

Abstract
Endothelin-1 (ET-1) is unequivocally elevated in the kidney with ischemic acute renal failure (ARF), whereas ET receptors (ETAR and ETBR) are variably expressed. Although renal functional and structural changes are similar between ischemic and nephrotoxic ARF, there are few reports on the alteration in the ET system in nephrotoxic ARF. This study was, therefore, undertaken to investigate changes in renal expression of ET-1 and its receptors in nephrotoxic ARF induced by cisplatin. Mice were intraperitoneally injected with 16 mg of cisplatin/kg at a single dose, and the expression of mRNA and protein was then quantified by real-time RT-PCR and Western blot, respectively. Immunohistochemistry was conducted for localization. Three days after treatment, ET-1 transcript in cisplatin-treated mice was thirteen times higher than that in controls, whereas ET-1 peptide was increased by 1.5-fold. Cisplatin caused a 2-fold increase in the levels of ETAR mRNA and protein. Most of the increased immunoreactive ET-1 and ETAR were localized in damaged tubules. Neither the expression of ETBR mRNA nor the abundance and immunoreactive level of ETBR protein were changed. The findings suggest that the individual components of the renal ET system are differentially regulated in cisplatin-induced nephrotoxic ARF.
Keywords: Cisplatin, Acute renal failure, Endothelin, Endothelin receptor
INTRODUCTION
The endothelin (ET) system includes ET isotypes (ET-1, ET-2, ET-3), ET converting enzyme (ECE), and its receptors (ETAR, ETBR). ET exerts its effects by binding to the cell-surface ET receptors, which belong to G protein-coupled receptor. Vasoconstriction, mitogenesis, and anti-apoptosis are mediated through ETAR, whereas vasodilation, Na and water balance, ET clearance, and inflammatory pain through ETBR (Nelson et al, 2003). Evidence indicates that ET plays a role in the pathophysiology of various forms of renal disease, including renal hypertension (Hoffman et al, 1994; Ahn et al, 2004), renal fibrosis (Chatziantoniou & Dussaule, 2005), transplant rejection (Braun et al, 1999) and renal failure (Takaoka et al, 2000).
Acute renal failure (ARF) is characterized by an abrupt and reversible kidney dysfunction. Two spectra of ARF from intrarenal origin are ischemic and nephrotoxic. Ischemic ARF primarily affects renal hemodynamics with afferent arteriolar vasoconstriction. Reduced renal blood flow increases vasoconstrictors such as ET and angiotensin II, and decreases vasodilators such as nitric oxide and prostaglandin I-2 (Sheridan & Bonventre, 2001). The imbalance in intrarenal vasoactive hormones causes persistent intrarenal hypoxia, thereby exacerbating tubular injury (Sheridan & Bonventre, 2001). Nephrotoxic ARF usually causes primary tubular injuries, and an initial decrease in RBF is not necessary for the development of renal functional impairment as in ischemic ARF. Rather, a specific cellular insult may precede in this type of ARF. Depending on nephrotoxicants, injury sites may be glomeruli, proximal tubules, medulla, renal vessels, and interstitium. Tubular dysfunction by nephrotoxicants leads to reduced Na reabsorption, backleak of glomerular filtrate, and tubular obstruction. Both types of ARF result in reduced glomerular filtration, tubular cell necrosis and apoptosis (Stein et al, 1978; Schellmann & Kelly, 1999). Therefore, ischemic and nephrotoxic ARF are likely to show similar functional and histological changes.
Although renal expression of ET-1 in ischemic ARF is elevated in most cases, that of ETAR and ETBR varies depending on ischemic models and investigators (Roubert et al, 1994; Shimizu et al, 1998; Forbes et al, 2001). Prior to evaluating the usefulness of ET receptor antagonists, we examined the expressions of renal ET-1, ETAR and ETBR in nephrotoxic ARF. For that purpose we used cisplatin, which is well-known to cause nephrotoxic ARF (Stein et al, 1978; Ramesh & Reeves, 2002).
