

Abstract
Objective
To determine whether changes in the DNA methylation status in the promoter region of the gene encoding interleukin-1β (IL-1β) account for expression of IL1B mRNA after long-term treatment of human articular chondrocytes with inflammatory cytokines.
Methods
IL-1β or TNFα combined with oncostatin M (OSM), or 5-aza-deoxycytidine (5-aza- dC) were added twice weekly for 4–5 weeks to primary cultures of normal human articular chondrocytes, obtained from patients with a femoral neck fracture. Expression of MMP13, IL1B TNFA and DNMT1 were determined by SybrGreen-based qRT-PCR on genomic DNA and total RNA extracted from the same sample before and after culture. Bisulfite modification was used to identify which CpG sites in the IL1B promoter showed differential methylation between expressing and non-expressing cells. The percentages of cells that were methylated at that critical CpG site (−299bp) were quantified by a method that depended on methylation-sensitive restriction enzymes and real-time PCR. Secretion of IL-1β was assessed by enzyme-linked immunoabsorbent assay (ELISA) of the culture media.
Results
Healthy chondrocytes did not express IL1B mRNA, but the levels were increased 5-fold by 5-aza-dC and 100- to 1000-fold by or TNFα/OSM. The % CpG methylation was decreased by 5-aza-dC, but reduced considerably more by IL-1β and almost abolished by TNFα/OSM. The mRNA was translated into protein in cytokine-treated chondrocytes.
Conclusion
These novel findings indicate that inflammatory cytokines can change DNA methylation status at key CpG sites, resulting in long-term induction of IL1B in human articular chondrocytes.
INTRODUCTION
Idiopathic osteoarthritis (OA) is a late-onset, complex disease of the joint. It is characterized by progressive failure of the extracellular cartilage matrix and changes in the synovium and subchondral bone (1,2). Susceptibility genes for OA have been indentified (3), which represents potential risks, whereas getting the disease is due to interaction with the environment. This interaction may be mediated by epigenetically induced changes in gene expression that are then transmitted to generations of daughter cells (4). As in all adult somatic cells, the phenotype of normal adult chondrocytes is stabilized by epigenetic mechanisms, such as DNA methylation of CpG (Cytosine-P-Guanine) sites, modifications of histone tails and changes in chromatin structure. DNA methylation is generally stable in somatic cells throughout adult life (5,6), whereas histone modifications are readily reversible by specific enzymes (7). During DNA replication, the methylation pattern is rapidly reproduced on the nascent strand by DNMT1, the maintenance DNA methyl transferase. (8,9). The histone code can be re-established after cell division by interactions of methyl binding domains and DNMTs with histone methyltransferases and histone deacetylases (10,11). As a result, both the DNA methylation pattern and the histone code are reproduced during mitosis.
In normal adult articular cartilage, chondrocytes synthesize cartilage matrix proteins, such as collagens II, IX, and XI as well as aggrecan. These molecules impart tensile strength and compressive resistance to the joint. Normally, chondrocytes maintain a low turnover of cartilage matrix proteins, but in OA many chondrocytes undergo a phenotypic change and acquire a gene expression repertoire that is characterized by the aberrant expression of numerous catabolic genes, including matrix metalloproteases (MMPs), aggrecanases (ADAMTS-4 and -5), inducible nitric oxide synthethase (iNOS) and prostaglandins (12), IL1B and other cytokines (13,14), and many more genes (15,16). This change in gene expression does not take place in all OA chondrocytes, but predominantly in chondrocytes of the surface zone and near weight-bearing regions (17). To distinguish these chondrocytes from OA chondrocytes with a normal gene expression repertoire, they will be referred to as `degradative' chondrocytes. Disease progression correlates with an increase in the number of `degradative' chondrocytes.
Gene expression is regulated by both epigenetic and non-epigenetic mechanisms. Genes that are part of the repertoire of a particular cell type show DNA hypo-methylation and chromatin has an open structure. This permits binding of specific transcription factors in combination with cofactors, e.g. histone acetylases and methylases, that mediate rapid responses (minutes to hours) to inductive or repressive factors. Genes that are not part of a given repertoire tend to be permanently silenced by DNA hypermethylation; this prevents access of the relevant transcription factors to their promoter. Epigenetic silencing is essential to insure genomic stability throughout life. Epigenetic disruption may activate normally silent genes (18) or silence normally expressed genes. This is precisely the situation in degradative OA chondrocytes, where many non-chondrocytic genes are permanently activated. So far there only a few studies have dealt with the question whether the aberrant gene expression in OA is linked to loss of DNA methylation. Our group (19)(20) has demonstrated hypo-methylation at specific CpG sites in the promoters of MMP-3, MMP-9, MMP-13 and ADAMTS-4 in `degradative' OA chondrocytes. Iliopoulos et al. (21) have showed that the aberrant induction of leptin in OA is associated with loss of DNA methylation. On the other hand, loss of OP-1 expression in aged chondrocytes is correlated with hypermethylation of the OP-1 promoter (22).
