

Abstract
Protoporphyrin IX ferrochelatase (EC 4.99.1.1) catalyzes the terminal step in the heme biosynthetic pathway, the insertion of ferrous iron into protoporphyrin IX. Ferrochelatase shows specificity, in vitro, for multiple metal ion substrates and exhibits substrate inhibition in the case of zinc, copper, cobalt, and nickel. Zinc is the most biologically significant of these; when iron is depleted, zinc porphyrins are formed physiologically. Examining the kcat/Kmapp ratios for zinc and iron reveals that, in vitro, zinc is the preferred substrate at all concentrations of porphyrin. This is not the observed biological specificity, where zinc porphyrins are abnormal; these data argue for the existence of a specific iron delivery mechanism in vivo. We demonstrate that zinc acts as an uncompetitive substrate inhibitor, suggesting that ferrochelatase acts via an ordered pathway. Steady-state characterization demonstrates that the apparent kcat depends on zinc and shows substrate inhibition. Although porphyrin substrate is not inhibitory, zinc inhibition is enhanced by increasing porphyrin concentration. This indicates that zinc inhibits by binding to an enzyme-product complex (EZnDIX) and is likely to be the second substrate in an ordered mechanism. Our analysis shows that substrate inhibition by zinc is not a mechanism that can promote specificity for iron over zinc, but is instead one that will reduce the production of all metalloporphyrins in the presence of high concentrations of zinc.
Introduction
Ferrochelatase (EC 4.99.1.1), the final enzyme of heme biosynthesis, catalyzes the insertion of ferrous iron into protoporphyrin IX (1); this reaction is commonly proposed to involve a distorted porphyrin intermediate (1, 2). In vitro, ferrochelatase has a broad metal ion specificity with insertion of Zn2+, Co2+, and Cu2+ having also been observed (1, 3). Metal ion specificity is species-dependent; the ferrochelatase from Bacillus subtilis accepts Cu2+ but not Co2+ as a substrate (4), in contrast to the widely reported specificity of most other ferrochelatases. Interestingly, it has recently been demonstrated that the poor activity toward Cu2+ as a substrate, in the case of the murine and yeast ferrochelatases at least, arises from substantial substrate inhibition (3). Of the competing metal ions, Zn2+ is perhaps the most biologically significant; under conditions of iron depletion or lead poisoning, zinc porphyrins can be formed physiologically (5), and the presence of zinc porphyrins in human blood can be used as a diagnostic test for lead poisoning (6). It has been suggested that the metal ion specificity of ferrochelatase is determined by the extent of porphyrin distortion in the active site of the enzyme (2). More recent crystallography has shown that some metal ion inhibitors are inserted into porphyrins, and the inhibition therefore arises from reduced product release (7).
Crystal structures of free enzyme (8, 9), as well as enzyme with metal (10, 11), inhibitors (12, 13), porphyrin substrate (14), and a range of porphyrin products (7, 15) are available. Ferrochelatase is a peripheral membrane protein, and in vitro biochemical studies generally require the use of detergent; this is most often cholate. When crystallized, the human enzyme had cholate bound in the active site (8).
Transient kinetic studies have shown that the metal ion insertion step in the ferrochelatase-catalyzed reaction is at least 10 times faster than the overall reaction (16). Recent studies of the metal specificity have shown that the presence of metal ion chelators in ferrochelatase assays can have a significant impact on the observed kinetics when using nonphysiological metal ion substrates (3). Extending these observations, we have demonstrated that the apparent substrate inhibition caused by metal can also be alleviated by increasing the concentration of detergent in the assay.
We show here that zinc acts as an uncompetitive substrate inhibitor, strongly suggesting a model where human ferrochelatase acts in a steady-state ordered fashion. The substrate inhibition most likely arises from zinc binding to an enzyme-product complex (EZnDIX).2 This inhibition is not observed in the presence of higher concentrations of the detergent cholate, and it is likely that increasing detergent concentrations also increases KmDIX and so reduces the concentration of enzyme-product species for the inhibitor to bind.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise stated, chemicals were obtained from Sigma. Detergents were obtained from Merck.
