

Abstract
Bivalent morphinan compounds containing ester linkers were synthesized and their binding affinities at the μ, δ, and κ opioid receptors determined. Addition of methyl groups adjacent to the hydrolytically labile ester linkage increased stability while only partially affecting binding affinity. The resulting bivalent ligands with optimized spacer-length and structure show potent binding profiles with the most potent compound (4b) having Ki values of 0.47 nM for both the μ and κ opioid receptors, and 4a having Ki values of 0.95 and 0.62 nM for the μ and κ receptors, respectively. Both 4a and 4b were partial agonists at the κ and μ receptors in the [35S]GTPγS binding assay.
Introduction
Previous reports from our laboratory have shown that bivalent morphinan (butorphan) ligands, where two identical (“homobivalent”) or two distinct (“heterobivalent”) morphinan ligands are connected by a molecular spacer, can lead to compounds with improved binding affinities and selectivities.1,2,3 The binding profile of the bivalent ligand is strongly dependent on the molecular structure of the opioid pharmacophore and on the length of the connecting spacer, with highest affinities obtained with ten carbon atoms separating the morphinan structure.1,2 In our previous studies, various methods to connect the two opioid pharmacophores were investigated, the most active compounds at the opioid receptors were connected via 10 carbon linking ester chain (Table 1, 10 (MCL-144) Ki = 0.090 nM at μ; Ki = 0.49 nM at κ).2 It has been suggested that such bivalent ligands containing ester groups produced their affinities by the hydrolysis to the active pharmacophore butorphan during the assay.
Table 1.
Ki Values for the inhibition of μ, δ and κ opioid binding to CHO membranes by uni- and bivalent ligands
| Compound/Structure | Ki (nM) ± SE | Selectivity | |||
|---|---|---|---|---|---|
| μ | δ | κ | μ/δ/κ | ||
| Butorphana |  |
0.23 ± 0.01 | 5.9 ± 0.55 | 0.079 ± 0.003 | 3/75/1 |
| 9b | 0.71 ± 0.02 | 18 ± 0.6 | 0.29 ± 0.02 | 2.4/62/1 | |
| 7a | 0.46 ± 0.024 | 20 ± 0.074 | 0.42 ± 0.0081 | 1/48/1 | |
| 6a | 1.7 ± 0.11 | 42 ± 2.8 | 0.94 ± 0.02 | 1.8/45/1 | |
| 7b | 47 ± 3.1 | 390 ±11 | 18 ± 0.48 | 3/22/1 | |
| 6c | 8.6 ± 0.63 | 210 ±30 | 4.7 ± 0.68 | 1.8/45/1 | |
| 10c | 0.090 ± 0.004 | 4.2 ± 0.4 | 0.049 ± 0.0001 | 2/90/1 | |
| 4a | 0.95 ± 0.057 | 37 ± 1.8 | 0.62 ± 0.071 | 1.5/60/1 | |
| 4b | 0.47 ± 0.027 | 15 ± 0.86 | 0.47 ± 0.046 | 1/32/1 | |
| 4c | 2.9 ± 0.091 | 50 ± 2.6 | 2.2 ± 0.096 | 1.3/23/1 | |
| 11a | 0.43 ± 0.001 | 39 ±2 | 0.13 ± 0.03 | 3.3/300/1 | |
| 8 | 3.7 ± 0.36 | 130 ± 5.4 | 3.4 ± 0.22 | 1/38/1 | |
We thus wished to further investigate the factors that lead to high affinity in this series of bivalent morphinan ligands. To decrease the rate of hydrolysis, we modified the linker by introducing methyl groups in the α-position (and α′-position, respectively) of the linking dicarboxylic acid, this based in-part on the observation that steric hindrance decreases the rate of hydrolysis of esters.4 A similar observation has been observed for a series of phenyl esters which underwent aminolysis, in which the rate decreased dramatically with α methyl substitution.5 A more recent study investigated methods for increasing the oral bioavailability of otherwise potent orally unavailable drugs by synthesizing prodrugs with ester functionalities.6 These investigators found that increased half-lives of esters branched with methyl-groups in the acid moiety could be observed both at pH = 1 (t1/2 CH3/iso-propyl/tert-butyl = 1/2/10) and pH = 10 (t1/2 CH3/iso-propyl/tert-butyl =1/4.6/16).6
The possible decrease in the hydrolytic reactivity of esters of bivalent ligands containing branched spacers with esterases is important for such bivalent morphinans. By reducing the reactivity toward esterases the effect of the ligand in contrast to the hydrolysis product butorphan could be assessed. In one study the enzymatic rates of hydrolysis of a series of phenyl esters were determined using α-chymotrypsin and carboxylesterase. It was found that phenyl acetate is hydrolyzed much faster than phenyl trimethylacetate.7 Studies showing increased half-lives of highly substituted phenyl esters of phenolic drugs with different degrees of substitution in the acid moiety at physiological pH (7.4), in human plasma, and in pig liver homogenate may enable potential usage as prodrugs.8
We have chosen to modify the high affinity opioid agonist butorphan9 (MCL-101), which was prepared from commercially available (−)-3-hydroxy-N-methyl-morphinan (levorphanol). Butorphan possesses mixed κ agonist and μ agonist/antagonist binding properties with low affinity to the δ subtype,10 potentially useful for application in cocaine abuse therapy.11,12
In order to investigate the in vitro sensitivity toward hydrolysis, the stability of the most potent bivalent ligand in buffer and in the presence of an esterase was determined. Furthermore, the affinity of compounds consisting of one butorphan unit plus the spacer (which would also be the first hydrolysis product) was determined to obtain information on how the affinity of these compounds might contribute to the affinity in the binding assay, and especially to see how the spacer contributes to the binding at the receptor.
Chemistry
For the synthesis of α,α′-dimethylated dicarboxylic acid (3a, 3c), several synthetic approaches were investigated, e.g. hydrocarboxylation of α,ω-dienes in the presence of copper and palladium salts.13 The most straightforward method to obtain these dicarboxylic acids was to deprotonate the α-carbon of 2 moles of propionic acid (α,α′-dimethyldicarboxylic acids 3a, 3c) or isobutyric acid (α,α,α′,α′-tetramethyldicarboxylic acid 3b14) using lithium diisopropylamide, followed by alkylation with dibromoalkanes (Scheme 1).
Scheme 1.

