

Abstract
Histamine regulates functions via four receptors (HRH1, HRH2, HRH3, and HRH4). The d-myo-inositol 1,4,5-trisphosphate (IP3)/Ca2+/protein kinase C (PKC)/mitogen-activated protein kinase pathway regulates cholangiocarcinoma growth. We evaluated the role of HRH3 in the regulation of cholangiocarcinoma growth. Expression of HRH3 in intrahepatic and extrahepatic cell lines, normal cholangiocytes, and human tissue arrays was measured. In Mz-ChA-1 cells stimulated with (R)-(α)-(−)-methylhistamine dihydrobromide (RAMH), we measured (a) cell growth, (b) IP3 and cyclic AMP levels, and (c) phosphorylation of PKC and mitogen-activated protein kinase isoforms. Localization of PKCα was visualized by immunofluorescence in cell smears and immunoblotting for PKCα in cytosol and membrane fractions. Following knockdown of PKCα, Mz-ChA-1 cells were stimulated with RAMH before evaluating cell growth and extracellular signal–regulated kinase (ERK)-1/2 phosphorylation. In vivo experiments were done in BALB/c nude mice. Mice were treated with saline or RAMH for 44 days and tumor volume was measured. Tumors were excised and evaluated for proliferation, apoptosis, and expression of PKCα, vascular endothelial growth factor (VEGF)-A, VEGF-C, VEGF receptor 2, and VEGF receptor 3. HRH3 expression was found in all cells. RAMH inhibited the growth of cholangiocarcinoma cells. RAMH increased IP3 levels and PKCα phosphorylation and decreased ERK1/2 phosphorylation. RAMH induced a shift in the localization of PKCα expression from the cytosolic domain into the membrane region of Mz-ChA-1 cells. Silencing of PKCα prevented RAMH inhibition of Mz-ChA-1 cell growth and ablated RAMH effects on ERK1/2 phosphorylation. In vivo, RAMH decreased tumor growth and expression of VEGF and its receptors; PKCα expression was increased. RAMH inhibits cholangiocarcinoma growth by PKCα-dependent ERK1/2 dephosphorylation. Modulation of PKCα by histamine receptors may be important in regulating cholangiocarcinoma growth.
Introduction
Cholangiocarcinoma is the second most common type of cancer in the liver after hepatocellular carcinoma (1). Biliary tract cancers are challenging to diagnose and treat (1, 2). Chronic inflammation and obstruction of bile ducts may play an important role in the progression of this deadly disease (3). Cholangiocarcinoma, once diagnosed, is commonly treated by surgical resection; however, this option is not always viable (1).
Histamine acts by interacting with four G-protein–coupled receptors, HRH1, HRH2, HRH3, and HRH4 (4-7); these four G-protein–coupled receptors exert actions on numerous G-proteins (8). We have shown that the HRH3 agonist, (R)-(α)-(−)-methylhistamine dihydrobromide (RAMH), decreases hyperplastic cholangiocyte growth in bile duct–ligated cholestatic rats by inhibition of cyclic AMP (cAMP)–dependent signaling (6). The actions of RAMH likely occur by activation of the G-coupled protein, GαI, which inhibits adenylyl cyclase and consequently cAMP activation (9, 10); however, the H3 histamine receptor can signal through Gαo (11). We have shown that HRH1 acts on Gαq to mobilize Ca2+, thus increasing the proliferation of small mouse cholangiocytes by activation of the d-myo-inositol 1,4,5-trisphosphate (IP3)/calmodulin-dependent protein kinase I/cAMP-responsive element binding protein–dependent signaling pathway (7). Histamine receptors have also been shown to be involved in the regulation of numerous cancers (12).
Protein kinase C (PKC) is a broad family of protein kinases made up of approximately 10 to 12 isozymes (13). Conventional isoforms of the PKC family, including PKCα, PKCβI, PKCβII, and PKCγ (13), require calcium, diacylglycerol, and a phospholipid-like phosphatidylcholine for activation. These conventional PKCs act through the same signal transduction pathway as phospholipase C (PLC; ref. 13). PKC is involved in tumor growth (14) and represents a tool for therapeutic strategies in cancer progression. PKCα activation has been shown to be required for activation of extracellular signal–regulated kinase (ERK)-1/2 signaling in human lung cancer cells (15).
Without current successful therapies for the treatment of cholangiocarcinoma, the purpose of this research is to evaluate the role of the H3 histamine receptor in the regulation of cholangiocarcinoma growth.
Results
Receptor Expression
By immunofluorescence, all the cell lines express HRH3; specific immunoreactivity is shown in red, and the cell nuclei are counterstained with 4′,6-diamidino-2-phenylindole (Fig. 1A, blue). Real-time PCR analyses revealed that HRH3 mRNA expression [expressed as ratio to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA] increased in Mz-ChA-1, SG231, and CCLP-1 compared with H69 cells (Fig. 1B). Immunohistochemical analysis of human liver biopsy samples (using commercially available tissue arrays) showed that there was increased HRH3 immunoreactivity in cholangiocarcinoma samples compared with control (Fig. 1C). Evaluation of HRH1, HRH2, and HRH4 by real-time PCR revealed that all cell types express these receptors (data not shown).
Figure 1.