Methods
Human ferrochelatase (R115L) was prepared as described previously (16, 17). Briefly, Escherichia coli JM109 cells containing recombinant ferrochelatase were suspended in binding buffer (50 mm Tris-HCl, 0.1 m KCl, 1% (w/v) sodium cholate, pH 8.0) containing 1 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride, sonicated on ice for 3 × 30 s, and centrifuged at 50,000 × g for 30 min at 4 °C. The supernatant was loaded onto a 2-ml Talon column (Clontech), previously equilibrated with binding buffer. The column was washed with 20 ml of binding buffer and 10 ml of binding buffer containing 1 m KCl. The column wash containing 1 m KCl was used to remove any endogenous porphyrin. Ferrochelatase was eluted with binding buffer containing 300 mm imidazole. Imidazole was removed by applying purified protein to a 30-ml column of Sephadex G25 (GE Healthcare) equilibrated in binding buffer. Pure protein was stored at −80 °C in 50 mm Tris-HCl, 0.1 m KCl, 1% (w/v) sodium cholate, pH 8.0. Protein concentration was determined spectrophotometrically using the calculated extinction coefficient ϵ278 46,900 m−1 cm−1 (17).
Assay Conditions
All buffers were treated with Chelex 100 (Bio-Rad) to remove trace metal ions. Control assays in the absence of added metal ion substrate showed no metalloporphyrin product formation. Steady-state rates were determined using a spectrophotometric assay (16, 18, 19) at 536 nm. All assays were performed at 30 °C, in 0.1 m Tris-HCl, pH 8.1, 0.5% Tween 20 with 10 nm enzyme. Assays contained either <0.002% or 0.5% (w/v) sodium cholate as indicated in the figure legends. DIX was prepared in assay buffer, and its concentration was determined in 0.1 m HCl using the ϵ399 433,000 m−1 cm−1 (20). ZnCl2 was prepared in water treated with Chelex 100, and stock concentrations were determined spectrophotometrically after reaction to completion with excess DIX. Assays were performed over a range of substrate concentrations detailed in the appropriate figure legends. Consistent results were obtained with distinct protein preparations.
Determining Extinction Coefficients
Extinction coefficients for the transformation of DIX into ZnDIX were determined from the ΔA measured on complete transformation of a known concentration of DIX. These reactions were performed at two different zinc concentrations to ensure that the equilibrium results in essentially complete formation of product and at two different enzyme concentrations to ensure that there was no contribution of enzyme-bound porphyrin to the observed absorbance change. A Δϵ536 of 10,400 m−1 cm−1 was used throughout.
Data Analysis
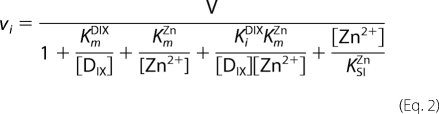
Kinetic parameters were evaluated by fitting the appropriate equation to initial rates using nonlinear regression analysis implemented in Igor Pro (Version 5.04B; Wavemetrics Inc., Lake Oswego, OR).
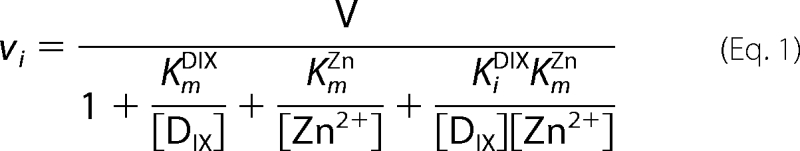 |
 |
RESULTS
Steady-state Kinetics
In the absence of added cholate, the response of initial rate of ZnDIX formation to the concentration of metal ion shows the characteristic pattern associated with substrate inhibition (Fig. 1A). Similar data have been reported recently for the murine and the yeast enzymes (3). In this case, however, it is apparent that the extent of substrate inhibition also depends on the concentration of porphyrin; high porphyrin concentrations noticeably increase the extent of zinc inhibition (Fig. 1A, compare filled circles at 22 μm DIX and open circles at 4 μm DIX). No inhibition by deuteroporphyrin was observed (Fig. 1B). In contrast, the presence of the detergent sodium cholate (0.5% w/v) abolishes the substrate inhibition by zinc (Fig. 1C). Under these conditions apparent Km values for DIX are substantially higher than those seen in the absence of added cholate (Fig. 1D).
FIGURE 1.