a. Synthesis of homobivalent butorphan ligands with spacers branched in α-position
a Reagents and conditions: (i) 2 equiv acid, 4 eq LiN(i-Pr)2, THF/heptane/ethylbenzene, −25°C to 50 °C; (ii) cool to −30 °C, addition of dibromide, warm to 40 °C for 1hr; (iii) 4 eq (COCl)2/CH2Cl2/cat. DMF, concentration in vacuo, then 2 equiv butorphan/CH2Cl2/Et3N, rt 24 h.
The dicarboxylic acids 3a, 3b, 3c (1 mole) were activated to the respective dicarboxylic acid chlorides, which were then coupled to butorphan (2 moles) to yield the homobivalent compounds 4a, 4b, 4c.2 To obtain compounds containing only linker plus butorphan (7a, 7b), (Scheme 2) reacting equimolar amounts was not an effective method since small amounts of bivalent ligands were always formed. Protecting one carboxylic group of the diacids with a benzyl-group to yield the monobenzylesters 5b, 5c, reaction with butorphan to yield 6b, 6c, followed by deprotection using catalytic hydrogenolysis gave the corresponding acids (7a, 7b). Monobenzylation of α,α′-dimethyldicarboxylic acids could be easily achieved using DCC/DMAP and benzyl alcohol.15 Monobenzylation of α,α,α′,α′-tetramethyldicarboxylic acid to yield 5c could not be achieved this way due to steric hinderance. Also activation of the acid with N,N′-carbonyldiimidazole (CDI) did not lead to product formation and activation as the acid chloride gave yields < 50 %. Quantitative monoesterifaction was achieved using benzylbromide in the presence of tetrabutylammonium fluoride in THF.16 The methyl ester of the α,α′-dimethyldicarboxylic acid with butorphan (6a) was also prepared and together with the corresponding benzylester (6c) was included in the pharmacological evaluation. Since the highest affinities for unhindered bivalent butorphan esters were observed for a C10 and a C4-spacer, respective hindered C4 bivalent ligands would need 2,3-dimethyl or tetramethyl succinic acid as spacers.1 Interestingly, neither coupling of 2,3-dimethyl nor tetramethylsuccinic acid chlorides with butorphan leads to any product formation.
Scheme 2.

a. Synthesis of univalent butorphan ligands 6a–c and 7a–b.
a Reagents and conditions: (i) MeOH, DCC, DMAP, CHCl3, rt, 24 h; (ii) C6H5CH2OH, DCC, cat. DMAP, CHCl3, rt, 24 h (iii) C6H5CH2Br, Bu4NF, THF, rt, 24 h; (iv) 2 equiv (COCl)2/CH2Cl2/cat. DMF, concentration in vacuo, then butorphan/CH2Cl2/Et3N, rt 24 h; (v) 10% Pd/C, H2, 1 atm, MeOH/EtOH, rt.
The butorphan ester 7a when coupled with the opioid antagonist naloxone via the in situ generated acid chloride as in the schemes above led to the heterobivalent ligand 8.17
Binding Affinity
The affinity and selectivity of the compounds synthesized were evaluated for their binding affinities at all three opioid receptor types (μ, δ, and κ) using a previously described procedure (Table 1).1
The introduction of methyl groups on the carbon atom adjacent to the carbonyl atom of the ester had an effect on the binding affinities and selectivity. Both compounds with one methyl groups at the α-position (the dimethyl-compound 4a and the tetramethyl-compound 4b) were approximately 10 times less active than the unsubstituted C10 bivalent ligand (10) with a binding affinity of 0.049 nM at the κ receptor.2 The dimethyl ligand 4a was marginally less selective for the κ receptor versus the μ receptor than 10.
The tetramethyl ligand 4b was equipotent at both κ and μ receptors. Binding affinities at the μ- and δ-receptors showed no coherent tendencies. At the μ receptor the binding affinity decreased for the dimethyl-compound (4a), but further increased slightly for the tetramethyl-compound (4b) with a binding affinity of 0.47 nM. Dramatic effects were observed with regard to binding affinity and selectivity at the δ opioid receptor. Introduction of a α-methyl group (4a) led to a four-fold decrease in binding affinity whereas introduction of a α-dimethyl group (4b) only slightly decreased binding affinity, relative to the unsubstituted bivalent ligand 10. The dimethyl ligand 4a lost some selectivity for μ and κ receptors versus the δ receptor but for the tetramethyl bivalent ligand 4b, 32-fold selectivity at κ (and μ) over δ was obtained. As in the homobivalent series, introduction of a α-methyl group also reduces the affinity of heterobivalent ligands (11 vs. 8) by a factor of 9 at the μ opioid receptor and 26 at the κ opioid receptor (Table 1).
We have shown that the second pharmacophore is not always necessary to achieve binding to the opioid receptors.1 This was confirmed in this study as the univalent compounds 6a and 7a have good binding affinities to the μ and κ opioid receptors when compared with the corresponding bivalent ligands 4a and 4b.
The presence of an acidic carboxylic acid in the linker chain does not appear to have any dramatic affect on the binding affinities as methylation of 7a to produce 6a only decreased binding affinity by a factor of four at μ and a factor of two at the κ and δ receptors. The poor binding of 7b could be only partially restored by the addition of a benzyl group at the terminal carboxylic acid. The benzyl ester, 6c, of 7b had five times stronger binding affinity at the μ opioid receptor than 7b.
Efficacy of Selected Ligands
The agonist and antagonist properties of these ligands in stimulating [35S]GTPγS binding mediated by the μ opioid receptor are shown in Table 2.
Table 2.
[35S]GTPγS Binding by uni- and bivalent ligands mediated by the μ opioid receptor
| Mu Agonist Effects | EC50 (nM) ± S.E.M. | Emax (% stimulation) ± S.E.M. | Functional Activity |
|---|---|---|---|
| Butorphan | 1.6 ± 0.15 | 50 ± 2.5 | Partial Agonist |
| 9 | 3.0 ± 0.64 | 110 ± 8.1 | Agonist |
| 7a | 13 ± 1.4 | 25 ± 3.9 | Partial Agonist |
| 10 | 1.3 ± 0.15 | 51 ± 6.5 | Partial Agonist |
| 4a | 7.1 ± 3.1 | 26 ± 1.9 | Partial Agonist |
| 4b | 11 ± 1.2 | 40 ± 6.1 | Partial Agonist |
| 11 | 2.0 ± 0.54 | 34 ± 1.2 | Partial Agonist |
| Mu Antagonist Effects | IC50 (nM) ± S.E.M. | Imax (% inhibition) ± S.E.M. | |
| Butorphan | 20 ± 2.7 | 50 ± 2.6 | |
| 9 | No inhibition | No inhibition | |
| 7a | NA | 48 ± 6.4 at 10 μM | |
| 10 | 16 ± 3.0 | 44 ± 5.1 | |
| 4a | 140 ± 17 | 50 ± 6.1 | |
| 4b | 81 ± 16 | 50 ± 0.59 | |
| 11 | 25 ± 1.6 | 73 ± 1.2 |
To determine agonist effects, 12 different concentrations of the compounds were incubated with CHO cells that expressed the μ opioid receptor. Antagonist activity was determined by measuring the inhibition of [35S]GTPγS binding by the compounds. For antagonist studies, [35S]GTPγS binding was stimulated with 200 nM DAMGO. The stimulation observed with DAMGO was set at 100% control binding.
