

Abstract
We previously reported that [[N-[3β-hydroxyl-lup-20(29)-en-28-oyl]-7-aminoheptyl]-carbamoyl]methane (A43D, 4) was a potent HIV-1 entry inhibitor. However, 4 was inactive against HIV-2 virus, suggesting the structural requirements for targeting these two retroviruses are different. In this study, a series of new betulinic acid derivatives were synthesized, and some of them displayed selective anti-HIV-2 activity at nanomolar concentrations. In comparison to compounds with anti-HIV-1 activity, a shorter C-28 side chain is required for optimal anti-HIV-2 activity.
Keywords: Betulinic acid, HIV-2, HIV-1
Introduction
HIV-2 is a retrovirus related to HIV-1, the causative agent of AIDS. HIV-2 infections can also cause AIDS-like diseases, but these usually progress more slowly than those caused by HIV-1.1 While HIV-1 has spread to almost all of the countries in the world, HIV-2 is mostly concentrated in countries of West Africa.1, 2 As the vaccine development against HIV-1 encounters tremendous challenges, progress in antiretroviral therapy (ART) has been encouraging.3 However, almost all drugs have been developed with a focus on their effectiveness against HIV-1. Drug regimes for HIV-1 therapy have been used for HIV-2 infections, but some of the drugs that were potent against HIV-1 replication appeared to be less active or inactive against HIV-2 virus. For example, HIV-2 is mostly insensitive to non-nucleoside reverse transcriptase inhibitors and HIV-1 entry inhibitor, Fuzeon.4, 5 Some HIV-1 protease inhibitors have also been shown to have reduced potency against HIV-2.6 Giving the fact that the genetic homology between HIV-1 and HIV-2 is less than 50%,7 it is no surprise that drugs currently used for HIV-2 are not as optimal as they are for HIV-1.
Our previous work has been focused on the synthesis of betulinic acid derivatives as potential anti-HIV-1 agents.8–10 Betulinic acid (BA, 1) is a pentacyclic triterpene found in abundance in many plant species.11 Unmodified 1 was inactive or exhibited very weak activity against HIV-1 replication. Chemical modification of 1 at C-28 and/or C-3 positions resulted in potent anti-HIV-1 compounds. We and others have synthesized C-28 modified BA or other pentacyclic triterpene derivatives that inhibited HIV-1 entry.10, 12–15 Compound 17 (IC9564)12 and compound 4 (A43D)10 are among the most potent BA derivatives that inhibit HIV-1 entry by targeting HIV-1 gp120.16, 17 Chemical modifications of 1 at C-3 resulted in compounds that inhibited HIV-1 maturation as opposed to entry.13, 18, 19 The HIV-1 maturation inhibitor, 3-O-(3’,3’-dimethylsuccinyl)-betulinic acid (bevirimat),20 is a C-3 BA derivative currently under clinical trial for anti-HIV-1 therapy. Chemical modifications at both C-3 and C-28 resulted in bi-functional BA derivatives that inhibited both HIV-1 entry and maturation.9, 10 [[N-[3β-O-3’,3’-Dimethylsuccinyl-lup-20(29)-en-28-oyl]-7-aminoheptyl]-carbamoyl]methane (A12-2) was one of the most potent BA derivatives that inhibited HIV-1 replication at both maturation and entry steps.10 Due to the relatively low genetic homology between HIV-1 and HIV-2, we hypothesized that the optimal pharmacophore of BA derivatives required for inhibiting HIV-2 is different from that for anti-HIV-1 activity. Since C-3 derivatives, such as 22 (bevirimat) did not inhibit HIV-2 replication in our assays (Table 1), chemical synthesis of this study is focused on the modification of the C-28 position of 1. The results of this study support the hypothesis in that a shorter C-28 side chain of BA, an equivalent of CONH-(CH2)nR with n being 4 or 5, is needed for optimal anti-HIV-2 activity.
Table 1.
Inhibitory activity of BA and its derivatives against HIV-1 and HIV-2.
