

Abstract
DNA damage is detected and repaired in the context of chromatin. In this issue of Cell, van Attikum et al. (2004) and Morrison et al. (2004) demonstrate that double-stranded break repair involves the recruitment of a specialized chromatin remodeling complex, INO80, through an interaction with phosphorylated histone H2A.
Chromosome breaks resulting from ionizing radiation or DNA replication defects pose a considerable threat to genome integrity, as broken chromosomal segments are susceptible to shortening and loss. The loss of chromosomes (or chromosome segments) can lead to the loss of tumor suppressor genes, a common feature of cancer cells. To help maintain genome integrity, cells have evolved two different pathways to heal breaks: nonhomologous end joining (NHEJ), which involves end religation, and homologous recombination (HR), which utilizes an undamaged homologue for template-guided repair (Paques and Haber, 1999). These repair systems must contend with the repressive aspects of chromatin and therefore may require the services of specialized chromatin remodeling machinery (Peterson and Cote, 2004). Two articles in this issue of Cell provide evidence that a chromatin remodeling complex important for repair is recruited to chromosome breaks by phosphorylated H2A.
Briefly, for double-stranded break (DSB) repair by NHEJ, the MRX complex and Ku proteins bind and protect the broken ends and recruit end processing factors and a DNA ligase to accomplish end ligation (Figure 1). For HR, additional processes and proteins are required. Here, 5′ to 3′ resection of the end occurs to create a 3′ ssDNA overhang that is coated with a ssDNA binding protein and other factors. Coated ends search the genome for a homologous donor, base pair with the donor to form Holliday structures, and then utilize the donor as a template for 3′ extension by DNA polymerase. The Holliday structure is migrated during this process and resolved into two duplexes by resolvase, and the resulting nicks are ligated to restore two intact duplexes.
Figure 1. Chromatin in DSB Repair in S. cerevisiae.
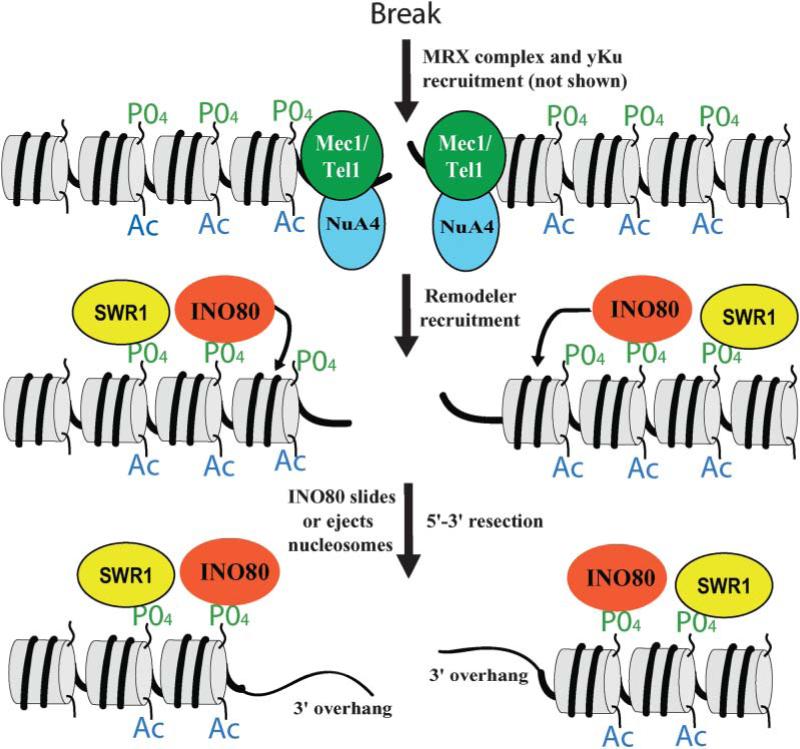
Following break initiation, DNA ends are bound by yKu and MRX complex (data not shown). The checkpoint kinases Mec1 and Tel1 are recruited to the break and phosphorylate H2A Ser129. This is followed by NuA4 recruitment (a histone acetyltransferase) and acetylation of the region. INO80 and SWR1 are then recruited. The chromatin remodeling activity of INO80 is required for the appearance of the processed 3′ ssDNA overhang, which may be facilitated by the removal of nucleosomes from the region. The presence of SWR1 may cause histone H2A replacement for the histone H2A variant Htz1at the DSB but has not been tested. INO80 and SWR1 may have chromatin remodeling roles during repair by HR (see text; data not shown due to space limitations).