Ligand 9 produced maximal stimulation of [35S]GTPγS binding mediatedby the μ receptor while it produced no inhibition (Imax) of the DAMGO stimulated [35S]GTPγS binding comparable to that of butorphan. These data indicate that 9 is a full μ agonist. Bivalent ligands 4a and 4b were partial agonists. Ligands 7a, 4a, 4b, and 11 produced less maximal stimulation of [35S]GTPγS binding (Emax) comparable to that of butorphan. The EC50 values of the ligands are slightly lower that butorphan (1) suggesting that these ligands are less potent than butorphan.
The agonist and antagonist properties of these ligands in stimulating [35S]GTPγS binding mediated by the κ receptor are shown in Table 3.
Table 3.
[35S]GTPγS Binding by uni- and bivalent ligands mediated by the kappa opioid receptor
| Kappa Agonist Effects | EC50 (nM) ± S.E.M. | Emax (% stimulation) ± S.E.M. | Functional activity |
|---|---|---|---|
| Butorphan | 1.3 ± 0.44 | 80 ± 6.8 | Agonist |
| 9 | 2.8 ± 0.43 | 75 ± 1.1 | Agonist |
| 7a | 4.9 ± 0.82 | 58 ± 7.4 | Agonist |
| 10 | 0.85 ± 0.053 | 65 ± 0.68 | Agonist |
| 4a | 7.3 ± 1.6 | 54 ± 3.5 | Agonist |
| 4b | 6.4 ± 0.87 | 60 ± 8.0 | Agonist |
| 11 | 1.6 ± 0.23 | 86 ± 7.3 | Agonist |
| Kappa Antagonist Effects | IC50 (nM) ± S.E.M. | Imax (% inhibition) ± S.E.M. | |
| Butorphan | NT | NT | |
| 9 | No inhibition | No inhibition | |
| 7a | No inhibition | No inhibition | |
| 10 | No inhibition | No inhibition | |
| 4a | No inhibition | No inhibition | |
| 4b | No inhibition | No inhibition | |
| 11 | No inhibition | No inhibition |
To determine agonist effects, 12 different concentrations of the compounds were incubated with CHO cells that expressed the κ opioid receptor. Antagonist activity was determined by measuring the inhibition of [35S]GTPγS binding by the compounds. For antagonist studies, [35S]GTPγS binding was stimulated with 100 nM U50,488. The stimulation observed with U50,488 was set at 100% control binding.
Ligands 4a, 4b, 7a, 9, and 11 produced similar minimal stimulation of [35S]GTPγS binding mediated by the κ receptor, while they produced no inhibition of [35S]GTPγS binding mediated by the κ receptor. These data indicate that these ligands are all κ agonists. The univalent ligand 9 was a full agonist at both the μ and κ opioid receptors.
Hydrolytic Stability
The hydrolysis results (Table 4) indicate that introduction of α-dimethyl groups does indeed increase the stability of the bivalent ligands towards in vitro hydrolysis by at least a factor of 2. The presence of the α-methyl groups appears to have no effect on the enzymatic rate of hydrolysis as the half-life of all bivalent ligands in the presence of porcine liver esterase is approximately the same. Typical binding experiments involve incubating the ligands at 25 °C in a CHO membrane homogenate for 60 minutes. The hydrolysis results, conducted at physiological-like conditions of 37 °C, show that the compounds investigated would be stable during the time course of the binding experiments. It is generally accepted that the rate of a reaction doubles with an increase in temperature of 10 °C. As such, it can be predicted that the rate of hydrolysis of the bivalent esters at 25 °C will be approximately one-half of that at 37 °C. It is clear that the activity measured during the binding affinity experiments is due to the bivalent ligands and not to butorphan produced by hydrolysis. While the introduction of steric hindrance can reduce the amount of hydrolysis, chain length also appears to be a factor as shown by 4c that was surprisingly more stable than its C10 congener 4a.
Table 4.
Hydrolysis of Bivalent Ligands
| Compound | t1/2 (minutes) 37 °C | ||
|---|---|---|---|
| Buffer pH 7.4 | Liver esterase | ||
| 10 | 46 | 110 | |
| 4a | 83 | 131 | |
| 4b | 101 | 100 | |
| 4c | 221 | 114 | |
The hydrolysis rate results agree with a previous study where the mono phenyl ester of 2-methylsuccinic acid was 43 times more stable at pH 7.4 than mono phenyl succinate.8 It has also been shown that the introduction of α gem-dimethyl groups onto the phenyl ester of 4-pentenoic acid increases its hydrolytic stability in acidic solutions by a factor of 2 and in alkaline solution by a factor of 215.18
In summary, our data suggests that the introduction of α-methyl groups increases the hydrolytic stability of the bivalent ligands and has the effect of decreasing the binding affinities to opioid receptors for the di- as well as for the tetra-substituted compound. The difference in binding affinities could be due to the inherent properties of the bivalent ligands themselves and not due to hydrolytic products that may develop during the binding assay.
Experimental Section
General Synthetic Methods
1H and 13C NMR spectra were recorded at 300 MHz using CDCl3 or DMSO-d6 on a Bruker AC300 spectrometer. Chemical shifts are given as δ value (ppm) downfield from tetramethylsilane as an internal reference. Melting points were determined on a Thomas-Hoover capillary tube apparatus and are reported uncorrected. LC-MS and HRMS was performed on an Agilent 6210 time-of-flight spectrometer using an ESI source with the LC being performed on an Agilent 1200 series system with an autosampler and the mobile phases A: H2O with 0.1 % formic acid and B: acetonitrile with 0.1 % formic acid. All target compounds showed one peak only in the LC prior to MS. Elemental analyses, performed by Atlantic Microlabs, Atlanta, GA, were within ±0.4% of theoretical values. Analytical thin-layer chromatography (TLC) was carried out on 0.2 μm Kieselgel 60F-254 silica gel plastic sheets (EM Science, Newark, NJ). Flash chromatography was used for the routine purification of reaction products. Eluent systems are described for the individual compounds.