Because of the association of aberrant expression of proteases with DNA demethylation, it is of interest to identify factors causing loss of DNA methylation. The aberrant expression of non-chondrocytic genes that occurs in OA can arguably be reproduced in vitro by treating healthy chondrocytes with either IL-1β (23) or TNFα, especially in combination with OSM (24). However, it is not known whether this aberrant induction depends solely on non-epigenetic regulation by transcription factors or whether epigenetic changes are also involved. In the first case, one would expect expression to be induced within hours and be readily reversible by cytokine withdrawal. In the second case, induction might take days or weeks, but expression would probably persist after cytokine withdrawal. We hypothesized that the latter situation is linked to de-methylation of specific CpG sites in the relevant promoter regions, whereas this is not the case for short-term induction. To test our hypothesis, mRNA expression of IL1B, an aberrantly induced gene, was compared with DNA methylation status in primary human chondrocytes cultured in the absence and presence of inflammatory cytokines.
MATERIALS AND METHODS
Chondrocyte isolation
Human articular cartilage was dissected from femoral heads obtained within six hours of operation with ethical permission and patients' consent. Cartilage was obtained after hemi-arthroplasty following a fracture (#) of the Neck of Femur (#NOF) or after total hip arthroplasty for OA. A previous study (25) had shown that the deep zones of #NOF patients contain healthy, albeit aged chondrocytes, which did not aberrantly express proteases and cytokines. However, the superficial zone did contain a few cells with aberrant expression. This zone typically displayed a pinkish colouring so that it was relatively easy to separate this zone with a scalpel from the deep zone. In OA patients, a significant amount of cartilage had already been degraded, especially at weight-bearing regions. To obtain enough cartilage, we collected the “surface zone” irrespective of whether this was superficial, intermediate or deep zone. To obtain proper deep-zone OA cartilage was only possible if thick cartilage had remained at non-weight bearing regions, which was not always the case. To liberate the cells, cartilage pieces were cut into small fragments and digested with 10% trypsin (LONZA, Wokingham, UK) in PBS for 30 minutes, 1mg/ml hyaluronidase (Sigma-Aldrich, Gillingham, UK) in PBS for 15 minutes and 10 mg/ml collagenase B (Roche, Lewes, UK) in α-Modified Eagle Medium (α -MEM) (Sigma-Aldrich, Gillingham, UK) for 12 to 15 hours at 37°C. In total, 21 #NOF patients and 12 OA patients were used in this study.
Chondrocyte culture
For culture only non-OA chondrocytes from the deep zone of #NOF patients (25) were used. Before treatment, chondrocytes were cultured for 48 hours at a density of 2 to 4 ×105 cells/25 cm2 flask in 5ml α-MEM supplemented with 10% fetal calf serum (FCS; Invitrogen, Paisley, UK), 1% ITS (Sigma-Aldrich, Gillingham, UK), 100 U/ml penicillin and 100 μg/ml streptomycin (LONZA, Wokingham, UK) and 100 μg/ml ascorbic acid (Sigma-Aldrich, Gillingham, UK) in an atmosphere of 5% CO2 at 37°C. In the first experiment, chondrocytes were cultured until confluence, passaged once, incubated with a single addition of 10 ng/ml IL-1β/10ng/ml OSM, and harvested after 24 or 72 hours. Other P1 cultures were treated with cytokines at each medium change for three weeks. Half of the cultures were then harvested by trypsinization, whereas the other half was passaged again and cultured without cytokine treatment until confluence at around 2 weeks.
In another experiment, chondrocytes were divided into five groups immediately after isolation: i) non-cultured; ii) cultured without treatment (control culture); iii) cultured with 2 μM 5-aza-deoxycytidine (5-aza-dC); iv) cultured with 10 ng/ml IL-1β and v) cultured with a mixture of 10 ng/ml TNFα/10ng/ml OSM. For group iii), the histone deacetylase inhibitor Trichostatin A (TSA, 300nM) was added just once at the first treatment to facilitate access of 5-aza-dC, a cytidine analogue that inhibits the activity of DNMT1 (26). This results in non-specific loss of DNA methylation during cell division. Media were changed twice per week, when reagents were also added. These primary cultures were maintained for 4 to 5 weeks until confluence.
DNA and RNA extraction
Genomic DNA and total RNA were extracted simultaneously from the harvested chondrocytes by AllPrep DNA/RNA Mini Kit (Qiagen, West Sussex, UK) according to the manufacturer's instructions. RNA was immediately reverse-transcribed with AMV reverse transcriptase and both oligo(dT)15 and random primers (27).
Quantitative polymerase chain reaction (qRT-PCR)
Relative quantification of gene expression was performed with Applied Biosystems (Warrington, UK) ABI Prism 7500 detection system. Reactions were performed in triplicate with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as internal control. Primer Express 3.0 software was used to design primers across exon-exon boundaries. The primers for IL1B were commercially designed by PrimerDesign Ltd (Southampton, UK) and the primers for COL2A1 were obtained from (28). mRNA expression was quantified by the 2−ΔΔCt method. The 25 μl reaction mixture contained 1 μl cDNA, 12.5 μl 2x Power SYBR® Green PCR Master Mix and 500 μM of each primer. Thermocycler conditions comprised an initial activation step at 95°C (10 min), followed by a 2-step PCR program of 95°C (15s) and 60°C (60s) for 40 cycles. A dissociation curve was obtained for each qPCR run.
Bisulfite Modification
Genomic DNA was modified with MethylDetector™ (Active Motif, Rixensart, Belgium), according to the manufacturer's instructions. The MethPrimer website (http://www.urogene.org/methprimer) was used to design primers which contained no CpG sites. 21 CpG sites located between −1300 and + 15bp were investigated by three pairs of nested PCR primers (Figure 1). The sequences of all primers can be found at http://www.som.soton.ac.uk/research/dohad/groups/bone_joint/Who/Roach/default.htm
Figure 1. CpG methylation map for the proximal IL1B promoter.