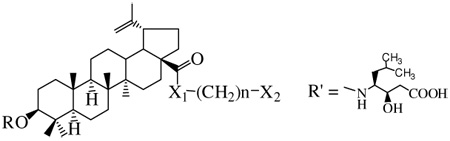 | |||||||
|---|---|---|---|---|---|---|---|
| Cpds. | X1 | n | X2 | R | IC50 (µM)a HIV-1 NL4-3 |
IC50 (µM)a HIV-2 KR.X3 |
TC50 (µM)a Cytotoxicity |
| 1 (BA) | OH | H | Inactive | Inactive | >80 | ||
| 2 | NH | 5 | -NHCO-CH3 | H | Inactive | Inactive | 13.9±3.3 |
| 3 | NH | 6 | -NHCO-CH3 | H | 0.027±0.013 | Inactive | 48.1±12.6 |
| 4 (A43D) | NH | 7 | -NHCO-CH3 | H | 0.038±0.011 | Inactive | >66 |
| 5 | NH | 8 | -NHCO-CH3 | H | 0.14±0.02 | Inactive | >64 |
| 6 | NH | 6 | -NHCO-CH2COOH | H | 0.44±0.02 | Inactive | 34.6±13.8 |
| 7 | NH | 3 | -COOH | H | Inactive | 2.8±1.8 | 31.2±1.3 |
| 8 | NH | 4 | -COOH | H | 32.8±6.5 | 0.17±0.14 | >72 |
| 9 | NH | 5 | -COOH | H | 2.0±0.6 | 0.15±0.04 | >70 |
| 10 | NH | 6 | -COOH | H | 11.1±9.8 | 4.5±4.3 | 36.2±1.2 |
| 11 | NH | 7 | -COOH | H | 0.84±0.52 | Inactive | 34.2±1.8 |
| 12 | NH | 3 | -CO-Gly | H | Inactive | 3.7±1.5 | >67 |
| 13 | NH | 4 | -CO-Gly | H | Inactive | 2.6±2.8 | >65 |
| 14 | NH | 5 | -CO-Gly | H | 34.7±8.5 | 0.35±0.25 | >64 |
| 15 | NH | 6 | -CO-Gly | H | 4.1±2.1 | 0.42±0.33 | >63 |
| 16 | NH | 7 | -CO-Gly | H | 0.12±0.04 | 0.64±0.02 | 15.8±4.1 |
|
17 (IC9564) |
NH | 7 | -CO-R’ | H | 0.042±0.013 | 2.9±1.3 | 33.2±3.3 |
| 18 | NH | 5 | -CO-Ala | H | 48.9±29.4 | 0.37±0.40 | >63 |
| 19 | NH | 5 | -CO-Tyr | H | Inactive | 0.23±0.10 | 54.6±28.3 |
| 20 | NH | 5 | -CO-Pro | H | 51.4±29.7 | 0.63±0.14 | >60 |
| 21 | NH | 5 | -CO-Gly-Gly | H | >58 | 1.7±0.9 | >58 |
|
22 (Bevirimat) |
OH | R = HOOC-C(CH3)2CH2CO- | 0.13±0.01 | Inactive | >30 | ||
| AZT | - | 0.069±0.01 | >78 | ||||
Data represent an average of 3 independent experiments; “Inactive” denotes the compounds with TC50/IC50 < 2.
Results and Discussion
Chemistry
Compounds 4, 17, 7–11, and 1 are known compounds that have been synthesized and reported for their activities against HIV-1 virus.10, 12 The C-28 modified BA derivatives tested in this study were synthesized using methods previously described (Scheme 1). Based on their C-28 side chain structures, four types of compounds were synthesized. The first type of compounds included the analogs 2–5. This type was synthesized by coupling di-amino alkane [NH2-(CH2)n-NH2], where n was 5 to 8, with the C-28 carboxylic acid of 3-O-acetate-BA. The resultant C-28 terminal amine intermediate was then acylated with acetic anhydride following a step of alkaline hydrolysis of the C-3 ester to form analogs 2–5. The amine intermediate 3a was coupled with malonic acid in the presence of EDC and Et3N to form the second type of compound (6) after the saponification mediated by sodium hydroxide. The third type of compounds was synthesized by coupling the ω-aminoalkanoates of varying lengths to the C-28 carboxylic acid of 3-O-acetate BA. Compounds 7–11 were obtained after alkaline hydrolysis of esters at C-3 and C-28 side chains. Further coupling of the C-28 terminal carboxylic acid of 7–11 with mono- or di-amino acids furnished the fourth type of compounds, which included 12–16 and 18–21.
Scheme 1.

Sythesis of type 1–4 BA derivatives.