General procedure for the preparation of α,α′-dimethyl- and α,α,α′,α′ -tetramethyldicarboxylic acids (3a,b,c)
The α,α′-dimethyldicarboxylic acids were prepared in a similar fashion as described for α,α,α′,α′-tetramethyldicarboxylic acids.14 In an ice/acetone bath 42 mL of a 2.0 M solution of lithium diisopropylamide (84 mmol) in heptane/THF/ethylbenzene was cooled to −25 °C. Very slowly (caution: vigorous reaction!) 35 mmol of propionic acid (for preparation of 3a and 3c) or isobutyric acid (for preparation of 3b) were added so that the temperature did not rise over −20 °C. The mixture was vigorously stirred and after addition of acid the mixture was slowly warmed to 50 °C. After ~30 min a yellow precipitate was formed. After warming for 2h at 50 °C, the mixture was cooled to −30 °C and 1,6-dibromohexane (for preparation of 3a and 3b) or 1,4-dibromobutane (for preparation of 3c) were added drop wise. After addition the temperature was raised to ~ 40 °C, while the suspension lost its brown color and became yellow. Stirring at 40 °C was continued for 1h, after which concentrated hydrochloric acid was added under ice-cooling until pH = 1. The mixture was extracted three times with dichloromethane. The combined organic phases were washed three times with 10% hydrochloric acid (20 mL) and twice with 100 mL of water. The organic phase was dried over Na2SO4 and the solvent evaporated. High vacuum was used to remove residues of ethylbenzene. The brown oily residue was recrystallized from ethyl acetate to yield colorless crystals.
2,9-dimethyldecanedioic acid (3a)
23% yield; mp: 104 °C; 1H (DMSO-d6): 12.00 (bs, 2H, COOH), 2.28 (sex, 2H, J = 7 Hz, CH), 1.52 (qui, 2H, CH2CH), 1.35 (qui, 2H, J = 7 Hz, CH2CH), 1.28 (bs, 8H, CH2), 1.03 (d, 6H, J = 7 Hz, CH3) ppm. 13C (DEPT, DMSO-d6): 178.19 (COOH), 39.37 (CH), 33.93 (CH2), 29.54, 27.29, 17.66 (CH3) ppm.
2,2,8,8-tetramethyldecanedioic acid (3b)
40% yield; mp: 117 °C (lit:8 118 °C). 1.52 (m, 4H), 1.28 (m, 8H), 1.18 (bs, 12H) ppm. 13C (CDCl3): 185.0 (COOH), 42.4, 40.7, 30.0, 25.2, 24.9 ppm.
2,7-dimethyloctanedioic acid (3c)
23% yield; mp: 121 °C; 1H (DMSO-d6): 12.02 (bs, 2H, COOH), 2.28 (sex, 2H, J = 7 Hz, CH), 1.52 (m, 2H, J = 7 Hz), 1.21–1.33 (m, 8H), 1.03 (d, 6H, J = 7 Hz, CH3) ppm.13C (DMSO-d6): 178.15 (COOH), 39.31 (CH), 33.80 (CH2), 27.28, 17.63 (CH3) ppm.
General procedure for the preparation of 10-methoxy- (5a) and 10-(benzyloxy) -2,9-dimethyl-10-oxodecanoic acid (5b)
To 2 mL of anhydrous chloroform were added 1 mmol of methanol (dried over Mg, for preparation of 5a) or benzyl alcohol (for preparation of 5b), followed by 230 mg (1 mmol) of 2,9-dimethyldecanedioic acid, then a catalytic amount of DMAP was added, followed by 240 mg (1.2 mmol) of DCC. The mixture was stirred at rt under nitrogen overnight. To the suspension were added 1 mL of water and additional chloroform and the mixture was vigorously stirred for an additional hour to hydrolyse excess DCC. The precipitated urea was filtered off, the solution dried over Na2SO4 and the solvent evaporated. The oil obtained was used for the next reaction step without further purification.
10-methoxy-2,2,9,9-tetramethyl-10-oxodecanoic acid (5a)
70% yield; colorless oil. 1H (CDCl3): 3.67 (s, 3H), 2.44 (m, 1H), 1.61–1.71 (m, 3H), 1.15–1.42 (m, 10H), 1.13 (d, J =7 Hz, 6H) ppm. 13C (CDCl3): 177.4, 156.2, 51.5, 39.4, 33.8, 33.7, 33.6, 29.3, 27.1, 25.5, 24.9, 17.1 ppm.
10-(benzyloxy)-2,9-dimethyl-10-oxodecanoic acid (5b)
79% yield; colorless oil. 1H (CDCl3): 7.37 (bs, 5H, arom.), 5.11 (s, 2H, Bn-CH2), 2.46 (qui, 2H, J = 7 Hz), 1.50–1.81 (m, 4H), 1.14–1.38 (m, 14H), ppm. 13C (CDCl3): 176.6 (CO), 136.04, 128.5, 128.1, 128.0, 65.9 (Bn-CH2), 39.5, 33.7, 29.3, 27.1, 27.0, 17.0 (CH3) ppm.
Synthesis of 10-(benzyloxy)-2,2,9,9-tetramethyl-10-oxodecanoic acid (5c)
To a solution of 0.57 mmol of 2,2,9,9-tetramethyldecanedioic acid in THF were added drop wise 0.57 mL of 1.0 M tetrabutylammonium fluoride in THF under stirring at rt, followed by addition of 0.57 mmol of benzylbromide in 1 mL of THF over 3h under vigorous stirring. The solution was allowed to stand overnight at rt, after which 3 mL of 2 N HCl were added and the mixture extracted 4× with EtOAc. The combined organic phases are subsequently washed 4× with a small amount of 1N HCl to remove TBA ions. The organic phase was dried over sodium sulfate to yield a colorless oil (143 mg, 72%), which was sufficiently pure for the following reaction step.1H (CDCl3): 7.34 (bs, 5H, arom.), 5.10 (s, 2H, BnCH2), 1.42–1.58 (m, 4H), 1.15–1.30 (m, 20H) ppm. 13C (CDCl3): 184.4 (COOH), 178.2 (COOBn), 136.7, 128.7, 128.2, 128.1, 128.0, 66.2 (BnCH2), 42.6, 42.3, 41.0, 40.7, 30.1, 29.9, 25.4, 25.3, 25.2, 25.1, 25.0, 24.8 ppm.