Normal chondrocytes were isolated from four #NOF patients and cultured for 4–5 weeks with or without the cytokines. Methylation status was determined by bisulfite modification. Each circle represents the average result of six sequenced clones. The CpG site at −299bp was selected for quantification.
Thermocycler conditions comprised an activation step at 94°C (2 min), and followed by a 3-step PCR program consisting of 94°C (30s), 55°C (60s), 72°C (60s) for 35 cycles, and final extension at 72°C (3 min) as. The PCR products were diluted 50x and inner reactions were run. The PCR products were cloned with the TOPO® TA Cloning® Kit with One Shot TOP10 Chemically Competent E. coli (Invitrogen, Plaisley, UK), followed by purification of the plasmids by PureLink™ Quick Plasmid MiniPrep Kit (Invitrogen, Paisley, UK). Six plasmids were sequenced for each sample by Eurofins MWG (Ebersberg, Germany).
Determining the % methylation at −299 bp in the IL1B promoter
This CpG site was selected for quantification because it was differentially methylated in control versus cytokine-treated cultured cells (see results, Figure 1) and because it is recognized by a methylation-sensitive restriction enzyme, namely HpyCH4IV (New England BioLabs), Fully methylated DNA and fully non-methylated DNA were used i) to estimate non-specific digestion and incomplete digestion respectively, and ii) to generate a standard curve. CpGenome-Universal Methylated DNA was obtained from Millipore (Eastleigh, UK), while fully non-methylated DNA was generated from the fully methylated DNA by whole genome amplification with the Illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare Life Sciences). DNA concentrations were measured using Nanodrop (Thermo Scientific, UK) and were adjusted to 4ng/μl (20ng DNA in 5μl). HpyCH4IV was diluted to contain 2 Units/2.5 μl enzyme solution. These concentrations had previously been determined as optimal (27). Each digestion sample consisted of a final volume of 8.3μl, containing 5μl of diluted DNA, 2.5μl diluted HpyCH4IV, and 0.8μl of 10x NE Buffer I. All prepared samples, together with controls, were incubated at 37°C for 12 hours, followed by heat inactivation at 65°C for 20 minutes.
PCR for % methylation analysis was performed as for RT-PCR. Each run included no-enzyme controls, fully methylated and non-methylated DNA controls, the standard curve and the enzyme-treated DNA samples. The DNA content of each enzyme-digested sample was normalized to its corresponding no-enzyme control and the % methylation was calculated from the standard curve (27).
When #NOF samples were compared with OA samples for mRNA expression or % DNA methylation, all samples were analyzed in the same assay.
ELISA for IL-1β in culture media
At the end of culture, the media containing the cytokines were removed and, after thorough washings to remove any remaining exogenous IL-1β, 5ml fresh medium with 1% FCS was added. After 48 hours, the media were harvested and stored at −80°C until analysis with the Human IL-1 beta/IL-1F2 Quantikine® ELISA Kit (R&D Systems, Abingdon, UK).
Statistical analysis
The data for IL-1β expression and % methylation were analyzed with Microsoft Excel® by theWilcoxon's assigned-rank test and a significance level of 0.05. Other data were calculated as the mean±standard deviation.
RESULTS
Identifying differentially methylated CpG sites
Of the 21 CpG sites in the 1300bp sequence upstream of exon 1 (Figure 1), 16 were methylated in all cultures. This suggests that epigenetic regulation does not involve the region between −1300 to −590 bp. The AvaI site at −511bp and the two CpG sites (−20 and +13) that encompass the transcription start site were essentially non-methylated in all groups. On the other hand, the two CpG sites at −299 and −256 bp were methylated in control samples, but had become demethylated in cytokine-treated chondrocytes. Because only the site at −299 bp was cleavable by a methylation-sensitive restriction enzyme, this site was selected for quantitative analysis in subsequent experiments.
IL1B, but not TNFA, is differentially expressed in control and OA cartilage
To investigate the zonal distribution of IL1B-expressing chondrocytes in vivo, mRNA expression was assessed separately for the superficial and deep zones of OA and control cartilage. Expression was 10- to 1000-fold higher in the surface zone of OA cartilage compared with #NOF cartilage (Figure 2A). The variability among patients bore some relation to age in that, with one exception, expression increased markedly with age. In contrast to the superficial zone, no expression of IL1B was apparent in the deep zone of OA cartilage. In the #NOF samples, very low expression was found in the surface, but not in the deep zone. These findings confirm that the deep zone of #NOF cartilage contains chondrocytes that do not express IL1B and are therefore suitable for culture experiments. A previous study of ADAMTS-4 expression showed similar differential expression (20). In contrast to IL1B, TNFA expression was not significantly different in the surface and deep zone (Figure 2B); this supports the notion that IL-1β, but not TNFα, is the major cytokine involved in OA pathology.
Figure 2.

Differential qPCR expression of IL1B (A) and TNFA (B). Articular cartilage was obtained from either #NOF patients or OA patients. The surface zones were removed and analyzed separately from the deep zones. Results are shown for individual patients, arranged in age order. For each gene, #NOF and OA samples were analyzed in the same assay. Low levels of IL1B in the superficial zone of #NOF patients contrasted the 100–1000 fold increased expression in the surface zone of OA cartilage. With one exception (72 year old male), expression increased with age. There was no expression of IL1B in the deep zones of #NOF (one exception) or OA cartilage. By contrast, TNFA was expressed in the control chondrocytes located in the deep zone of #NOF patients and expression did not increase significantly in surface-zone OA chondrocytes.