METHODS
Cisplatin administration and kidney preparation
C57BL/6 male mice of 10 to 11 week ages (20~25 g) were used and had free access to food and water. The animals received a single intraperitoneal injection of cisplatin (16 mg/kg, body weight) or saline. Seventy-two hrs after the treatment, mice were anesthetized with ketamine (50 mg/kg) and zylazine (16 mg/kg). Blood was drawn from cardiac puncture and then mice were sacrificed with cervical dislocation. Right kidneys and half of the left kidneys were frozen in liquid nitrogen and stored at -80℃ for RNA or protein isolation. The rest of left kidneys were fixed in 10% buffered saline, embedded in paraffin, sectioned at 4 um, and stained with Periodic acid-Shiff (PAS). Plasma creatinine concentration and blood urea nitrogen (BUN) were measured by an enzymatic method using commercially available kits (Asanpharm Co., Seoul, Korea).
Tissue ET-1 level
One preweighed kidney was immersed in a 2 ml solution containing 0.2 M acetic acid and 0.5 M NaCl and then homogenized for 2×10 sec on ice. The homogenate was centrifuged at 30,000 rpm for 30 min., and the supernatant was saved. A BondElut C18 column (Varian, Harbor City, CA, USA) was pre-equilibrated with 3 ml of 60% acetonitrile in 0.1% trifluoroacetic acid (TFA) and washed four times with 0.1% TFA. After applying the supernatant to the column, the column was eluted with 3×3 ml of pre-equilibration solution. The eluant was kept at -70℃ and then freeze-dried overnight. ET-1 was measured using EIA kit (IBL, Gunma, Japan) according to the supplier's instruction.
Real-time RT-PCR
Total RNA was extracted using RNAqueous-4PCR kit (Ambion, Austin, TX, USA) according to manufacturer's directions and treated with DNAse I included in the kit. After spectrophotometric determination of total RNA concentrations, reverse transcription was performed in a total volume of 40 ul containing 0.5 to 1 ug each of RNA and oligo (dT) using SuperScript II first-strand synthesis system (Invitrogen, Carlsbad, CA, USA). PCR was run in a ABI Prism 7000 system with a 25 ul reaction which contained 5 ul of 5-fold diluted cDNA, 1 nM primer, and 2× SYBR II master buffer (Applied Biosystems, Foster City, CA, USA). Reaction conditions are: 95℃ for 10 min followed by 45 cycles at 95℃ for 15 sec and 60℃ for 1 min. The relative mRNA expression was calculated using a comparative delta-delta method as described in ABI user bulletin #2. Relative PCR efficiencies of targets (ET-1, ETAR, and ETBR) and reference (β-actin) were approximately equal. Primers were generated using Primer 3 software (Rosen S and Skaletsky HJ, Whitehead Institute, MIT). All of them were designed to include one or two introns and their sequences are presented in Table 1.
Table 1.
Primer sequences for real time RT-PCR

ET-1, endothelin-1; ETAR, endothelin A receptor; ETBR, endothelin B receptor.
Western blot
The frozen kidney was thawed, and the entire kidney except the papillary region was homogenized in mannitol buffer (300 mM mannitol, 10 mM HEPES/Tris, 1 mM EDTA, pH 7.4) with 1× protease inhibitor cocktail (Sigma, St Louis, MO, USA). Protein concentration was determined using Bio-Rad assay kit (Hercules, CA, USA). For Western blot, an aliquot of homogenates was electrophoresed and then transferred to PVDF membranes (Amersham Pharmacia, Alington Height, IL, USA). The blot membranes were incubated overnight at 4℃ with either 1:200 dilution of rabbit anti-ETAR antibody (Alomone labs, Jerusalem, Israel) and mouse monoclonal anti-ET-1 antibody (Sigma, St Louis, MO, USA) or 1:500 dilution of rabbit anti-ETBR antibody (Alomone labs, Jerusalem, Israel). After washing the membranes, HRP-conjugated secondary antibody was added and signals were detected using ECL kit (Amersham Pharmacia, Alington Height, IL, USA). To confirm equal loading of sample proteins, blots were stripped for 30 min at 50℃ in stripping buffer (100 mM 2-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7) and then reprobed with primary antibody to β-actin. The intensity of bands was quantified and normalized to β-actin.