General procedure for the synthesis of butorphan univalent diesters (6a,b,c)
In 2 mL of anhydrous methylene chloride 0.75 mmol of the respective dicarboxylic monoester (5a,b,c) were dissolved, two drops of dimethylformamide were added followed by the addition of 1.5 mmol of oxalylchloride. Gas evolution could be observed and the solution was stirred for 4 h at rt under nitrogen. Then solvent and excess oxalyl chloride were removed under reduced pressure and high vaccuum, respectively. The residual acid chloride of the monoester was dissolved in 4 mL of anhydrous methylene chloride and 0.5 mmol of butorphan were added. Drop wise addition of 2 mmol of triethylamine lead to dissolution of butorphan and the solution was refluxed overnight. After cooling additional methylene chloride was added and the mixture washed with sat. NaHCO3 solution and brine. The organic phase was dried over Na2SO4 and solvent removed under reduced pressure. The residual oil was column chromatographed using hexane/ethyl acetate/triethylamine 10/10/1 as eluent system.
Methyl 17-((−)-N-cyclobutylmethyl)morphinan-3-yl 2,9-dimethyldecanedioate (6a)
colorless oil (21%); 1H (CDCl3): 7.11 (d, J = 8 Hz, 1H), 6.92 (d, J = 2 Hz, 1H), 6.85 (dd, J = 8, 2 Hz, 1H), 3.67 (s, 3H), 2.98 (m, 2H), 2.60–2.67 (m, 6H), 2.21–2.45 (m, 2H), 2.06–2.11 (m, 3H), 1.16–1.92 (m, 29H), 1.14 (d, J = 7 Hz, 3H) ppm. 13C (CDCl3): 177.3, 175.4, 149.3, 142.0, 135.1, 128.5, 118.4, 118.0, 61.5, 55.8, 51.4, 45.7, 44.8, 41.7, 39.6, 39.4, 37.7, 36.5, 34.9, 33.7, 33.6, 29.4, 29.3, 27.8, 27.1, 26.7, 26.5, 24.4, 22.1, 19.8, 17.1, 17.0, 16.9 ppm. MS (TOF ESI+): m/z = 538 ([M+H]+). HRMS Calcd for C34H52NO4: 538.3891. Found 538.4082. Anal. Calcd for C34H51NO4 × 2/3 H2O: C, 74.28; H, 9.59; N, 2.55. Found: C, 74.14; H, 9.45; N, 2.87.
Benzyl 17-((−)-N-cyclobutylmethyl)morphinan-3-yl 2,9-dimethyldecanedioate (6b)
Colorless oil (30%);. 1H (CDCl3): 7.12 (d, 1H, J = 8.4 Hz), 6.92 (d, 1H, J = 2.4 Hz), 6.87 (dd, 1H, J = 8.4, 2.4 Hz), 2.98 (d, 2H, J = 18 Hz), 2.50–2.78 (m, 8H), 1.01–2.18 (m, 36H) ppm. 13C (CDCl3): 176.9, 175.7, 149.7, 142.6, 136.5, 128.8, 128.7, 128.3, 128.29, 118.7, 118.3, 66.2, 61.6, 56.1, 46.0, 39.9, 39.4, 37.9, 36.7, 34.0, 29.6, 28.1, 27.4, 27.0, 26.7, 24.7, 22.3, 19.1, 17.3 ppm.
Benzyl 17-((−)-N-cyclobutylmethyl)morphinan-3-yl 2,2,9,9-tetramethyldecane-dioate (6c)
Colorless oil (9%). 1H (CDCl3): 7.33 (bs, 5H), 7.10 (d, J = 8 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8, 2.4 Hz, 1H), 5.11 (s, 2H), 2.89 (d, J = 42 Hz, 1H), 2.70 (m, 1H), 2.63-2.41 (m, 4H), 2.26 (m, 1 H), 2.09-2.01 (m, 2H), 1.91-1.63 (m, 7H), 1.61-1.50 (m, 4H), 1.40-1.15 (m, 29H) ppm. 13C (CDCl3): 178.1, 177.0, 149.8, 142.2, 136.7, 128.7, 128.2, 128.1, 118.7, 118.3, 66.2, 61.7, 56.1, 45.9, 45.2, 44.0, 42.8, 42.6, 42.0, 41.0, 40.5, 38.0, 36.8, 35.2, 30.3, 30.2, 28.1, 27.0, 26.8, 25.4 ppm. MS (TOF ESI+): m/z = 642 ([M+H]+). HRMS Calcd for C42H60NO4: 642.4517. Found 642.4546. Anal. Calcd for C42H59NO4 × 1/3 EtOAc: C, 77.53; H, 9.26; N, 2.09. Found: C, 77.55; H, 9.48; N, 1.89.
General procedure for the deprotection of butorphan univalent benzyl esters (7a,b)
The respective benzyl ester (0.12 mmol; 6b,c) was dissolved in 3 mL of a 1/1 mixture of methanol and ethanol, and 10 wt% of Pd/C was added. The atmosphere over the mixture was evacuated and flushed with hydrogen twice, after that the mixture was vigorously stirred at rt under hydrogen overnight. On the next day further 10 wt% of Pd/C was added and the procedure repeated until no starting material could be seen by TLC control (hexane/ethyl acetate/triethylamine 10/10/1). The catalyst was filtered off, washed with methylene chloride and the solvents evaporated to yield the pure products.
2,9-Dimethyldecanedioic acid 10-((−)-N-cyclobutylmethylmorphinan-3-yl) ester (7a)
Colorless oil (30%); 1H (CDCl3): 7.16 (d, 1H, J = 8.5 Hz), 6.96 (s, 1H, J = 2.4 Hz), 6.95 (dd, 1H, J = 8.5, 2.4 Hz), 2.85–3.18 (m, 5H), 2.21–2.87 (m, 8H), 1.07–2.47 (m, 33H) ppm. 13C (CDCl3): 181.8, 175.6, 150.6, 140.1, 134.4, 129.1, 120.1, 118.8, 59.0, 56.5, 40.5, 39.8, 38.7, 36.8, 35.6, 34.2, 33.9, 31.4, 29.8, 29.6, 28.2, 28.0, 27.6, 27.4, 27.2, 26.1, 25.9, 21.8, 18.9, 17.7, 17.2 ppm. MS (TOF ESI+): m/z (% rel. Int.)= 524 ([M+H]+, 100). Anal. Calcd for C33H50NO4Cl (hydrochloride) × H2O: C, 68.55; H, 9.06; N, 2.42. Found: C, 68.62; H, 9.01; N, 2.62.