Initial rates of zinc porphyrin formation catalyzed by 10 nm ferrochelatase. Assay conditions were 0.1 m Tris-HCl, pH 8.1, 0.5% Tween 20, without (A and B) or with (C and D) 0.5% cholate at 30 °C. A, zinc dependence of the initial rate of zinc chelation. Filled circles represent experimental points at 21.8 μm DIX, and the line is theoretical with characterizing parameters Vapp = 30.8 ± 8.0 nm s−1, Kmapp = 0.74 ± 0.45 μm, Kiapp = 10.0 ± 6.2 μm. Open circles represent experimental points at 4 μm DIX and a theoretical line with characterizing parameters Vapp = 16.2 ± 0.9 nm s−1, Kmapp = 1.2 ± 0.2 μm, Kiapp = 87.9 ± 15.6 μm. B, DIX dependence of initial rate at 1.5 μm zinc with experimental points fitted to a theoretical line with characterizing parameters Vapp = 25.7 ± 4.5 nm s−1 and Kmapp = 9.0 ± 5.3 μm. C, zinc dependence of initial rate in the presence of 0.5% cholate. Filled circles represent experimental points at 20 μm DIX and a theoretical line with characterizing parameters Vapp = 11.9 ± 1.7 nm s−1 and Kmapp = 10.3 ± 3.1 μm. Open circles represent experimental points at 5 μm DIX and a theoretical line with characterizing parameters Vapp = 2.5 ± 0.5 nm s−1 and Kmapp = 7.9 ± 4.0 μm. D, DIX dependence of initial rate at 1 μm zinc in the presence of 0.5% cholate, the theoretical line is described by the characterizing parameters Vapp = 1.2 ± 0.5 nm s−1 and Kmapp = 25.9 ± 20.0 μm.
To characterize fully the behavior of ferrochelatase with respect to zinc we undertook a full steady-state characterization in the presence and absence of added cholate (Table 1). In the absence of added cholate, the apparent kinetic parameters kcatZn-app and both kcat/KmDIX-app and kcat/KmZn-app show hyperbolic dependences on the concentration of porphyrin (Fig. 2). In contrast, the kinetic parameter kcatDIX-app has a nonhyperbolic dependence on zinc concentration and clearly shows substrate inhibition. In the presence of 0.5% cholate, the profiles are rather different, and three of the apparent kinetic parameters show a hyperbolic dependence on substrate concentration (Fig. 3). Two main differences between the kinetics collected with and without cholate are seen: first, no substrate inhibition is seen in the plot of kcatDIX-app against [Zn2+] in the presence of cholate; and second, the responses of kcatZn-app and kcat/KmZn-app to [DIX] do not saturate in the presence of cholate. The data in these secondary plots arise from fitting single-substrate equations to the plots of vi against [S] (examples in Fig. 1), whereas the theoretical lines are obtained from the parameters estimated from a fit of Equation 1 or 2 to the entire dataset. The observation that substrate inhibition by zinc can be abolished at high concentrations of detergent is not only seen when that detergent is cholate but also when we use the detergent CHAPS (Fig. 4).
TABLE 1.
Kinetic parameters for the human ferrochelatase (R115L)-catalyzed insertion of zinc into deuteroporphyrin
The center column shows parameters obtained from global analysis of data in the presence of no added cholate assuming an ordered ternary complex mechanism with substrate inhibition. The right column shows those obtained from data collected in the presence of 0.5% cholate assuming a ternary complex mechanism. Standard deviations are given for the fitted parameters.
| Parameters | No added cholate | 0.5% cholate |
|---|---|---|
| kcat/s−1 | 3.2 ± 0.35 | 3.5 ± 1.1 |
| kcat/KmDIX/m−1s−1 | 1.2 × 106 | 76 × 103 |
| kcat/KmZn/m−1s−1 | 19 × 106 | 2.2 × 106 |
| KiDIXKmZn/m2 | 12.1 | 521 |
| KiDIX/μm | 71.2 ± 74.7 | 337.6 |
| KmDIX/μm | 2.60 ± 1.43 | 46.1 ± 26.7 |
| KmZn/μm | 0.17 ± 0.16 | 1.5 ± 4.0 |
| KSIZn/μm | 14.1 ± 3.3 | N.A.a |
a N.A., not applicable.
FIGURE 2.