Immunohistochemistry
The kidneys were perfusion-fixed with 4% paraformaldehyde and postfixed overnight in the same solution. Paraffin sections were mounted on probe-on slides (Fisher Scientific Korea, Seoul, Korea), deparaffinized, hydrated, and treated with 3% H2O2 for 6 min to inhibit intrinsic H2O2 of the tissue. H2O2 was removed by extensive washing in immunoassay buffer (Biomeda, Fostar City, CA, USA). The tissue sections were then treated with blocking buffer (Zymed, South San Francisco, CA, USA) for 15 min and incubated with the primary antiserum of ET-1 (1:40), ETAR (1:20), or ETBR (1:40) as instructed by the manufacturer (HistoMouse-Max kit, Zymed, South San Francisco, CA, USA). The immunoreactive signals were observed through 3-amino-9-ethylcarbazole staining.
Statistical analysis
Data are expressed as mean±SD. Unpaired Student's t-test was performed and a p value less than 0.05 was considered statistically significant.
RESULTS
Induction of ARF
As shown in Table 2, plasma creatinine and BUN were significantly increased at 72 hr after cisplatin treatment, indicating deterioration of renal function. PAS staining showed that cisplatin administration caused necrosis of proximal tubules and produced tubular luminal casts, however, no effects on glomeruli, distal tubules, and medullary regions (data not shown).
Table 2.
Changes in BUN and plasma creatinine concentration

Blood was obtained 72-hr after saline or cisplatin (16 mg/kg) injection. Values are mean±SD of 6 animals. *p<0.05 vs. control
Changes in transcript and protein levels
Quantitative real-time PCR was performed to follow changes in mRNA expression of the ET system. Fig. 1 depicts a relative renal expression of ET-1, ETAR, and ETBR mRNAs. Cisplatin administration upregulated ET-1 mRNA by 13-fold and ETAR mRNA by 2-fold compared with controls. However, the expression of ETBR mRNA was not different between cisplatin-treated mice and controls. To examine whether the changes in mRNA level are correlated with protein expression, ELISA and Western blot were conducted. Fig. 2 shows that the content of ET-1 peptide in kidney homogenates was increased by 1.5-fold after cisplatin treatment (p<0.05). Similar increase was also observed in Western blot (Fig. 3). The primary antibody to ET-1 recognized the band at about 21 kDa, which is the expected size of unprocessed preproET-1 (Gumz et al, 2003). ETAR expression was increased by 1.7-fold, but ETBR expression was not different between cisplatin-treated mice and controls.
Fig. 1.

Relative expression of ET-1, ETAR, and ETBR genes in the kidney of the control and cisplatin-treated mice. Real-time RT-PCR was performed as described in Materials and Methods. Relative expression was calculated by a comparative CT method. Values are mean±SD of 3 separate experiments. *p<0.05 vs. matched control.
Fig. 2.

ET-1 contents in kidney homogenates of the control and cisplatin-treated mice. Values are mean±SD of 6 animals. *p<0.05 vs. control.
Fig. 3.

Representative immunoblots (upper panel) and densitometric analysis (lower panel) of ET-1, ETAR, and ETBR proteins. Each blot was loaded with 50 ug of protein. Band density was normalized to β-actin. Values are mean±SD of 3 separate experiments. *p<0.05 vs. respective control.