2,2,9,9-Tetramethyldecanedioic acid 10-((−)-N-cyclobutylmethylmorphinan-3-yl) ester (7b)
Colorless oil (23%). 1H (CDCl3): 7.15 (d, J = 8 Hz, 1H), 6.95 (d, J = 2 Hz, 1H), 6.89 (dd, 1H, J = 8, 2 Hz), 3.43 (bs, 1 H), 2.93–3.10 (m, 5H), 2.05–2.34 (m, 4H), 1.81 (m, 3H), 1.66 (m, 3H), 1.10–1.52 (m, 34H) ppm. 13C (CDCl3): 182.9, 176.6, 150.4, 128.8, 127.8, 119.7, 118.4, 58.8, 56.0, 42.6, 42.2, 42.1, 40.8, 40.7, 36.6, 35.4, 31.3, 30.1, 30.0, 27.9, 27.7, 26.0, 25.7, 25.3, 25.2, 25.0, 24.5, 21.6, 18.6 ppm. MS (TOF ESI+): m/z (% rel. Int.)= 552 ([M+H]+, 100). HRMS (TOF ESI+): Calcd for C35H54NO4: 552.4053. Found: 552.4044. Anal. Calcd for C35H54NO4HCl × H2O: C, 69.34; H, 9.31; N, 2.31. Found: C, 69.37; H, 9.20; N, 2.53.
General procedure for the preparation of homobivalent ligands of butorphan (4a,b,c)
To a solution of 0.3 mmol of the respective dicarboxylic acid (3a,b,c) in anhydrous methylene chloride (part of the diacid keeps suspended) were added two drops of dimethylformamide, followed by the addition of 1.2 mmol of oxalylchloride. Gas evolution was observed and the solution is stirred for 4 h at r.t. under nitrogen. Then solvent and excess oxalyl chloride were removed under reduced pressure and high vaccuum, respectively. The residual diacid dichloride was dissolved in a small amount of anhydrous methylene chloride and 0.9 mmol of butorphan were added and a clear solution was formed. Drop wise addition of 1.2 mmol of triethylamine were added and the solution was stirred at rt overnight. Additional methylene chloride was added and the mixture was washed with sat. NaHCO3 solution and brine. The org. phase was dried over Na2SO4 and solvent removed under reduced pressure. The residual oil was column chromatographed using hexane/ethyl acetate/triethylamine 10/10/1 as eluent system.
Bis((−)-N-cyclobutylmethylmorphinan-3-yl) 2,9-dimethyldecanedioate (4a)
Colorless oil (71%). 1H (CDCl3): 7.10 (d, 2H, J = 8.4 Hz), 6.91 (d, 2H, J = 2.4 Hz), 6.87 (dd, 2H, J = 8.4, 2.4 Hz), 3.01 (d, 2H, J = 19 Hz), 2.80 (m, 2H), 2.22–2.73 (m, 16H), 1.05–2.17 (m, 50H) ppm. 13C (CDCl3): 175.7, 149.5, 142.3, 135.5, 128.7, 118.7, 118.3, 61.8, 56.0, 45.9, 45.2, 42.0, 39.9, 38.0, 36.8, 35.2, 34.0, 29.7, 28.1, 28.1, 27.5, 27.0, 26.8, 24.6, 22.4, 19.1, 17.3 ppm. MS (TOF ESI+): m/z (% rel. Int.)= 816 ([M]+, 11), 409 ([M+2H]2+, 100). Anal. Calcd for C54H78N2O4Cl2 (dihydrochloride) × H2O: C, 71.42; H, 8.88; N, 3.02. Found: C, 71.37; H, 8.98; N, 3.00.
Bis((−)-N-cyclobutylmethylmorphinan-3-yl) 2,2,9,9-tetramethyldecanedioate (4b)
Colorless oil (70%). 1H (CDCl3): 7.10 (d, 2H, J = 8.4 Hz), 6.91 (d, 2H, J = 2.4 Hz), 6.87 (dd, 2H, J = 8.4, 2.4 Hz), 3.01 (d, 2H, J = 19 Hz), 2.90 (m, 2H), 2.22–2.73 (m, 16H), 1.05–2.17 (m, 54H) ppm. 13C (CDCl3): 177.0, 149.7, 142.2, 135.3, 128.7, 118.7, 118.3, 61.7, 56.0, 45.9, 45.2, 42.8, 42.0, 41.0, 37.0, 36.8, 35.2, 30.3, 28.1, 27.0, 26.8, 25.4, 25.4, 25.3, 24.6, 22.4, 19.1 ppm. MS (TOF ESI+): m/z (% rel. Int.)= 846 ([M+H]+, 11), 423.5 ([M+2H]2+, 100). HRMS (TOF ESI+): Calcd for C56H80N2O4: 845.6196. Found: 845.6228. Anal. Calcd for C56H82N2O4Cl2 (dihydrochloride) × 1.3 H2O: C, 71.43; H, 9.06; N, 2.98. Found: C, 71.38; H, 8.95; N, 3.00.
Bis((−)-N-cyclobutylmethylmorphinan-3-yl) 2,7-dimethyloctanedioate (4c)
Colorless oil (47%). 1H (CDCl3): 7.10 (d, 2H, J = 8.4 Hz), 6.91 (d, 2H, J = 2.4 Hz), 6.85 (dd, 2H, J = 8.4, 2.4 Hz), 2.98 (d, 2H, J = 19 Hz), 2.87 (m, 2H), 2.22–2.74 (m, 14H), 1.01–2.10 (m, 48H) ppm. 13C (CDCl3): 175.6, 149.5, 142.3, 135.4, 128.7, 118.7, 118.3, 61.7, 56.1, 45.9, 45.1, 41.9, 39.8, 38.0, 36.8, 35.1, 33.8, 28.1, 27.4, 27.0, 26.8, 24.6, 22.4, 19.1, 17.2 ppm. MS (TOF ESI+): m/z (% rel. Int.)= 789 ([M+H]+, 13), 395 ([M+2H]2+, 100). Anal. Calcd for C52H72N2O4 × 1/3 EtOAc: C, 78.21; H, 9.23; N, 3.54. Found: C, 78.34; H, 9.34; N, 3.45.