Secondary plots showing the substrate concentration dependence of the apparent steady-state parameters kcatapp and kcat/Kmapp in the absence of added cholate. Points represent the calculated kinetic parameters of initial rate versus substrate curves, and the fitted lines are described as follows: A, kcat = 3.6 ± 0.4 s−1, Km-Zn = 0.4 ± 0.2 μm, and KSI = 8.9 ± 2.0 μm; B, kcat/Km-DIX = 0.9 ± 0.4 × 106 m−1 s−1 and KiZn = 2.9 ± 1.3 μm; C, kcat = 3.6 ± 0.4 s−1 and Km-DIX = 3.9 ± 1.5 μm; D, kcat/Km-Zn = 9.6 ± 4.2 × 106 m−1 s−1 and KiDIX = 30.5 ± 16.1 μm.
FIGURE 3.

Secondary plots showing the substrate concentration dependence of the apparent steady-state parameters kcatapp and kcat/Kmapp in the presence of 0. 5% cholate. Points represent the calculated kinetic parameters of initial rate versus substrate curves, and the fitted lines are described as follows: A, kcat = 3.5 ± 1.1 s−1 and Km-Zn = 1.6 ± 4.0 μm; B, kcat/Km-DIX = 0.08 ± 0.05 × 106 m−1 s−1 and KiZn = 11.4 ± 5.8 μm; C, kcat = 3.5 ± 1.1 s−1 and Km-DIX = 46.1 ± 26.7 μm; D, kcat/KmKi = 6.7 ± 5.6 × 109 m−2 s−1.
FIGURE 4.

Initial rates of zinc porphyrin formation catalyzed by 10 nm ferrochelatase. Assay conditions were 0.1 m Tris-HCl, pH 8.1, 0.5% Tween 20, with 0.5% (w/v) CHAPS at 30 °C. Filled circles represent experimental points at 35 μm DIX with a theoretical line described by kinetic parameters Vapp = 2.6 ± 0.2 nm s−1 and Kmapp = 1.8 ± 0.8 μm. Open circles represent experimental points at 5 μm DIX with a theoretical line described by kinetic parameters Vapp = 1.3 ± 0.2 nm s−1 and Kmapp = 4.5 ± 2.4 μm.
DISCUSSION
Zinc has been widely used as a convenient substrate for ferrochelatase because of zinc porphyrin spectral properties (21) and because it avoids any concern over substrate oxidation (22). However, reasonable objections can be made to the use of zinc; it is not the physiological substrate, and conclusions are hard to translate between metal ion substrates (1). This concern is strongly supported by the recent demonstrations with a variety of metal ions showing that alternative metal ion binding sites can inhibit or activate the reaction (3, 23). In addition, the relatively fast uncatalyzed insertion of zinc into porphyrins can complicate the observed kinetics. The work presented here with the human enzyme and recently with the murine and yeast enzymes (3) shows the difficulties with the simple assumption that data collected with one metal ion substrate is interpretable in terms of another. However, changing the metal ion used as a substrate is a relatively modest structural variation and arguably less extreme than mutagenesis of active site residues. In this report we consider the mechanistic variation on going from a reasonable model of the physiological substrate, which is most likely a Fe-(iron carrier protein) complex (24, 25), to an abnormal, but biologically relevant, substrate.
Steady-state kinetic parameters can be used to describe enzyme specificity, and it is well known that in the case of a single substrate enzyme the appropriate parameter is the ratio of specificity constants (kcat/Km values) of the two substrates (26). We are concerned here with a two-substrate enzyme, and the situation is slightly more complex (27–31). In general, the ratio of rates toward two alternative substrates depends on the ratio of apparent kcat/Km values (31) and so on the concentration of the other substrate. Under some conditions and with some mechanisms, this is the same as the kcat/Km ratio (29, 31). As a consequence, inhibitors and activators can only change the specificity if they perturb the ratio of apparent kcat/Km values.
In general, the ratio of apparent kcat/Km values varies with the concentration of the common substrate, A, from the ratio of kcat/KiAKmB values at low concentrations of A to the ratio of kcat/KmB values at high concentrations of A; this is low and high compared with KmA, and clearly KmA may be different in the presence of alternative substrates.