Only long-term cytokine treatment results in persistent aberrant expression
A single treatment with IL-1β/OSM induced the aberrant expression of MMP3 (not shown), MMP13 (Fig. 3A) and IL1B (Fig. 3B) within 24 hours (Figure 3A). By 72 hours enzyme expression had declined or disappeared. By contrast, when expression was induced by repeated cytokine addition during 3 weeks of incubation, the expression persisted after 2 more weeks without further cytokines treatment (Figure 3D,E). COL2A1 expression was reduced after a single cytokine addition and remained low throughout (Fig. 2C,F)
Figure 3.

Effects of cytokine withdrawal following short-term (A–C) or long-term (D–F) treatment with IL-1β/OSM on expression of the indicated genes. The catabolic genes MMP13 and IL1B are up-regulated several 100-fold after 24 hours following a single addition of IL-1β/OSM, whereas COL2A1 is down-regulated. After 72 hours with no further cytokine treatment the expression of the catabolic genes reduced again to near-normal, but the expression of COL2A1 was not re-gained.
(D–F) When IL-1β/OSM was added twice a week for 3 weeks to passage 1 (P1+ IL-1) cells, the catabolic genes MMP13 and IL1B were again up-regulated considerably. Cells were then passaged and cultured for a further 3 weeks during which no cytokines were added (P2 no IL-1). This time expression was maintained even in the absence of cytokines, consistent with a permanent induction of gene expression. COL2A1 expression was abolished by the cytokines and remained low throughout. Representative examples of four experiments.
Experimental de-methylation increases expression of IL1B
If DNA de-methylation underpins aberrant IL1B expression in chondrocytes, then experimentally-induced de-methylation should result in increased expression. When normal chondrocytes were cultured with 5-aza-dC, the expression of IL1B increased 3.6- to 8.6-fold (mean = 5.5±2.2) compared with control cultures (Figure 4A). To check that 5-aza-dC treatment had actually resulted in loss of DNA methylation in the IL1B promoter, the % of methylation was quantified. As shown in Figure 4B, 61.0±6.1 % of chondrocytes were methylated in uncultured chondrocytes. In cultured samples, this was reduced to 44.8±9.9 %, indicating culture per se could cause limited loss of DNA methylation. However, 5-azadC further reduced the % DNA to 33.6±5.7 %. The results show that experimentally induced loss of DNA methylation results in increased gene transcription, thus confirming a cause-effect relationship between DNA de-methylation and transcription.
Figure 4.

Long-term treatment with cytokines causes DNA de-methylation together with aberrant gene expression. Chondrocytes were cultured with 5-aza-dC (A,B); or IL-1β or TNF-α/OSM (C,D,E,F). Relative mRNA expression of IL1B was compared with the % DNA methylation at −299bp in the IL1B promoter.
(A,B) Culture per se induced some expression and a 20% loss in methylation, 5-aza-dC increased expression 5-fold compared to control culture (A) and caused a further loss of DNA methylation (B). Means and SDs of six samples; * = P<0.05. (C) Relative expression of IL1B in chondrocytes from six individual patients, labelled with the patients' numbers and treated as indicated in the key. No IL1B was detectable in non-cultured chondrocytes. Culture itself induced low expression (set to =1), but IL-1β, and TNF-α/OSM increased expression considerably. (D) The % DNA methylation in the same samples as above. Before culture ~60% of cells were methylated. Culture alone could reduce DNA methylation, which was significant in patient 406 and 421. However, cytokine treatment caused greater loss of DNA methylation, particularly the combined treatment. (E,F) Means and S.D. of the samples shown in C and D. The differences between all groups are significant with P<0.05.
Long-term exposure to cytokines increases expression of IL1B 100 to 1000-fold
Healthy chondrocytes were cultured for 4 to 5 weeks with twice-weekly additions of either IL-1β or TNF-α/OSM (Figure 4C). Absence of expression in non-cultured chondrocytes was confirmed, and the low expression induced by culture alone was set to 1. IL-1β increased its own expression 15.8- to 197-fold, while the combination of TNF-α/OSM increased expression of IL1B by 306- to 1750-fold compared with control cultures (P < 0.01). This showed that the capacity of the cytokines to increase expression was several orders of magnitude greater than that of 5-aza-dC.
Long-term exposure to cytokines causes loss of DNA methylation
Quantitative analysis showed that culture alone reduced DNA methylation in two cases (patients Nr 406 and 421), but this decrease was not significant in the other patients. By contrast, the effects of the cytokines were very prominent. Only 4 to 25% (mean = 17.9±6.9 %) of IL-1β-treated chondrocytes were still methylated at −299 bp and with the combined TNFα/OSM treatment only 0–8 % (average 4.5±3.7 %) of cells remaining methylated. Although there is not a direct linear correlation between the fold increases in gene expression and the loss of DNA methylation, the trends are very clear: those samples with the greatest loss of DNA methylation showed, on the whole, the greatest induction of gene transcription.