(5α)-17-Allyl-14-hydroxy-6-oxo-4,5-epoxymorphinan-3-yl-17-((−)-N-cyclobutylmethyl)morphinan-3-yl) 2,9-dimethyldecanedioate (8)
The compound was synthesized from equimolar amounts of 2,9-dimethyldecanedioic acid 10-((−)-N-cyclobutylmethylmorphinan-3-yl) ester (7a) and naloxone free base according to the general procedure for the synthesis of butorphan univalent diesters (6a,b,c) The compound was purified using hexane/EtOAc/NEt3 10/10/1 as eluent.
Colorless oil (11%). 1H (CDCl3): 7.08 (d, 2H, J = 8.4 Hz), 6.87 (d, 2H, J = 2.1 Hz), 6.82 (dd, 2H, J = 8.4, 2.1 Hz), 6.68 (d, 1H, J = 8.4 Hz), 5.76–5.89 (m, 1H), 5.18–5.25 (m, 2H), 4.67 (s, 1H), 2.98–3.18 (m, 6H), 1.03–2.82 (m, 51 H) ppm. 13C (CDCl3):206.5, 174.5, 173.5, 148.3, 146.8, 141.0, 134.2, 134.0, 131.7, 129.0, 127.7, 127.5, 121.8, 118.3, 117.5, 117.2, 117.1, 89.5, 69.1, 61.0, 60.5, 56.6, 54.8, 49.6, 44.7, 43.9, 42.2, 40.7, 38.6, 38.3, 36.7, 35.5, 35.0, 33.9, 32.8, 30.1, 29.6, 28.4, 28.3, 26.9, 26.2, 26.0, 25.8, 25.5, 23.4, 22.0, 21.1, 17.8, 16.0 ppm. MS (TOF ESI+): m/z (% rel. Int.)= 833 ([M+H]+, 27), 418 ([M+2H]2+, 100). Anal. Calcd for C52H68N2O7 × 1.5 H2O: C, 72.61; H, 8.32; N, 3.26. Found: C, 72.79; H, 8.21; N, 3.41.
HPLC analysis
HPLC analysis was performed on a Varian Prostar HPLC modular system operated by Star Chromatography Workstation software, Version 5. Chromatographic separations were performed on a Supelco Discovery C18 column (4.6 mm × 25 cm, 5 micron) operated at ambient temperature. The samples were injected using a Rheodyne 7725 manual sample injector equiped with a 20 μl injection loop. The mobile phase of 0.1% TFA in acetonitrile and 0.1% TFA in water was operated at a gradient of 60–100% acetonitrile over 10 min at 1.0 mL/min. Detection was at 265 nm.
Determination of Hydrolysis of Bivalent Ligands
The trifluoroacetate salts of the bivalent ligands were dissolved in acetonitrile and diluted in pH 7.4 20 mM phosphate buffer to give a final concentration of 100 μg/μL of ligand at a final acetonitrile concentration of 20%. The samples were incubated in duplicate in a 37 °C water bath and at appropriate times removed and injected directly for analysis. The half-life for each ligand was calculated assuming pseudo first-order kinetics from the rate constant measured from a plot of ln(%ester from t=0) versus time (minutes). The esterase mediated hydrolysis was conducted as above using approximately 0.15 units of porcine liver esterase (esterase reported as a specific activity of 24 units/mg where 1 unit will hydrolyze 1.0 μmole of ethyl butyrate to butyric acid and ethanol per min at pH 8.0 at 25 °C).
Opioid Binding to Human μ, δ, and κ Opioid Receptors
To determine the affinity and selectivity of the peptides for the μ, δ, and κ opioid receptors, Chinese hamster ovary (CHO) cells that stably expressed one type of human opioid receptor were used as previously described.19 Cell membranes were incubated at 25°C with the radiolabeled ligands in a final volume of 1 ml of 50 mM Tris-HCl, pH 7.5. Incubation times of 60 min were used for the μ-selective peptide [3H]DAMGO and the κ-selective ligand [3H]U69,593, and a 3-hr incubation was used with the d-selective antagonist [3H]naltrindole. The final concentrations of [3H]DAMGO, [3H]naltrindole, and [3H]U69,593 were 0.25 nM, 0.2 nM, and 1 nM, respectively. Nonspecific binding was measured by inclusion of 10 μM naloxone for μ and κ binding and 100 μM for δ binding. The binding was terminated by filtering the samples through Schleicher & Schuell No. 32 glass fiber filters using a Brandel 48-well cell harvester. The filters were washed three times with 3 ml of cold 50 mM Tris-HCl, pH 7.5, and were counted in 2 ml of ScintiSafe 30% scintillation fluid (Fisher Scientific, Fair Lawn, NJ). For [3H]U69,593 binding, the filters were soaked in 0.1% polyethylenimine for at least 30 min before use. IC50 values were calculated by least squares fit to a logarithm-probit analysis. Ki values of unlabeled compounds were calculated from the equation Ki = (IC50)/1 + S where S = (concentration of radioligand) (Kd of radioligand).20
Figure 1.
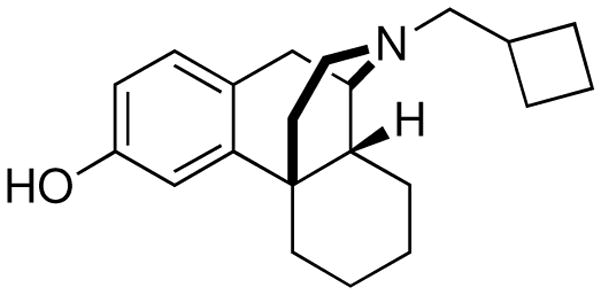
Structure of the opioid ligand butorphan (MCL-101)
Acknowledgments
A fellowship for M. D. from the Deutsche Akademie der Naturforscher Leopoldina (BMBF-LPD 9901/8-147) is gratefully acknowledged. This work was supported in part by NIH grants R01-DA14251 (J. L. N.), K05-DA 00360 (J. M. B.) and T32 DA007252 (B.S.F.). Levorphanol tartrate was generously donated by Mallinckrodt Inc.