In principle, resident nucleosomes could interfere with any or all of these repair steps. By analogy to transcriptional regulation, special ATP-dependent chromatin remodeling complexes (termed remodelers) slide, eject, or reconfigure repressive nucleosomes, revealing the underlying DNA. As cells have evolved specialized remodelers for transcriptional regulation (and other processes), one might expect specialized remodelers to assist DSB repair: enter INO80.
INO80 is a 12 protein complex that includes the Ino80 protein, which bears the signature ATPase/translocase domain of remodelers (Shen et al., 2000). ino80Δ mutants display transcriptional defects, and INO80 directly occupies certain targets (Jonsson et al., 2004; Shen et al., 2000); however, this preview will focus solely on its role in DNA repair. Mutations in INO80 complex members render cells sensitive to DNA damaging agents, and here van Attikum et al. (2004) show they are likewise defective in NHEJ. Furthermore, HR in Arabidopsis utilizes an ortholog of Ino80 (Fritsch et al., 2004). In this issue, Morrison et al. (2004) and van Attikun et al. (2004) provide evidence that the repair defects are not an indirect consequence of transcriptional or checkpoint defects, and they provide strong evidence for a direct role, recruitment to the break.
Both Morrison et al. (2004) and van Attikum et al. (2004) initiate a single DSB at a unique site in the S. cerevisiae genome using controlled expression of the HO endonuclease. Remarkably, chromatin immunoprecipitation analyses show the recruitment of INO80 to the DSB region within 30−60 min of break induction. This observation prompted both groups to determine which factors/marks are required for INO80 recruitment.
In yeast, breaks recruit many proteins, including two checkpoint kinases of the ATM/ATR family, Tel1 and Mec1, which phosphorylate substrates that promote cell cycle arrest and facilitate repair. In yeast, Mec1/Tel1 phosphorylate histone H2A at serine 129 on nucleosomes that reside near the DSB. This phosphorylation is important for repair, as strains bearing an Ser129Ala substitution (which prevents phosphorylation) are sensitive to DNA damaging agents. Both groups demonstrate that S129 phosphorylation is required for INO80 recruitment. Strains bearing mutations in the kinases (Mec1 and Tel1) or substrate (H2A lacking S129) fail to recruit INO80 to the break. The interaction between INO80 and phospho-H2A appears direct, as Morrison et al. (2004) show copurification of INO80 with phospho-H2A, along with other core histones. Remarkably, an INO80 derivative that lacks two components (Nhp10 and Ies3) fails to interact with phospho-H2A but retains interaction with unphosphorylated H2A and other core histones. This result suggests that Nhp10 and/or Ies3 promote selectivity for phospho-H2A, a surprising result, as these proteins lack the BRCT (BRCA1 C-terminal) domains that mediate this interaction in higher cells. Consistent with a role for Nhp10, INO80 fails to be recruited to the DSB in nhp10Δ mutants. However, wild-type INO80 persists at the DSB after phospho-H2A diminishes, suggesting an alternative protein/mark for INO80 retention.
Van Attikum et al. (2004) reveal a defect in the formation of the 3′ single-stranded overhang in strains bearing mutations in INO80 complex or in strains lacking H2A-S129. The authors suggest the exciting possibility that INO80 may remove and/or slide nucleosomes from the broken ends to help facilitate 5′−3′ resection. However, this observation does not rule out additional roles for INO80 in other stages of DNA repair.
The composition of INO80 suggests additional roles in repair through HR. INO80 bears two AAA+ family ATPases, Rvb1/Rvb2, that are similar to RuvB, a protein required for DSB repair and recombination in bacteria. RuvB is a double hexameric DNA helicase that binds to and migrates the Holliday structure. Similarly, Rvb1/Rvb2 appear to be present as a double hexamer, and this holo-INO80 complex possesses ATP-dependent helicase activity in vitro (though Holliday migration has not been tested) (Shen et al., 2000). Rvb1/Rvb2 could use their DNA helicase/tracking function to disrupt nucleosomes proximal to the break. Alternatively, Rvb1/Rvb2 might help migrate the Holliday structure, while the remodeling function of INO80 slides or transfers nucleosomes encountered during migration.