Increased IL1B is associated with protein release into the medium
Significant amounts of IL-1β protein were produced by chondrocytes that had been cultured with either IL-1β or TNFα/OSM, but no IL-1β was detected in the control cultures or those treated with 5-aza-dC. This indicates that the small increases in transcription by 5-aza-dC did not produce measurable amounts of IL-1β protein. Treatment with exogenous IL-1β resulted in the release of 0.011 to 0.225 pg IL-1β/μg DNA/hr, while TNFα/OSM treatment caused release of 0.066 to 0.169 pg IL-1β/μg DNA/hr. These findings indicate that the increased expression of IL1B, a likely result of transcriptional promoter activation, resulted in increased synthesis of IL-1β protein.
Cytokine treatment does not increase TNFA expression in chondrocytes
Because IL-1β induced its own expression in vitro, we explored whether the same was true for TNFα. Non-cultured chondrocytes expressed TNFA (Figure 2B) and culture had no further effect. Expression of TNFA in chondrocyte cultures treated with IL-1β or TNFα/OSM was not significantly different from that of cultured controls, with mean increases in expression of 0.59±0.44 and 0.91±0.49 fold, respectively. Thus, in contrast to IL1B, inflammatory cytokines did not increase TNFA expression in vitro.
Culture and cytokine treatment decreases DNMT1 expression
To determine whether decreased expression of DNMT1 contributed to the observed loss of DNA methylation, we investigated expression by qRT-PCR. Indeed, DNMT1 expression in surface-zone OA chondrocytes was half that in the #NOF control chondrocytes (Figure 5A). Culture alone also reduced DNMT1 expression to around half of that in non-cultured chondrocytes, which may, in part, explain the loss of DNA methylation in control cultures. IL-1β or TNFα/OSM addition caused only a small further decrease (Figure 5B).
Figure 5.
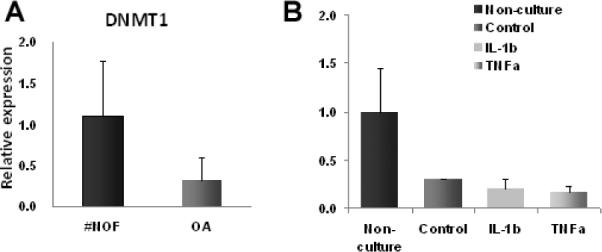
Expression of DNMT1 in articular chondrocytes in vivo (A) and in vitro (B). DNMT1 expression was less than half in OA chondrocytes compared with control (#NOF) chondrocytes (P<0.05). In vitro, culture alone had the greatest effect (P< 0.01) and the decrease in expression was of a similar in magnitude to the decrease in OA. In addition, TNF-α/OSM decreased expression further (P< 0.01). Means and SDs of n=5 for all groups.
DISCUSSION
The novel findings of the present study are that inflammatory cytokines have the capacity not only to induce changes in gene expression, but also de-methylation of specific CpG sites in the proximal IL1B promoter. IL-1β is at the apex of several inflammatory cascades and is arguably the most important cytokine in OA, although the debate is ongoing. Because IL-1β has a very short half-life and mRNA transcript levels correspond to protein levels, it is important to understand transcriptional regulation of this protein. Non-epigenetic regulation of the IL1B promoter has been studied extensively in monocytes/macrophages. In these cells, IL1B is readily induced by a variety of stimuli, including IL-1β itself (29,30). The CpG region between −131 and +12 bp is sufficient to direct expression of a reporter gene (31), but an inducible enhancer, located between −3134 and −2729 bp (31,32), is also involved in signal-dependent expression. In monocytes, this promoter is packaged into a non-transcribed, but “poised” promoter structure that is characterized by a histone-free transcription start site and constitutive association with the transcription factors PU.1 and C/EBPβ (30). Monocytes are thus primed for rapid activation in response to pathological or other stimuli.
Articular chondrocytes do not normally express IL1B, as confirmed here by qPCR. The low level of expression observed in the superficial zone of #NOF patients probably indicates an age-related change to `degradative' chondrocytes in a few cells. By contrast, the chondrocytes located near the surface of OA cartilage showed very high levels of IL1B expression, whereas this was undetectable in deep-zone OA cartilage. We hypothesized that IL1B was silenced by DNA methylation in normal chondrocytes and that demethylation had occurred in OA, as previously reported for MMPs and ADAMTS-4 (19). To investigate the epigenetic regulation of IL1B, we studied the proximal promoter because its activity regulates cell-type specific expression (33). When the CpG locations are compared with the transcription factor binding sites (Figure 6), the latter are concentrated in two CpG-free regions, each bracketed by CpG sites. Interestingly, the site at −511 bp and the two CpG sites around the transcription start site (−20 and +10 bp) were un-methylated, whether or not IL1B was expressed. The two CpG sites that are differentially methylated and where methylation status inversely correlated with expression (−256 and −299 bp) were located between the two regions that contain multiple transcription factor binding sites and bracket a NF-κB binding site. Both IL-1β and TNFα activate NF-κB (34–36), which, in turn, activates many genes (37), including IL1B and other catabolic genes induced in OA. Perhaps more relevant in the present context is the fact that NF-κB may be involved in demethylation (38), although details are not clear. The observation (39) that, in intestinal inflammation, the TNFα-induced NF-κB expression was associated with demethylation of CpG islands supports the inference that NF-κB may play a role in demethylation in the experiments reported here.
Figure 6.