Results
These new BA derivatives, along with 1 and some known BA derivatives, were tested for their anti-HIV-1 and HIV-2 activity (Table 1). The unmodified 1 did not exhibit anti-HIV-1 or anti-HIV-2 activity. For anti-HIV-1 activity, compounds 4, 17, and the newly synthesized 3 were three of the most potent inhibitors against HIV-1 NL4-3 replication. Compound 3 was the most potent compound with its IC50 at 27 nM. Compounds with side chains less than six methylene groups exhibited a significantly weaker anti-HIV-1 activity as demonstrated by 2. Compound 5, with eight methylenes in its C-28 side chain, was less potent than 4 against HIV-1 replication. In contrast to type 1 analogs, which have acetamide termini at their C-28 side chains, 17 and type 2–4 compounds possessed carboxylic acid at the terminus of their C-28 side chain. Among them, derivatives with C-28 methylene groups equal to 7 exhibited the most potent anti-HIV-1 activity when compared with analogs with shorter linker in their respective compound types (Table 1).
Although 4 and 17 were two of the most potent anti-HIV-1 BA derivatives, 17 exhibited weak anti-HIV-2 activity (about 70 times less potent than its anti-HIV-1 activity), and 4 was inactive against HIV-2 KR.X3 infection. The striking difference in their potencies against HIV-1 and HIV-2 suggested that the optimal pharmacophore against HIV-2 is different from those that required for anti-HIV-1 activity. To obtain potent anti-HIV-2 compounds, BA derivatives with a variety of C-28 side chains were synthesized and tested for their anti-HIV-2 activity. Similar to 4, the newly synthesized analogs 2, 3 and 5 with acetamide termini were inactive against HIV-2 replication. This result suggested that a free terminal carboxylic acid at C-28 side chain of BA might be needed for HIV-2 inhibition as shown by 17. Therefore, type 1 compounds were further modified by substituting the C-28 acetamide terminus with malonic acid. Except for compound 6, the resultant type 2 compounds were mostly unstable due to decomposition of the terminal malonamide R-NHCO-CH2-COOH into acetamide R-NHCO-CH3. However, 6 was less potent than 3 against HIV-1 and was inactive against HIV-2 (Table 1). Thus, a free terminal carboxylic acid at C-28 alone was not sufficient for anti-HIV-2 activity.
To systematically investigate the optimal length of the C-28 side chain required for anti-HIV-2 activity, a series of BA derivatives with C-28 side chains in the form of -NH-(CH2)n-COOH, where n ranged from 3 to 7, were synthesized. As shown in Table 1, type 3 compounds 7–11 were either inactive or displayed weak anti-HIV-1 activity with IC50s at low micromolar concentrations. Most of these compounds were more potent against HIV-2 than HIV-1 except 11, which was inactive against HIV-2 replication. Within this type, 8 and 9 exhibited the most potent anti-HIV-2 activity with IC50s at 0.17 µM and 0.15 µM, respectively. These results suggested that the optimal anti-HIV-2 activity was achieved when n = 4 or 5 for the length of the C-28 side chain. In contrast, the optimal anti-HIV-1 activity was achieved when n = 7 or 8 for the length of the C-28 side chain.10,12
In an effort to explore the effect of additional terminal moieties at the C-28 side chain on anti-HIV-2 activity, type 4 compounds 12–16 were synthesized by coupling a glycine to the carboxylic acid of 7–11. The addition of a glycine to C-28 side chain of 7 did not significantly change the anti-HIV-2 activity as shown by 12. However, the same glycine addition resulted in compounds 13 and 14 with reduced anti-HIV-2 activity (approximately 15 and 2-fold reduction) when compared to their precursors 8 and 9, respectively. On the other hand, a 10-fold or more increases in anti-HIV-2 activities were observed for compounds 15 and 16 when compared to their precursors without the glycine addition. Overall, 14 and 15 were the two most potent anti-HIV-2 compounds with a glycine terminus. However, 14 and 15 were 2- to 3-fold less potent compared to the type 3 compounds 8 and 9. Among the above potent anti-HIV-2 derivatives, 8 and 14 preferentially inhibited HIV-2 with IC50s approximately 2 log10 lower than that against HIV-1 (Table 1).