Cells may utilize a second remodeler to assist with repair in chromatin, SWR1 complex, which shares several proteins with INO80, including Rvb1/Rvb2 (Mizuguchi et al., 2004). SWR1 is a histone exchange complex that replaces H2A with the histone H2A variant Htz1. Interestingly, SWR1 complex is also recruited to the DSB (Downs et al., 2004). It is tempting to speculate that SWR1 complex might exchange phospho-H2A for Htz1 (which is not phosphorylatable) during Holliday structure migration (or during NHEJ) to revert chromatin back to the undamaged/unphosphorylated state. Consistent with this model, the Drosophila ortholog of SWR1 complex, Domino/p400, replaces phospho-H2Av with unmodified H2Av in vitro (Kusch et al., 2004). In addition, the Domino/p400 complex bears Tip60, a histone acetyltransferase, and acetylation stimulates histone exchange in vitro (Kusch et al., 2004). Tip60 is virtually identical to the Esa1 subunit of the yeast NuA4 complex, which is required for efficient DNA repair. Interestingly, NuA4 recruitment precedes INO80 and SWR1 recruitment, and esa1 mutants are defective in damage repair, suggesting that acetylation promotes remodeler association (Downs et al., 2004). Together, these studies suggest that histone modification, remodeling, and replacement are coordinated at the DSB to facilitate both the DNA repair process and restoration of the undamaged chromatin state.
A conspicuous feature of INO80, SWR1, Domino/p400, and NuA4 complexes is the presence of actin-related proteins (ARPs). The importance of ARPs for chromatin remodeling was first demonstrated in studies of SWI/SNF family remodelers, and they are now recognized as components of multiple remodeler and histone modification complexes. For INO80, ARPs are absolutely required for ATPase activity and nucleosome remodeling (Shen et al., 2003). The Arp5 component binds to the Rvb proteins and also to Ino80p, raising the interesting possibility that Arp5 might coordinate the functions of these ATPases. Interestingly, Downs et al. (2004) show that Arp4 (a component of INO80, SWR1, and NuA4) binds directly to phospho-H2A. This result appears at odds with those of Morrison et al. (2004); INO80 complex purified from nhp10Δ mutants retains Arp4 but loses the ability to interact with phospho-H2A. However, one way to reconcile these observations is that Nhp10 (or Ies3) may assist Arp4 in phospho-H2A recognition. Finally, it will be of interest to determine how NuA4 is recruited prior to INO80 or SWR1, considering that all three bear Arp4.
Taken together, these studies provide interesting new information about how histone modification, remodeling, and replacement are coordinated to facilitate repair and restore the undamaged chromatin state. Given the current pace of discovery, INO80 days we will know much more.
Selected Reading
- Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Côté J. Mol. Cell. 2004;16 doi: 10.1016/j.molcel.2004.12.003. in press. Published online December 16, 2004. 10.1016/S1097276504007580. [DOI] [PubMed] [Google Scholar]
- Fritsch O, Benvenuto G, Bowler C, Molinier J, Hohn B. Mol. Cell. 2004;16:479–485. doi: 10.1016/j.molcel.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Jonsson ZO, Jha S, Wohlschlegel JA, Dutta A. Mol. Cell. 2004;16:465–477. doi: 10.1016/j.molcel.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates Iii JR, Abmayr SM, Washburn MP, Workman JL. Science. 2004 doi: 10.1126/science.1103455. in press. Published online November 4, 2004. 0: 11034551. [DOI] [PubMed] [Google Scholar]
- Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. Science. 2004;303:343–348. doi: 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. Cell. 2004;119(this issue):767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Cote J. Genes Dev. 2004;18:602–616. doi: 10.1101/gad.1182704. [DOI] [PubMed] [Google Scholar]
- Shen X, Mizuguchi G, Hamiche A, Wu C. Nature. 2000;406:541–544. doi: 10.1038/35020123. [DOI] [PubMed] [Google Scholar]
- Shen X, Ranallo R, Choi E, Wu C. Mol. Cell. 2003;12:147–155. doi: 10.1016/s1097-2765(03)00264-8. [DOI] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Hohn B, Gasser SM. Cell. 2004;119(this issue):777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]