Proximal promoter of IL1B. Vertical bars indicate the position of CpG sites. The CpG sites at -299 and -256bp bracket a NF-κB binding site
Having identified the differentially methylated CpG sites, we determined the percentage of cells that were methylated at −299 bp (27). Even in control chondrocytes, which do not express IL1B, the % methylation was only ~60% and not the expected 100%. This raises the question why 40% of non-methylated cells did not express IL1B. Hypomethylation is a necessary, but not sufficient, condition for gene activation, inasmuch as histone modifications are also required. Deacetylated histones or methylated H3K9 and H3K27 can in theory silence a gene even in the absence of DNA methylation. An alternative possibility is that normal articular chondrocytes do not contain the transcription factors required for IL1B expression. Thus the rapid induction after 24 hours could depend on cytokine-induced histone modifications and/or activation of transcription factors in those cells with pre-existing de-methylation. In support of this notion, IL-1α has been shown to translocate to the nucleus and to interact with histone acetyltransferase in yeast strains (40). Whether this also applies to IL-1β is not known. Upregulation of IL1B after short-term treatment did not persist, yet after long-term treatment the expression of IL1B persisted for at least two weeks after the inducer had been withdrawn. This is consistent with permanent epigenetic changes. Because the pathology of OA involves long-term aberrant induction of IL-1β and proteases, short-term in vitro experiments are unlikely to model the in vivo situation.
Further evidence for a cause-effect relationship between DNA methylation and gene expression was obtained in 5-aza-dC treated cultures, in which the experimentally induced loss of DNA methylation increased IL1B expression by 5-fold. Treatment with 5-aza-dC reduced, yet did not abolish, the % methylation. One reason may be that 5-aza-dC, whose half-life is around four hours (26), can act only on those cells that undergo cell division during that time. Because 5-aza-dC was added 3 times per week, it is likely that active 5-aza-dC was not present at all times when cell division took place. Treatment with cytokines rather than with 5-aza-dC was much more effective in inducing gene expression. This was especially true for the combination of the two cytokines (TNFα/OSM). TNFα with or without OSM has frequently been used to induce expression of degradative proteases in cultured chondrocytes (34,41). The findings presented here show that the TNFα/OSM combination is also highly effective in stimulating IL1B in chondrocytes, increasing its expression up to 1000-fold. OSM belongs to the IL-6 family, acts via JAK/STAT signalling, and can, on its own, induce cartilage degradation in vitro (42), but is usually used together with TNFα (43) or IL-1β (24,41) to enhance the cytokine effect. The % DNA methylation correlated negatively with increases in IL1B expression, although this varied considerably among patients. Samples treated with TNFα/OSM had the highest expression level and the greatest loss of DNA methylation. Indeed DNA methylation was completely abolished in some samples. On the basis of these findings it is evident that long-term aberrant expression of IL1B was a consequence of DNA de-methylation, presumably caused by inflammatory cytokines. The question then arises of how cytokines can achieve greater DNA demethylation than 5-aza-dC. Because 5-aza-dC inhibits DNMT1, we asked whether cytokines also alter DNMT1 expression. This was indeed reduced in vitro by the cytokines and in OA chondrocytes compared with #NOF chondrocytes in vivo. However, since culture per se significantly reduced expression, lower expression of DNMT1 alone could not explain the results. We hypothesize that activation of NF-κB plays a major role in the cytokine-induced DNA de-methylation (38), but further studies are required.
There are several caveats that should be borne in mind. When chondrocytes are grown in monolayer for several weeks, their phenotype is lost. However, explants of articular cartilage in organ culture, in which the chondrocytic phenotype is maintained, exhibit little or no loss of DNA methylation or gene induction. This suggests that the extracellular matrix protects the cells from loss of phenotype and possibly loss of DNA methylation. To maintain genomic stability in vivo, one would expect resistance to changes in DNA methylation status in healthy fully differentiated cells and the extracellular matrix might provide increased resistance in explant cultures. Monolayer cultures probably sensitize chondrocytes to epigenetic changes so that one can obtain within weeks the phenotypic changes that probably take years in OA. To what extent the protease-producing `degradative' cells are still chondrocytes is a matter for debate.
One should also be careful about extending these in vitro findings to the in vivo situation. In vitro, we observed decreased expression of COL2A1 after a single cytokine addition, whereas in vivo we actually found increased COL2A1 expression in surface-zone OA chondrocytes compared with #NOF chondrocytes (not shown), suggesting that the cultures do not model the vivo situation as far as COL2A1 is concerned. There is also some disagreement about the importance of IL-1β in OA. We have observed significant expression of IL1B in chondrocytes of the superficial zone of OA cartilage, where cytokines have also been immunolocalized (44,45). By contrast, Fan et al. (46) found no significant upregulation of IL1B in OA patients, but had used early degenerate cartilage of the knee joint, with the deep zone included. In IL1B knock-out mice, experimentally-induced OA was either enhanced or reduced (47,48) depending on genetic background, whereas over-expression of human TNFA induced severe inflammatory arthritis (49). However the mouse models may not be representative of human OA.
Summary
Our novel findings support the conclusion that the long-term cytokine-stimulated induction of IL1B in human articular chondrocytes in vitro involves loss of DNA methylation. If applicable in vivo, then the mechanisms for OA progression may involve the following: An initial inflammatory episode in the synovium, perhaps as a consequence of mechanical stress, activates synovial macrophages to produce IL-1β and TNFα. These diffuse into the articular cartilage. There the cytokines induce aberrant expression of proteases and IL1B in the chondrocytes (50). If this induction leads to the loss of DNA methylation, IL1B will now be included in the expression repertoire of the OA chondrocytes, even after synovial inflammation has abated. This scenario may explain why protease inhibitors, such as TIMPs, have little effect when injected into the joint, and why, once degradative processes have been operative, OA progression cannot be halted.