In the case of human ferrochelatase, kcat/Km-Zn is 19 × 106 m−1 s−1, and kcat/Km-Fe is 16 × 103 m−1 s−1 (16), so with saturating DIX the preferred substrate is zinc; kcat/KI-DIXKm-Zn is 0.26 μm−2 s−1, kcat/KI-DIXKm-Fe is 3.9 × 10−3 μm−2 s−1, so with limiting DIX the preferred substrate remains zinc. Similar comparisons for the murine enzyme at 3 μm PIX show kcat/Km-Znapp > 10 × 106 m−1 min−1, kcat/Km-Coapp is 10.8 × 106 m−1 min−1, kcat/Km-Feapp is 8.3 × 106 m−1 min−1, and kcat/Km-Niapp is 1.2 × 106 m−1 min−1; whereas for the yeast enzyme kcat/Km-Znapp is 121 × 106 m−1 min−1, kcat/Km-Coapp is 55 × 106 m−1 min−1, kcat/Km-Feapp is 8.3 × 106 m−1 min−1, and kcat/Km-Niapp is 3.6 × 106 m−1 min−1 (3). Although it appears reasonable that the substrate inhibition seen with metals other than iron acts to increase the specificity for iron (3), this is only correct if binding to the inhibitory site changes the kcat/Kmapp ratio. In the case of uncompetitive substrate inhibition,
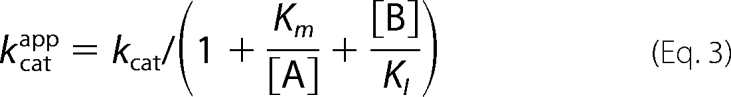 |
and as
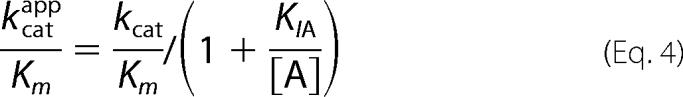 |
it is clear that this inhibition cannot affect the distribution of metalloporphyrin products. The inhibition may still be of physiological benefit because it will reduce the amount of inappropriate metalloporphyrins formed, although at a cost of reducing the amount of heme formed. So, in the presence of high concentrations of zinc we would expect the production of both heme and zinc protoporphyrin to be substantially reduced.
In all of these cases, there is no substantial specificity for iron over zinc; this is not, however, the observed biological specificity where zinc porphyrins are abnormal (5). The physiological distribution of products also depends on the relative concentration of the two substrates, and it is reasonable that iron delivery systems act to increase the local iron concentration (24, 25) or change the specificity ratio on binding to ferrochelatase.
It is noticeable that zinc is an effective inhibitor of the reaction as well as being a substrate. The primary plots (Fig. 1) show that the zinc inhibition is promoted by the porphyrin substrate. This argues that zinc binds to the EZnDIX complex to inhibit (Scheme 1) and that Zn2+ is the second substrate (32). This behavior requires that significant amounts of the EZnDIX complex exist in the steady state and therefore that ZnDIX disassociation is partially rate-limiting. This result is thus in agreement with previous transient kinetic studies using iron (16) and zinc (33, 34) as metal ion substrates. This conclusion can be contrasted to previous work with the bovine enzyme (35) where steady-state inhibition studies using both product and alternative substrate demonstrated an ordered mechanism where iron bound first. As discussed below, the alternative detergents used in this study could modify the observed behavior.
SCHEME 1.

A number of potential metal ion binding sites in ferrochelatase have been proposed (for a recent summary, see reference 3). As a consequence, it is difficult to make a reasonable proposal to explain how binding an additional metal ion impedes product release from the enzyme-product complex in the absence of a structure of the enzyme-product-inhibiting metal complex. Nevertheless, the substrate inhibition described here requires an additional zinc binding site, in contrast to the inhibition that arises from poor release of product after chelation of inhibitory metals (7).