Footnotes
Contribution to celebrate the 100th anniversary of the Division of Medicinal Chemistry of the American Chemical Society
Abbreviations DCC, dicyclohexyl carbodiimide; DMAP, 4-dimethylaminopyridine; DMF, dimethylformamide; HPLC, high performance liquid chromatography; TFA, trifluoroacetic anhydride; THF, tetrahydrofuran
References
- 1.Neumeyer JL, Zhang A, Xiong W, Gu X, Hilbert JE, Knapp BI, Negus SS, Mello NK, Bidlack JM. Design and Synthesis of Novel Dimeric Morphinan Ligands for κ and μ Opioid Receptors. J Med Chem. 2003;46:5162–5170. doi: 10.1021/jm030139v. [DOI] [PubMed] [Google Scholar]
- 2.Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Synthesis and Preliminary In vitro Investigation of Bivalent Ligands Containing Homo- and Heterodimeric Pharmacophores at μ, δ, and κ Opioid Receptors. J Med Chem. 2006;49:256–262. doi: 10.1021/jm050577x. [DOI] [PubMed] [Google Scholar]
- 3.Peng X, Neumeyer JL. Kappa Receptor Bivalent Ligands. Curr Top Med Chem. 2007;7:363–373. doi: 10.2174/156802607779941251. [DOI] [PubMed] [Google Scholar]
- 4.Newman MS. Some Observations Concerning Steric Factors. J Am Chem Soc. 1950;72:4783–4786. [Google Scholar]
- 5.Shawali ASAS, Biechler SS. Aminolysis of Esters. I. Kinetics and Mechanism in Anhydrous Dioxane. J Am Chem Soc. 1967;89:3020–3026. doi: 10.1021/ja00988a039. [DOI] [PubMed] [Google Scholar]
- 6.Bender DM, Peterson JA, McCarthy JR, Gunaydin H, Takano Y, Houk KN. Cyclopropanecarboxylic Acid Esters as Potential Prodrugs with Enhanced Hydrolytic Stability. Org Lett. 2008;10:509–511. doi: 10.1021/ol702892e. [DOI] [PubMed] [Google Scholar]
- 7.Stoops JK, Horgan DJ, Runnegar MTC, de Jersey J, Webb EC, Zerner B. Carboxylesterase (EC 3.1.1). Kinetic Studies on Carboxylesterases. Biochemistry. 1969;8:2026–2033. doi: 10.1021/bi00833a037. [DOI] [PubMed] [Google Scholar]
- 8.Fredholt K, Mørk N, Begtrup M. Hemiesters of Aliphatic Dicarboxylic Acids as Cyclization-activated Prodrug Forms for Protecting Phenols against First-pass Metabolism. Int J Pharm. 1995;123:209–216. [Google Scholar]
- 9.Neumeyer JL, Bidlack JM, Zong R, Bakthavachalam V, Gao P, Cohen DJ, Negus SS, Mello NK. Synthesis and Opioid Receptor Affinity of Morphinan and Benzomorphan Derivatives: Mixed κ Agonist and μ Agonists and Antagonists as Potential Pharmacotherapeutics for Cocaine Dependance. J Med Chem. 2000;43:114–122. doi: 10.1021/jm9903343. [DOI] [PubMed] [Google Scholar]
- 10.Neumeyer JL, Gu XH, van Vliet LA, DeNunzio NJ, Rusovici DE, Cohen DJ, Negus SS, Mello NK, Bidlack JM. Mixed κ Agonists and μ Agonists/antagonists as Potential Pharmacotherapeutics for Cocaine Abuse: Synthesis and Opioid Binding Affinity of N-substituted Derivatives of Morphinan. Bioorg Med Chem Lett. 2001;11:2735–2740. doi: 10.1016/s0960-894x(01)00543-1. [DOI] [PubMed] [Google Scholar]
- 11.Negus SS, Mello NK, Portoghese PS, Lin CE. Effects of Kappa Opioids on Cocaine Self-administration by Rhesus Monkeys. J Pharmacol Exp Ther. 1997;282:44–55. [PubMed] [Google Scholar]
- 12.Mello NK, Negus SS. Effects of Kappa Opioid Agonists on Cocaine- and Food-maintained Responding by Rhesus Monkeys. J Pharmacol Exp Ther. 1998;286:812–824. [PubMed] [Google Scholar]
- 13.Alper H, Woell JB, Despeyroux B, Smith DJH. The Regiospecific Palladium Catalyzed Hydrocarboxylation of Alkenes under Mild Conditions. J Chem Soc, Chem Commun. 1983:1270–1271. [Google Scholar]
- 14.Newkome GR, Baker B, Caruso A, Greenwald MM, Hanson PG, Mangogna GA, Mathes PD, Pascal A, Rigby HO, Riser JM, Schnabel JJ, Sonnier JA, Steinkampf MP, Johnson JL. An Improved Synthesis of α,α,α′,α′-Tetramethyldicarboxylic Acids. Synthesis. 1975;8:517–518. [Google Scholar]
- 15.Babler JH, Moy RK. A Facile Method for Monoesterification of α,ω-Dicarboxylic Acids: Application to the Synthesis of Traumatic Acid, a Prostaglandin Synthon. Synth Commun. 1979;9:669–675. [Google Scholar]
- 16.Wu CY, Brik A, Wang SK, Chen YH, Wong CH. Tetrabutylammonium Fluoride-mediated Rapid Alkylation Reaction in Microtiter Plates for the Discovery of Enzyme Inhibitors in Situ. ChemBioChem. 2005;6:2176–2180. doi: 10.1002/cbic.200500295. [DOI] [PubMed] [Google Scholar]
- 17.(a) Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Pharmacological Properties of Bivalent Ligands Containing Butorphan linked to Nalbuphine, Naltrexone and Naloxone at μ, δ, and κ Opioid Receptors. J Med Chem. 2007;50:2254–2258. doi: 10.1021/jm061327z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mathews JL, Fulton BS, Negus SS, Neumeyer JL, Bidlack JM. In Vivo Characterization of (−)(−)MCL-144 and (+)(+)MCL-193: Isomeric, Bivalent Ligands with kappa/mu Agonist Properties. Neurochem Res. 2008;33:2142–2150. doi: 10.1007/s11064-008-9752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almeida MI, Amaral AT, Amaral L. Effects of α Substitution on the Carbonyl Stretching Frequencies of Phenyl Carboxylates. J Org Chem. 1982;47:1567–1571. [Google Scholar]
- 19.Parkhill AL, Bidlack JM. Several δ-Opioid Receptor Ligands Display No Subtype Selectivity to the Human δ-Opioid Receptor. Eur J Pharmacol. 2002;451:257–264. doi: 10.1016/s0014-2999(02)02241-0. [DOI] [PubMed] [Google Scholar]
- 20.Cheng YC, Prusoff WH. Relationship Between the Inhibition Constant (Ki) and the Concentration of Inhibitor Which Causes 50 Percent Inhibition (1C50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]