ACKNOWLEDGEMENTS
We are grateful to the Orthopaedic Surgeons of Southampton General Hospital for supplying us with femoral heads following joint replacements. The critical reading of the manuscript by Dr. Felix Bronner (Connecticut) was much appreciated.
Supported by NIH grant R21-AR054887 to MBG and HIR, and Wessex Medical Research grant No M19 to HIR
REFERENCES
- 1.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 2.Aigner T, Sachse A, Gebhard PM, Roach HI. Osteoarthritis: Pathobiology-targets and ways for therapeutic intervention. Adv Drug Deliv Rev. 2006;58:128–149. doi: 10.1016/j.addr.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Valdes AM, Doherty M, Spector TD. The additive effect of individual genes in predicting risk of knee osteoarthritis. Ann Rheum Dis. 2008;67:124–127. doi: 10.1136/ard.2007.075838. [DOI] [PubMed] [Google Scholar]
- 4.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 5.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 6.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 7.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 8.Attwood JT, Yung RL, Richardson BC. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci. 2002;59:241–257. doi: 10.1007/s00018-002-8420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr Opin Cell Biol. 2007;19:266–272. doi: 10.1016/j.ceb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, et al. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J Biol Chem. 2003;278:24132–24138. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 11.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, et al. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 12.Goldring MB, Berenbaum F. The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res. 2004:S37–S46. doi: 10.1097/01.blo.0000144484.69656.e4. [DOI] [PubMed] [Google Scholar]
- 13.Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004:S27–S36. doi: 10.1097/01.blo.0000144854.66565.8f. [DOI] [PubMed] [Google Scholar]
- 14.Aida Y, Maeno M, Suzuki N, Namba A, Motohashi M, et al. The effect of IL-1beta on the expression of inflammatory cytokines and their receptors in human chondrocytes. Life Sci. 2006;79:764–771. doi: 10.1016/j.lfs.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 15.Sato T, Konomi K, Yamasaki S, Aratani S, Tsuchimochi K, et al. Comparative analysis of gene expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 2006;54:808–817. doi: 10.1002/art.21638. [DOI] [PubMed] [Google Scholar]
- 16.Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54:3533–3544. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- 17.Roach HI, Tilley S. The Pathogenesis of Osteoarthritis. In: Bronner F, Farach-Carson MC, editors. Bone and Osteoarthritis, Volume 4, Topics in Bone Biology 2007. pp. 1–18. [Google Scholar]
- 18.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 19.Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52:3110–3124. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- 20.Cheung KS, Hashimoto K, Yamada N, Roach HI. Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol Int. 2009;29:525–534. doi: 10.1007/s00296-008-0744-z. [DOI] [PubMed] [Google Scholar]
- 21.Iliopoulos D, Malizos KN, Tsezou A. Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann Rheum Dis. 2007;66:1616–1621. doi: 10.1136/ard.2007.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeser RF, Im HJ, Richardson B, Lu Q, Chubinskaya S. Methylation of the OP-1 promoter: potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthritis Cartilage. 2009;17:513–517. doi: 10.1016/j.joca.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saas J, Haag J, Rueger D, Chubinskaya S, Sohler F, et al. IL-1beta, but not BMP-7 leads to a dramatic change in the gene expression pattern of human adult articular chondrocytes--portraying the gene expression pattern in two donors. Cytokine. 2006;36:90–99. doi: 10.1016/j.cyto.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Barksby HE, Hui W, Wappler I, Peters HH, Milner JM, et al. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 2006;54:540–550. doi: 10.1002/art.21574. [DOI] [PubMed] [Google Scholar]
- 25.da Silva MA, Yamada N, Clarke NM, Roach HI. Cellular and epigenetic features of a young healthy and a young osteoarthritic cartilage compared with aged control and OA cartilage. J Orthop Res. 2009;27:593–601. doi: 10.1002/jor.20799. [DOI] [PubMed] [Google Scholar]
- 26.Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: implications for methylation-associated cellular processes. Pharmacol Ther. 1995;65:19–46. doi: 10.1016/0163-7258(94)00053-6. [DOI] [PubMed] [Google Scholar]
- 27.Hashimoto K, Kokubun S, Itoi E, Roach HI. Improved quantification of DNA methylation using methylation-sensitive restriction enzymes and real-time PCR. Epigenetics. 2007;2:86–91. doi: 10.4161/epi.2.2.4203. [DOI] [PubMed] [Google Scholar]
- 28.Preradovic A, Kleinpeter G, Feichtinger H, Balaun E, Krugluger W. Quantitation of collagen I, collagen II and aggrecan mRNA and expression of the corresponding proteins in human nucleus pulposus cells in monolayer cultures. Cell Tissue Res. 2005;321:459–464. doi: 10.1007/s00441-005-1116-6. [DOI] [PubMed] [Google Scholar]