Since both hexanoic moiety-containing compounds 9 and 14 exhibited the most potent anti-HIV-2 activity from type 3 and the type 4 compounds, the hexanoic acid was again modified in an attempt to further diversify the C-28 terminus. As a result, additional type 4 compounds 18–21 were synthesized with their C-28 side chains in the form of -NH-(CH2)5-CO-aa, where aa was a mono- or di-amino acid. Among the single amino acid coupled hexanoic derivatives, 19, with a tyrosine terminus, displayed the most potent anti-HIV-2 activity with IC50 at 0.23 µM. Although all the mono-amino acid derivatives remained quite active against HIV-2 (IC50 ranging from 0.23 – 0.63 µM), these compounds were less potent than 9 (IC50 = 0.15 µM). For the BA derivatives with hexanoic moiety, their anti-HIV-2 activity based on their terminus, was in the order of none > tyrosine > glycine = alanine > proline. The dipeptide derivative 21 was approximately 10-fold less potent than 9.
We have previously shown that the BA derivatives 4 and 17 inhibited HIV-1 entry by targeting the V3 loop of HIV-1 gp120.17 To determine whether the V3 loop of gp120 is responsible for HIV-2 inhibition, the sensitivities of HIV-2 KR.X3 and HIV-2 KR.X3-YU-2/V3 were determined in the presence of compound 14. HIV-2 KR.X3-YU-2/V3 is a chimeric virus containing the V3 loop of HIV-1 YU-2 instead of the original V3 of HIV-2 KR.X3. Compound 14 was used for this study, because it was the most potent anti-HIV-2 BA derivative available before compound 9 was synthesized. Compound 14 inhibited HIV-2 KR.X3 in a dose-dependent manner with an IC50 of 0.35 µM (Figure 1). The HIV-2 chimeric virus with YU-2 V3 loop, HIV-2 KR.X3-YU-2/V3, was approximately 5-fold less sensitive to 14 when compared to HIV-2 KR.X3. Since the only difference between the two viruses was the V3 loop, the results suggested that the V3 loop of HIV-2 was a possible target of 14.
Figure 1. Differential sensitivity of HIV-2 KR.X3 and HIV-2 KR.X3-YU-2/V3 to compound 14.

A dose-dependent inhibition of the two viruses was determined in the presence of various concentrations of 14 as indicated. Each data point represents the mean +/− SD of 4 independent experiments.
Conclusion
In summary, several important SARs for anti-HIV-2 activity of BA derivatives were observed in this study. First, for anti-HIV-2 activity, the optimal length of C-28 side chain is achieved when n = 5 in the form of -NH-(CH2)n-COR, as seen for compounds 9 and 14 (Table 1). A gradual drop of anti-HIV-2 activity was observed when n is greater or smaller than 5. Second, in contrast to the anti-HIV-1 BA derivatives, a carboxylic acid terminus of C-28 side chain was important for anti-HIV-2 activity. The potent anti-HIV- 1 derivative 3, along with analogs with various lengths of C-28 side chains that did not possess a carboxylic acid terminus, was completely inactive against HIV-2. Third, the addition of one amino acid to the carboxylic terminus of C-28 side chain, moderately compromised the anti-HIV-2 activity of the BA derivatives, especially when the C-28 side chain was at the optimal length with n = 5. On the other hand, the addition of one amino acid increased anti-HIV-2 activity when n was 6 or 7. These results provided clues for further characterization of ligand-target interaction and synthesis of next generation of anti-HIV-2 BA derivatives. In addition to the novel anti-HIV-2 BA derivatives, this study also synthesized a potent HIV-1 entry inhibitor, compound 3, with improved anti-HIV-1 activity (IC50 = 0.027 µM) when compared to the previously synthesized most potent BA derivative 4 with an IC50 of 0.038 µM.
When comparing anti-HIV-1 and anti-HIV-2 activities, most of the very potent HIV-1 inhibitors, such as 4 and its analogs, appeared to have poor or no activity against HIV-2, and vice versa for most active anti-HIV-2 compounds, such as 8, 14, 18–20. These observations support our hypothesis that the optimal pharmacophores required to target HIV-1 and HIV-2 are different. In terms of mechanism of action, compound 14 appeared to block HIV-2 replication by targeting the V3 loop of gp120. The homology between the V3 loops of HIV-1 (YU-2) and HIV-2 (KR) is approximately 30%.21 It is likely that the drug binding pocket in HIV-1 V3 loop is structurally and conformationally different from that in HIV-2 V3 loop.