Localization of immunoreactivity
To examine the localization of protein expression of the ET system, immunohistochemistry was applied (Fig. 4). Immunoreactive ET-1 was strongly detected in some of proximal tubules and vessels, but not in glomeruli. Although it was not as intense as in controls, ET-1 expression was more diffuse in cisplatin-treated mice than in controls. The ETAR immunoreactivity showed a distribution similar to ET-1 and tended to increase in cisplatin-treated mice. It was predominantly localized in damaged proximal tubules, but was sparsely detected in peritubular capillaries and arteries. In spite of its relatively high background, ETBR immunoreactivity was not changed by cisplatin treatment.
Fig. 4.
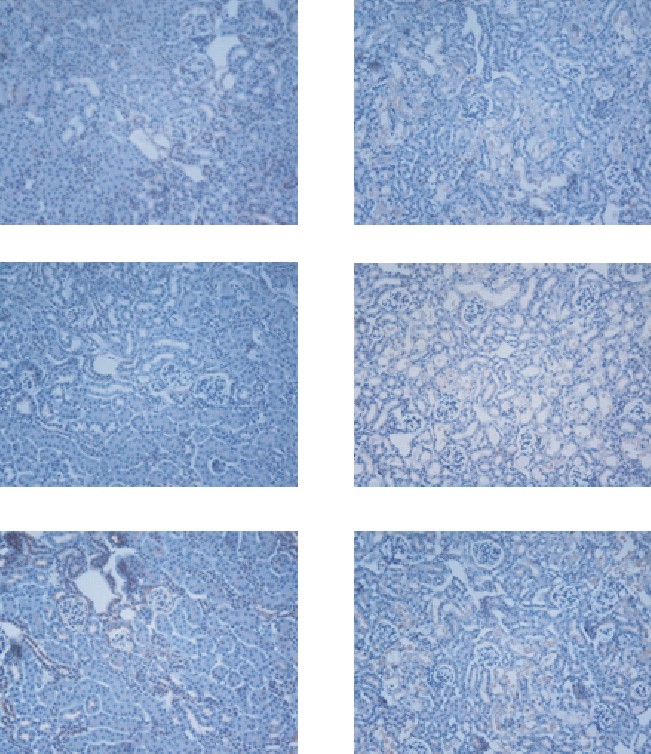
Immunostaining for ET-1, ETAR, and ETBR in the renal cortex of control and cisplatin-treated mice. ET-1, ETAR, and ETBR are diffusely expressed in tubular segments. Note that intense immunostaining for ET-1 and ETAR is found in the cisplatin-treated group. Magnification: ×100.
DISCUSSION
Nephrotoxic ARF is usually caused by heavy metals, anti-cancer agents, and aminoglycoside antibiotics. There are few reports on the expression of the ET system in nephrotoxic ARF. In cisplatin-induced nephrotoxic ARF, we found that renal damages were confined to the cortex, and ET-1 and ETAR were up-regulated without change of ETBR. Yanagisawa et al (1993) showed that HgCl2 treatment increased ET-1 mRNA in the kidney. Elevated ET-1 expression has been also found in other renal diseases such as ischemic ARF (Roubert et al, 1994; Shimizu et al, 1998; Forbes et al, 2001), chronic transplant rejection (Deng et al, 2000), polycystic kidney disease (Ong et al, 2003), mesangial proliferative nephritis (Yoshimura et al, 1995), and aminonucleoside-induced nephrosis (Nakamura et al, 1995). ET-1 directly inhibits Na-K-ATPase of renal tubular cells (Garvin & Sander, 1991) and modulates a variety of signal transduction pathways through its receptors as well. Therefore, while there is little doubt that ET-1 plays a certain role in the pathogenesis of renal diseases, it is not yet clear whether ET-1 acts as a causative factor in various renal diseases including ARF. An unusual finding in the present study was that ET-1 mRNA was increased in cisplatin-treated mice by 13-fold compared with controls, whereas ET-1 peptide was increased by less than 2-fold (Figs. 1 and 2). Since ET-1 gene is known to be transcriptionally regulated, such a discrepancy remains to be clarified.