The inhibition by zinc is, however, dependent on the concentration of detergent present in the assay. A number of explanations present themselves for this change in observed kinetic behavior. Most seriously, this change could arise if the order of substrate addition changes with detergent concentration. Although this may appear unlikely, it is possible if porphyrin release from detergent is slow compared with porphyrin uptake by ferrochelatase. So, increasing the detergent concentration disfavors the pathway shown in Scheme 1, and the enzyme proceeds through a substantially slower path where zinc binds first. Alternatively, detergent binding to the protein could prevent Zn2+ binding to the EZnDIX complex. Finally, most simply, inhibition by zinc could be reduced by reducing the concentration of the EZnDIX species; this could easily happen if increasing detergent concentration also increased the porphyrin Km and thus reduced the concentration of enzyme-product complex at a given set of substrate concentrations. The relative concentration of the EZnDIX complex could also be reduced if cholate preferentially solubilizes the metallated porphyrin product. This effect is not detergent-specific because CHAPS is also capable of relieving the zinc inhibition (Fig. 4). Although CHAPS is, like cholate, a bile salt-derived detergent, it is zwitterionic rather than anionic, and it forms rather larger micelles (36).
Adding cholate has a significant effect on the kinetic parameters. In particular, kcat/KmDIX decreases substantially on increasing the cholate concentration to 0.5% (w/v). In the simplest ordered mechanism, where there is no isomerization of either the free enzyme or the EDIX complex, this parameter reflects the rate constant for porphyrin binding. The drop in kcat/Km can either be attributed to cholate binding the porphyrin and making it less accessible to the active site or cholate binding in the enzyme active site and making it less accessible to porphyrin. Because crystallographic data demonstrate that cholate can bind in the active site of ferrochelatase (8), we prefer the second explanation of this observation. Additionally, kcat/KmZn decreases 10-fold on increasing the cholate concentration. In both cases, it remains on the order of 106 to 107 m−1 s−1; this is approaching diffusion values, suggesting that there is no slow conformational change before metal ion chelation.
In conclusion, we have shown that ferrochelatase shows kinetics consistent with an ordered mechanism under relatively low detergent concentrations, and changing the detergent environment has a noticeable effect on the behavior of ferrochelatase. Nonphysiological metals or metals associated with abnormal physiology have multiple binding modes. In the mechanism outlined here, the inhibitory binding cannot be associated with a change in specificity, but it can result in an overall inhibition of the enzyme; reducing the amount of the incorrect metalloporphyrin formed albeit at the cost of also reducing heme formation.
Acknowledgment
We thank Prof. Harry A. Dailey (University of Georgia, Athens) for the ferrochelatase expression construct.
This work was supported by the Biotechnology and Biological Sciences Research Council, United Kingdom.
- EZnDIX
- enzyme-zinc deuteroporphyrin complex
- DIX
- deuteroporphyrin IX
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]- 1-propanesulfonate
- ZnDIX
- zinc-deuteroporphyrin.
REFERENCES
- 1.Dailey H. A., Dailey T. A. (2003) in The Porphyrin Handbook (Kadish K. M., Smith K. M., Guildard R. eds), pp. 87–114, Academic Press, New York [Google Scholar]
- 2.Al-Karadaghi S., Franco R., Hansson M., Shelnutt J. A., Isaya G., Ferreira G. C. (2006) Trends Biochem. Sci. 31, 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter G. A., Sampson M. P., Ferreira G. C. (2008) J. Biol. Chem. 283, 23685–23691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson M., Hederstedt L. (1994) Eur. J. Biochem. 220, 201–208 [DOI] [PubMed] [Google Scholar]
- 5.Lamola A. A., Yamane T. (1974) Science 186, 936–938 [DOI] [PubMed] [Google Scholar]