Experimental Section
General experimental procedures
Positive or negative HR-FABMS were recorded on a Shimadzu LCMS-IT-TOF or a Joel SX-102 mass spectrometer. 1H and other NMR spectra were measured on a Varian Mercury 300 or 500 spectrometer. Samples were dissolved in CDCl3 or pyridine-d5 with TMS as an internal standard. Silica gel chromatography was carried out on a Biotage Horizon Flash chromatograph system with pre-packed Si gel column. The purity of BA derivatives was analyzed by using a Varian ProStar HPLC system with a PDA detector and Agilent Zorbax ODS or C-8 columns. The mobile phase was composed of solution A (5% acetonitrile in water with 0.045% trifluoroacetic acid) and solution B (water : methanol : acetonitrile = 5 : 10 : 85 with 0.045% trifluoroacetic acid). A linear gradient of 80% to 100% of solution B with a flow rate at 1 ml/min, or with flow rate at 4 ml/min was used to elute the compounds. The compounds were analyzed with the UV absorption displayed at 220 nm and recorded at a range from 200 to 250 nm. All the tested compounds have purity of 95% or above except for the inactive unmodified 1 (BA), which was purchased from Sigma-Aldrich with 90% purity.
Procedure for synthesizing type 1 compounds 2–5: A stirring solution of 3-O-Ac-BA (0.3 – 0.7 mmol) in dichloromethane (DCM, 2 ml) was added with oxalyl chloride (7 – 12 eq.). After 10 min of stirring, the organic solvent was removed under vacuum. The residue was dissolved in DCM and added with corresponding 1,ω-di-amino alkane (4 – 5 eq.) in DCM. The reaction was stirred overnight and then concentrated. The residue was washed with water and dissolved in ethanol. After filtration, the ethanolic solution was concentrated and the residue was chromatographed on Si-gel to yield the corresponding amine intermediate 2a–5a. To the corresponding amine intermediate 2a–5a in pyridine (anhydrous, 1 ml), acetic anhydride (0.5 ml) was added and stirred at room temperature overnight. The reaction mixture was concentrated and re-dissolved in DCM. After being washed with 1N HCl, water, and brine, the organic layer was concentrated and chromatographed on Si-gel to yield 2b–5b.
Procedure for saponification of ester at C-3 or/and C-28: Esters were hydrolyzed in a mixture of MeOH/THF/4N NaOH aq. (2:2:1). The intermediate 2b–5b was dissolved in organic solution (1 – 2 ml) and was added with aqueous NaOH solution (0.5 – 1 ml). After stirring overnight, the reaction mixture was neutralized with aqueous HCl (1 N). The resulted precipitate was washed with water and dried in vacuum to yield the final compound 2–5.
Procedure for synthesizing type 2 compound 6: The mixture of the above amine intermediate 3a (0.2 mmol) in DCM (2 ml) was added with malonic acid (10 eq.), EDC (2 eq.), and Et3N (10 eq.). After stirring at room temperature overnight, the reaction mixture was concentrated and re-dissolved in DCM, washed with water, brine, and was dried over Na2SO4. The organic layer was concentrated and chromatographed on Si-gel to yield the intermediate 6a. After the saponification procedure, as described above, 6 was obtained as a solid.
Procedure for synthesizing type 3 compounds 7–11: A stirring solution of 3-O-Ac-BA (0.1 – 0.6 mmol) in DCM (4 ml) was added with oxalyl chloride (10 eq.). After 10 min of stirring, the organic solvent was removed under vacuum. The residue was dissolved in DCM, then reacted with corresponding ω-amino alkanoate (1.2 – 1.5 eq.) in DCM and Et3N (6 eq.) overnight. The reaction mixture was diluted with DCM before washed with water, brine, and then dried over Na2SO4. After concentration in vacuum, the residue was chromatographed on Si-gel to yield the corresponding 7a–11a, which yielded the corresponding 7–11 after saponification.
Procedure for synthesizing type 4 compounds 12–16: To the solution of corresponding intermediates 7a–11a (0.2 mmol) in DCM (2 ml), was added glycine methyl ester hydrochloride (2.7 eq.), Et3N (7.5 eq), and EDC (3.0 eq.). After stirring overnight at room temperature, the reaction mixture was diluted with DCM, washed with water, brine, and was dried over Na2SO4. The organic layer was concentrated under vacuum and the residue was chromatographed on Si-gel. The pure intermediate was collected and subjected to saponification as described above to furnish the corresponding compounds 12–16.