Although ET-1 exerts its action through ETAR and/or ETBR, expression of ET receptors varies depending on ischemic ARF models used. In ischemic ARF induced by renal artery occlusion, Nambi et al (1993) showed that the affinity of ET receptor to ET-1 increased, accompanied with no change in the number of ETAR, whereas Forbes et al (2001) demonstrated that the level of ETAR decreased in the cortex. In glycerol-induced ischemic ARF, in which the pronounced fall of renal blood flow has been considered to be an important pathogenic event, ETAR mRNA in the cortex was up-regulated in one study but not in the other study (Roubert et al, 1994; Shimizu et al, 1998). In our model of nephrotoxic ARF, ETAR mRNA and protein was up-regulated by 1.5 to 2.0-fold compared with controls. Considering that ETAR mediates vasoconstrictive action of ET-1, an increase in ETAR expression could have reduced blood supply to the cortex, thus leading to aggravate the renal injury. It is, however, interesting to note that administration of selective ETAR antagonists has partly ameliorated renal tubular injuries and functions, in spite of decreased expression of ETAR in ischemic ARF (Gellai et al, 1994; Birck et al, 1998; Kuro et al, 2000).
ETBR level has been shown to be reduced or increased in ischemic ARF (Roubert et al, 1994; Shimizu et al, 1998; Forbes et al, 2001). The present study showed that both ETBR mRNA and protein expression levels were not different between controls and cisplatin-treated mice. Using a similar ETBR antibody that we have used, Francis et al (2004) reported that ETBR was detected mainly in tubular structures and was preferably expressed in the inner medulla. We found that the immunoreactivity of ETBR was much greater in the inner medulla than in the cortex and its immunoreactivity in the medulla seemed to be not different between the two groups (data not shown). Because renal damages by cisplatin administration was mainly confined to proximal tubules of the cortex, we speculate that ETBR expression would be least affected, if any.
In conclusion, this study demonstrated that the expressions of ET-1 and ETAR, but not ETBR, were increased, suggesting that the individual components of the renal ET system are differentially regulated in cisplatin-induced nephrotoxic ARF. Therefore, drug targeting to ETAR could help lessen nephrotoxic ARF induced by cisplatin.
ABBREVIATIONS
- ETAR
endothelin A receptor
- ETBR
endothelin B receptor
- ECE
endothelin-converting enzyme
- ARF
acute renal failure
References
- 1.Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, Kohan DE. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest. 2004;114:504–511. doi: 10.1172/JCI21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birck R, Knoll T, Braun C, Kirchengast M, Munter K, van der Woude FJ, Rohmeiss P. Improvement of postischemic acute renal failure with the novel orally active endothelin-A receptor antagonist LU 135252 in the rat. J Cardiovasc Pharmacol. 1998;32:80–86. doi: 10.1097/00005344-199807000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Braun C, Conzelmann T, Vetter S, Schaub M, Back WE, Yard B, Kirchengast M, Tullius SG, Schnulle P, van der Woude FJ, Rohmeiss P. Prevention of chronic renal allograft rejection in rats with an oral endothelin A receptor antagonist. Transplantation. 1999;68:739–746. doi: 10.1097/00007890-199909270-00005. [DOI] [PubMed] [Google Scholar]
- 4.Chatziantoniou C, Dussaule JC. Insights into the mechanisms of renal fibrosis: is it possible to achieve regression? Am J Physiol Renal Physiol. 2005;289:F227–F234. doi: 10.1152/ajprenal.00453.2004. [DOI] [PubMed] [Google Scholar]