- 6.Lamola A. A., Joselow M., Yamane T. (1975) Clin. Chem. 21, 93–97 [PubMed] [Google Scholar]
- 7.Medlock A. E., Carter M., Dailey T. A., Dailey H. A., Lanzilotta W. N. (2009) J. Mol. Biol. 393, 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C. K., Dailey H. A., Rose J. P., Burden A., Sellers V. M., Wang B. C. (2001) Nat. Struct. Biol. 8, 156–160 [DOI] [PubMed] [Google Scholar]
- 9.Al-Karadaghi S., Hansson M., Nikonov S., Jansson B., Hederstedt L. (1997) Structure 5, 1501–1510 [DOI] [PubMed] [Google Scholar]
- 10.Lecerof D., Fodje M. N., Alvarez León R., Olsson U., Hansson A., Sigfridsson E., Ryde U., Hansson M., Al-Karadaghi S. (2003) J. Biol. Inorg. Chem. 8, 452–458 [DOI] [PubMed] [Google Scholar]
- 11.Karlberg T., Lecerof D., Gora M., Silvegren G., Labbe-Bois R., Hansson M., Al-Karadaghi S. (2002) Biochemistry 41, 13499–13506 [DOI] [PubMed] [Google Scholar]
- 12.Karlberg T., Hansson M. D., Yengo R. K., Johansson R., Thorvaldsen H. O., Ferreira G. C., Hansson M., Al-Karadaghi S. (2008) J. Mol. Biol. 378, 1074–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lecerof D., Fodje M., Hansson A., Hansson M., Al-Karadaghi S. (2000) J. Mol. Biol. 297, 221–232 [DOI] [PubMed] [Google Scholar]
- 14.Medlock A., Swartz L., Dailey T. A., Dailey H. A., Lanzilotta W. N. (2007) Proc. Natl. Acad. Sci. U.S.A 104, 1789–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medlock A. E., Dailey T. A., Ross T. A., Dailey H. A., Lanzilotta W. N. (2007) J. Mol. Biol. 373, 1006–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoggins M., Dailey H. A., Hunter C. N., Reid J. D. (2007) Biochemistry 46, 8121–8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burden A. E., Wu C., Dailey T. A., Busch J. L., Dhawan I. K., Rose J. P., Wang B., Dailey H. A. (1999) Biochim. Biophys. Acta 1435, 191–197 [DOI] [PubMed] [Google Scholar]
- 18.Porra R. J., Vitols K. S., Labbe R. F., Newton N. A. (1967) Biochem. J. 104, 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sellers V. M., Wu C.-K., Dailey T. A., Dailey H. A. (2001) Biochemistry 40, 9821–9827 [DOI] [PubMed] [Google Scholar]
- 20.Falk J. E. (1964) Porphyrins and Metalloporphyrins, p. 236, Elsevier, London [Google Scholar]
- 21.Goldin B. R., Little H. N. (1969) Biochim. Biophys. Acta 171, 321–332 [DOI] [PubMed] [Google Scholar]
- 22.Abbas A., Labbe-Bois R. (1993) J. Biol. Chem. 268, 8541–8546 [PubMed] [Google Scholar]
- 23.Hansson M. D., Lindstam M., Hansson M. (2006) J. Biol. Inorg. Chem. 11, 325–333 [DOI] [PubMed] [Google Scholar]
- 24.Lange H., Kispal G., Lill R. (1999) J. Biol. Chem. 274, 18989–18996 [DOI] [PubMed] [Google Scholar]
- 25.Shaw G. C., Cope J. J., Li L., Corson K., Hersey C., Ackermann G. E., Gwynn B., Lambert A. J., Wingert R. A., Traver D., Trede N. S., Barut B. A., Zhou Y., Minet E., Donovan A., Brownlie A., Balzan R., Weiss M. J., Peters L. L., Kaplan J., Zon L. I., Paw B. H. (2006) Nature 440, 96–100 [DOI] [PubMed] [Google Scholar]
- 26.Fersht A. (1977) Enzyme Structure and Mechanism, W. H. Freeman, San Francisco [Google Scholar]
- 27.Cornish-Bowden A. (1993) Biochem. J. 291, 323–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engel P. C. (1993) Biochem. J. 291, 324–325 [Google Scholar]
- 29.Barnsley E. A. (1993) Biochem. J. 291, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel P. C. (1992) Biochem. J. 284, 604–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornish-Bowden A. (1984) J. Theor. Biol. 108, 451–457 [DOI] [PubMed] [Google Scholar]
- 32.Cook P. F., Cleland W. W. (2007) Enzyme Kinetics and Mechanism, p. 175, Garland Science, London [Google Scholar]
- 33.Franco R., Pereira A. S., Tavares P., Mangravita A., Barber M. J., Moura I., Ferreira G. C. (2001) Biochem. J. 356, 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Z., Ferreira G. C. (2006) Biochem. J. 399, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dailey H. A., Fleming J. E. (1983) J. Biol. Chem. 258, 11453–11459 [PubMed] [Google Scholar]
- 36.Neugebauer J. M. (1990) Methods Enzymol. 182, 239–253 [DOI] [PubMed] [Google Scholar]