Procedure for synthesis of type 4 compounds 18–21: The synthesis of these compounds was achieved by the same procedure as described above for 12–16, using intermediate 9 (for synthesis of 18–20) or 14 (for synthesis of 21) and the corresponding amino acid methyl ester hydrochloride.
HIV-1 and HIV-2 virus infection assay
Inhibition of HIV-1NL4-3, HIV-2 KR.X3 or HIV-2 KR.X3-YU2/V3 infection was measured through reduction in luciferase gene expression after a single round of virus infection of TZM-bl cells, as previously described (17). HIV-2 KR.X3 and HIV-2 KR.X3-YU2/V3 were generously provided by Dr. George Shaw, University of Alabama. For the anti-viral assays, 200 TCID50 of virus was used to infect TZM-bl cells in the presence of various concentrations of compounds. Two days after infection, the culture medium was removed from each well and 100 µl of Bright Glo reagent (Promega, Luis Obispo, CA) was added to the cells for measurement of luminescence using a Victor 3 luminometer. The 50% inhibitory concentration (IC50) was defined as the concentration that caused a 50% reduction of luciferase activity (Relative Light Units) compared to virus control wells.
Cytotoxicity assay
A CytoTox-Glo™ cytotoxicity assay (Promega) was used to determine the cytotoxicity of the synthesized BA derivatives. TZM-bl cells were cultured in the presence of various concentrations of the compounds for 2 days. Percent of viable cells was determined by following the protocol provided by the manufacturer. The 50% cytotoxic concentration (TC50) was defined as the concentration that caused a 50% reduction of cell viability.
Supplementary Material
Acknowledgment
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) Grant AI-65310 awarded to C. H. Chen, National Institutes of Drug Abuse (NIDA) Grant DA-024589 awarded to L. Huang, and in part by Grant AI-077417 from NIAID awarded to K. H. Lee. The authors are grateful to Dr. George Dubay of the Department of Chemistry and Dr. Anthony Ribeiro of the NMR Spectroscopy Center of Duke University for their assistance on mass and NMR spectroscopy data collection. We also thank Dominique Soroka for her help on preparation of this manuscript.
Abbreviations
- BA
betulinic acid
- ART
antiretroviral therapy
- EDC
N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride
- SARs
structure-activity-relationships
- THF
tetrahydrofuran
- DCM
dichloromethane
- Cpds
compounds
- AZT
3’-azido-3’-deoxythymidine.
Footnotes
The authors congratulate the 100th anniversary of the Division of Medicinal Chemistry.
Supporting Information Available: Spectroscopic and HPLC analytical data were included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jaffar S, Grant AD, Whitworth J, Smith PG, Whittle H. The natural history of HIV-1 and HIV-2 infections in adults in Africa: a literature review. Bull. World Health Organ. 2004;82:462–469. [PMC free article] [PubMed] [Google Scholar]
- 2.da Silva ZJ, Oliveira I, Andersen A, Dias F, Rodrigues A, Holmgren B, Andersson S, Aaby P. Changes in prevalence and incidence of HIV-1, HIV-2 and dual infections in urban areas of Bissau, Guinea-Bissau: is HIV-2 disappearing? AIDS. 2008;19:1195–1202. doi: 10.1097/QAD.0b013e328300a33d. [DOI] [PubMed] [Google Scholar]
- 3.Wainberg MA, Jeang KT. 25 years of HIV-1 research – progress and perspectives. BMC Med. 2008;6:31. doi: 10.1186/1741-7015-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witvrouw M, Pannecouque C, Van Laethem K, Desmyter J, De Clercq E, Vandamme AM. Activity of non-nucleoside reverse transcriptase inhibitors against HIV-2 and SIV. AIDS. 1999;13:1477–1483. doi: 10.1097/00002030-199908200-00006. [DOI] [PubMed] [Google Scholar]
- 5.Isaka Y, Miki S, Kawauchi S, Suyama A, Sugimoto H, Adachi A, Miura T, Hayami M, Yoshie O, Fujiwara T, Sato A. A single amino acid change at Leu-188 in the reverse transcriptase of HIV-2 and SIV renders them sensitive to non-nucleoside reverse transcriptase inhibitors. Arch. Virol. 2001;146:743–755. doi: 10.1007/s007050170143. [DOI] [PubMed] [Google Scholar]