- 5.Deng DX, Jiang J, Garcia B, Zhong R, Chakrabarti S. Endothelin-1, endothelin-3 and their receptors (endothelin (A) and endothelin (B)) in chronic renal transplant rejection in rats. Transpl Int. 2000;13:175–182. [PubMed] [Google Scholar]
- 6.Forbes JM, Jandeleit-Dahm K, Allen TJ, Hewitson TD, Becker GJ, Jones CL. Endothelin and endothelin A/B receptors are increased after ischaemic acute renal failure. Exp Nephrol. 2001;9:309–316. doi: 10.1159/000052626. [DOI] [PubMed] [Google Scholar]
- 7.Francis BN, Abassi Z, Heyman S, Winaver J, Hoffman A. Differential regulation of ETA and ETB in the renal tissue of rats with compensated and decompensated heart failure. J Cardiovasc Pharmacol. 2004;44:S362–S365. doi: 10.1097/01.fjc.0000166302.56184.fa. [DOI] [PubMed] [Google Scholar]
- 8.Gellai M, Jugus M, Fletcher T, DeWolf R, Nambi P. Reversal of postischemic acute renal failure with a selective endothelinA receptor antagonist in the rat. J Clin Invest. 1994;93:900–906. doi: 10.1172/JCI117046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman A, Grossman E, Goldstein DS, Gill JR, Jr, Keiser HR. Urinary excretion rate of endothelin-1 in patients with essential hypertension and salt sensitivity. Kidney Int. 1994;45:556–560. doi: 10.1038/ki.1994.72. [DOI] [PubMed] [Google Scholar]
- 10.Kuro T, Kohnou K, Kobayashi Y, Takaoka M, Opgenorth TJ, Wessale JL, Matsumura Y. Selective antagonism of the ETA receptor, but not the ETB receptor, is protective against ischemic acute renal failure in rats. Jpn J Pharmacol. 2000;82:307–316. doi: 10.1254/jjp.82.307. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura T, Ebihara I, Fukui M, Osada S, Tomino Y, Masaki T, Goto K, Furuichi Y, Koide H. Modulation of glomerularendothelin and endothelin receptor gene expression in aminonucleoside-induced nephrosis. J Am Soc Nephrol. 1995;5:1585–1590. doi: 10.1681/ASN.V581585. [DOI] [PubMed] [Google Scholar]
- 12.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer. 2003;3:110–116. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- 13.Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exp Nephrol. 2003;93:e80. doi: 10.1159/000068518. [DOI] [PubMed] [Google Scholar]
- 14.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roubert P, Gillard-Roubert V, Pourmarin L, Cornet S, Guilmard C, Plas P, Pirotzky E, Chabrier PE, Braquet P. Endothelin receptor subtypes A and B are up-regulated in an experimental model of acute renal failure. Mol Pharmacol. 1994;45:182–188. [PubMed] [Google Scholar]
- 16.Schellmann RG, Kelly KJ. Pathophysiology of nephrotoxic acute renal failure. In: Schrier RW, editor. Atlas of Diseases of the Kidney. 1st ed. Philadelphia: Current medicine Inc.; 1999. pp. 15.1–15.14. [Google Scholar]
- 17.Sheridan AM, Bonventre JV. Pathophysiology of ischemic acute renal failure. Contrib Nephrol. 2001;(132):7–21. doi: 10.1159/000060075. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu T, Kuroda T, Ikeda M, Hata S, Fujimoto M. Potential contribution of endothelin to renal abnormalities in glycerol-induced acute renal failure in rats. J Pharmacol Exp Ther. 1998;286:977–983. [PubMed] [Google Scholar]
- 19.Stein JH, Lifschitz MD, Barnes LD. Current concepts on the pathophysiology of acute renal failure. Am J Physiol. 1978;234:F171–F181. doi: 10.1152/ajprenal.1978.234.3.F171. [DOI] [PubMed] [Google Scholar]
- 20.Takaoka M, Kuro T, Matsumura Y. Role of endothelin in the pathogenesis of acute renal failure. Drug News Perspect. 2000;13:141–146. doi: 10.1358/dnp.2000.13.3.858437. [DOI] [PubMed] [Google Scholar]
- 21.Yoshimura A, Iwasaki S, Inui K, Ideura T, Koshikawa S, Yanagisawa M, Masaki T. Endothelin-1 and endothelin B type receptor are induced in mesangial proliferative nephritis in the rat. Kidney Int. 1995;48:1290–1297. doi: 10.1038/ki.1995.413. [DOI] [PubMed] [Google Scholar]