- 29.Toda Y, Tsukada J, Misago M, Kominato Y, Auron PE, et al. Autocrine induction of the human pro-IL-1beta gene promoter by IL-1beta in monocytes. J Immunol. 2002;168:1984–1991. doi: 10.4049/jimmunol.168.4.1984. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Saccani S, Shin H, Nikolajczyk BS. Dynamic protein associations define two phases of IL-1beta transcriptional activation. J Immunol. 2008;181:503–512. doi: 10.4049/jimmunol.181.1.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirakawa F, Saito K, Bonagura CA, Galson DL, Fenton MJ, et al. The human prointerleukin 1 beta gene requires DNA sequences both proximal and distal to the transcription start site for tissue-specific induction. Mol Cell Biol. 1993;13:1332–1344. doi: 10.1128/mcb.13.3.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bensi G, Mora M, Raugei G, Buonamassa DT, Rossini M, et al. An inducible enhancer controls the expression of the human interleukin 1 beta gene. Cell Growth Differ. 1990;1:491–497. [PubMed] [Google Scholar]
- 33.Kominato Y, Galson D, Waterman WR, Webb AC, Auron PE. Monocyte expression of the human prointerleukin 1 beta gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol Cell Biol. 1995;15:58–68. [PMC free article] [PubMed] [Google Scholar]
- 34.Liacini A, Sylvester J, Li WQ, Huang W, Dehnade F, et al. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp Cell Res. 2003;288:208–217. doi: 10.1016/s0014-4827(03)00180-0. [DOI] [PubMed] [Google Scholar]
- 35.Granet C, Maslinski W, Miossec P. Increased AP-1 and NF-kappaB activation and recruitment with the combination of the proinflammatory cytokines IL-1beta, tumor necrosis factor alpha and IL-17 in rheumatoid synoviocytes. Arthritis Res Ther. 2004;6:R190–R198. doi: 10.1186/ar1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffin BD, Moynagh PN. Persistent interleukin-1beta signaling causes long term activation of NFkappaB in a promoter-specific manner in human glial cells. J Biol Chem. 2006;281:10316–10326. doi: 10.1074/jbc.M509973200. [DOI] [PubMed] [Google Scholar]
- 37.Mercurio F, Manning AM. Multiple signals converging on NF-kappaB. Curr Opin Cell Biol. 1999;11:226–232. doi: 10.1016/s0955-0674(99)80030-1. [DOI] [PubMed] [Google Scholar]
- 38.Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, et al. A role for nuclear NF-kappaB in B-cell-specific demethylation of the Igkappa locus. Nat Genet. 1996;13:435–441. doi: 10.1038/ng0895-435. [DOI] [PubMed] [Google Scholar]
- 39.Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Charrier-Hisamuddin L, et al. Nuclear factor-kappaB is a critical mediator of Ste20-like proline-/alanine-rich kinase regulation in intestinal inflammation. Am J Pathol. 2008;173:1013–1028. doi: 10.2353/ajpath.2008.080339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buryskova M, Pospisek M, Grothey A, Simmet T, Burysek L. Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. J Biol Chem. 2004;279:4017–4026. doi: 10.1074/jbc.M306342200. [DOI] [PubMed] [Google Scholar]
- 41.Rowan AD, Hui W, Cawston TE, Richards CD. Adenoviral gene transfer of interleukin-1 in combination with oncostatin M induces significant joint damage in a murine model. Am J Pathol. 2003;162:1975–1984. doi: 10.1016/S0002-9440(10)64330-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El Mabrouk M, Sylvester J, Zafarullah M. Signaling pathways implicated in oncostatin M-induced aggrecanase-1 and matrix metalloproteinase-13 expression in human articular chondrocytes. Biochim Biophys Acta. 2007;1773:309–320. doi: 10.1016/j.bbamcr.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 43.Hui W, Rowan AD, Richards CD, Cawston TE. Oncostatin M in combination with tumor necrosis factor alpha induces cartilage damage and matrix metalloproteinase expression in vitro and in vivo. Arthritis Rheum. 2003;48:3404–3418. doi: 10.1002/art.11333. [DOI] [PubMed] [Google Scholar]
- 44.Moos V, Fickert S, Muller B, Weber U, Sieper J. Immunohistological analysis of cytokine expression in human osteoarthritic and healthy cartilage. J Rheumatol. 1999;26:870–879. [PubMed] [Google Scholar]
- 45.Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44:585–594. doi: 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 46.Fan Z, Soder S, Oehler S, Fundel K, Aigner T. Activation of interleukin-1 signaling cascades in normal and osteoarthritic articular cartilage. Am J Pathol. 2007;171:938–946. doi: 10.2353/ajpath.2007.061083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clements KM, Price JS, Chambers MG, Visco DM, Poole AR, et al. Gene deletion of either interleukin-1beta, interleukin-1beta-converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy. Arthritis Rheum. 2003;48:3452–3463. doi: 10.1002/art.11355. [DOI] [PubMed] [Google Scholar]
- 48.Glasson SS. In vivo osteoarthritis target validation utilizing genetically-modified mice. Curr Drug Targets. 2007;8:367–376. doi: 10.2174/138945007779940061. [DOI] [PubMed] [Google Scholar]
- 49.Butler DM, Malfait A-M, Mason LJ, Warden PJ, Kollias G, et al. DBA/1 mice expressing the human TNF-a transgene develop a severe, erosive arthritis. J Immunol. 1997;159:2867–2876. [PubMed] [Google Scholar]
- 50.Goldring MB, Otero M, Tsuchimochi K, Ijiri K, Li Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann Rheum Dis. 2008;67(Suppl 3):iii75–iii82. doi: 10.1136/ard.2008.098764. [DOI] [PMC free article] [PubMed] [Google Scholar]