- 6.Witvrouw M, Pannecouque C, Switzer WM, Folks TM, De Clercq E, Heneine W. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir. Ther. 2004;9:57–65. [PubMed] [Google Scholar]
- 7.Guyader M, Emerman M, Sonigo P, Clavel F, Montagnier L, Alizon M. Genome organization and transactivation of the human immunodeficiency virus type 2. Nature. 1987;326:662–669. doi: 10.1038/326662a0. [DOI] [PubMed] [Google Scholar]
- 8.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Lee KH. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agent. J. Med. Chem. 1996;39:1016–1017. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, Yuan X, Aiken C, Chen CH. Bifunctional anti-HIV-1 small molecules with two novel mechanisms of action. Antimicrob. Agents Chemother. 2004;48:663–665. doi: 10.1128/AAC.48.2.663-665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang L, Ho P, Lee KH, Chen CH. Synthesis and anti-HIV activity of bi-functional betulinic acid derivatives. Bioorg. Med. Chem. 2006;14:2279–2289. doi: 10.1016/j.bmc.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Huang L, Chen CH. The Molecular targets of anti-HIV-1 triterpenes. Curr. Drug Targets Infect. Disord. 2002;2:33–36. doi: 10.2174/1568005024605936. [DOI] [PubMed] [Google Scholar]
- 12.Soler F, Poujad C, Evers M, Carry JC, Héin Y, Bousseau A, Huet T, Pauwels R, De Clercq E, Mayaux JF, Le Pecq JB, Dereu N. Betulinic acid derivatives: a new class of specific inhibitors of human immunodeficiency virus type 1 entry. J. Med. Chem. 1996;39:1069–1083. doi: 10.1021/jm950669u. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto F, Kashiwada Y, Cosentino LM, Chen CH, Garrett PE, Lee KH. Anti-AIDS agent–XXVII. Synthesis and anti-HIV activity of betulinic acid and dihydrobetulinic acid derivatives. Bioorg. Med. Chem. 1997;5:2133–2143. doi: 10.1016/s0968-0896(97)00158-2. [DOI] [PubMed] [Google Scholar]
- 14.Yu D, Sakurai Y, Chen CH, Chang FR, Huang L, Kashiwada Y, Lee KH. Anti-AIDS agents 69. Moronic acid and other triterpene derivatives as novel potent anti-HIV agents. J. Med. Chem. 2006;49:5462–5469. doi: 10.1021/jm0601912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang L, Yu D, Ho P, Lee KH, Chen C-H. Synthesis and anti-HIV activity of bi-functional triterpene derivatives. Lett. Drug Des. Discov. 2007;4:471–478. [Google Scholar]
- 16.Huang L, Lai WH, Ho P, Chen CH. Induction of a nonproductive conformational change in gp120 by a small molecule HIV-1 entry inhibitor. AIDS Res and Human Retroviruses. 2007;23:28–32. doi: 10.1089/aid.2006.0137. [DOI] [PubMed] [Google Scholar]
- 17.Lai W, Huang L, Ho P, Li ZJ, Montefiori D, Chen CH. Betulinic acid derivatives that target gp120 and inhibit multiple genetic subtypes of HIV-1. Antimicrob. Agents Chemother. 2008;52:128–136. doi: 10.1128/AAC.00737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J, Chen CH, Aiken C. The sequence of the CA-SP1 junction accounts for the differential sensitivity of HIV-1 and SIV to the small molecule maturation inhibitor 3-O-{3',3'-dimethylsuccinyl}-betulinic acid. Retrovirology. 2004;1:15. doi: 10.1186/1742-4690-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, Zoumplis D, Matallana C, Kilgore NR, Reddick M, Yunus AS, Adamson CS, Salzwedel K, Martin DE, Allaway GP, Freed EO, Wild CT. Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology. 2006;356:217–224. doi: 10.1016/j.virol.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Martin DE, Salzwedel K, Allaway GP. Bevirimat: a novel maturation inhibitor for the treatment of HIV-1 infection. Antivir. Chem. Chemother. 2008;19:107–113. doi: 10.1177/095632020801900301. [DOI] [PubMed] [Google Scholar]
- 21.Davis KL, Bibollet-Ruche F, Li H, Decker JM, Kutsch O, Morris L, Salomon A, Pinter A, Hoxie JA, Hahn BH, Kwong PD, Shaw GM. Human immunodeficiency virus type 2 (HIV-2)/HIV-1 envelope chimeras detect high titers of broadly reactive HIV-1 V3-specific antibodies in human plasma. J. Virol. 2009;83:1240–1259. doi: 10.1128/JVI.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.