RJ, Huter EN, Davidson TS, Glass D, Ward JM, Shevach EM. Th1, th2, and th17 effector T cell-induced autoimmune gastritis differs in pathological pattern and in susceptibility to suppression by regulatory T cells. J Immunol. 2008;181:1908–1916. doi: 10.4049/jimmunol.181.3.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiPaolo RJ, Glass DD, Bijwaard KE, Shevach EM. CD4+CD25+ T cells prevent the development of organ-specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J Immunol. 2005;175:7135–7142. doi: 10.4049/jimmunol.175.11.7135. [DOI] [PubMed] [Google Scholar]
- 7.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 9.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol. 2008;38:1814–1821. doi: 10.1002/eji.200838346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McHugh RS, Shevach EM, Margulies DH, Natarajan K. A T cell receptor transgenic model of severe, spontaneous organ-specific autoimmunity. Eur J Immunol. 2001;31:2094–2103. doi: 10.1002/1521-4141(200107)31:7<2094::aid-immu2094>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. 2002;196:1079–1090. doi: 10.1084/jem.20020991. [DOI] [PMC free article] [PubMed] [Google Scholar]