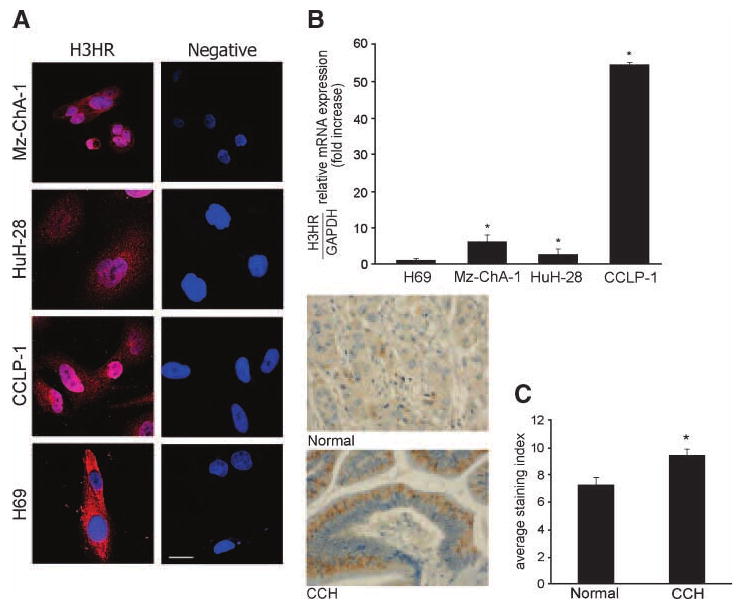
The expression of HRH3 was measured by immunofluorescence (A), real-time PCR (B), and tissue array analysis (C). A. By immunofluorescence, all the cholangiocarcinoma cell lines used as well as H69 expressed HRH3; specific immunoreactivity is shown in red, and the cell nuclei are counterstained with 4′,6-diamidino-2-phenylindole (blue). Bar, 50 μm. B. By real-time PCR, the HRH3 message was increased in cholangiocarcinoma cells compared with normal cells. Columns, mean of three experiments; bars, SEM. *, P < 0.05, versus HRH3 mRNA expression of H69 cells. C. HRH3 immunoreactivity was increased in cholangiocarcinoma samples compared with control. Original magnification, ×40. Columns, mean of three experiments; bars, SEM. *, P < 0.01, versus HRH3 expression of normal biopsy samples.
Evaluation of Cholangiocarcinoma Growth
By MTS assay, stimulation with RAMH (5-200 μmol/L) inhibited the growth of Mz-ChA-1, HuH-28, and CCLP-1 cells at 24 and 48 hours (Fig. 2A-C). RAMH did not change the growth of H69 cells (data not shown). 1,2-Bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid, tetraacetoxymethyl ester (BAPTA/AM), Gö6976, and U73122 blocked the inhibitory effect of RAMH on Mz-ChA-1 cell growth (Fig. 2D), but alone did not change Mz-ChA-1 cell growth (data not shown). Treatment of Mz-ChA-1 cells with the specific agonists for HRH1, HRH2, or HRH4 showed that HRH1 and HRH2 had slight proliferative effects on Mz-ChA-1 growth, whereas the HRH4 agonist decreases growth (data not shown).
Figure 2.
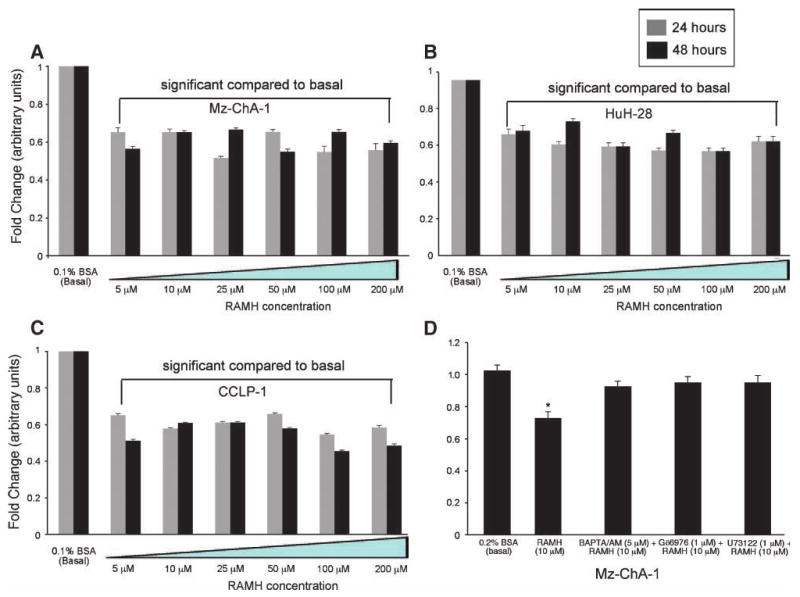
RAMH inhibited Mz-ChA-1 (A), HuH-28 (B), and CCLP-1 (C) growth at 24 and 48 h compared with their corresponding basal levels. Columns, mean of 12 experiments; bars, SEM. *, P < 0.05, versus the corresponding basal value. D. MTS assays in Mz-ChA-1 cells stimulated with 0.1% BSA or RAMH (10 μmol/L with 0.1% BSA at 24 h) in the absence or presence of preincubation with BAPTA/AM (5 μmol/L), Gö6976 (1 μmol/L), or U73122 (1 μmol/L). Pretreatment with BAPTA/AM, Gö6976, and U73122 prevented RAMH-induced inhibition of Mz-ChA-1 growth. *, P < 0.001, versus the corresponding basal value. Columns, mean of 12 experiments; bars, SEM.
Effect of RAMH on Intracellular IP3 and cAMP Levels
As RAMH was shown to decrease cholangiocarcinoma growth at varying concentrations, we chose to use a similar dose (10 μmol/L) that we have previously used to inhibit the growth of hyperplastic cholangiocytes (6). RAMH (10 μmol/L) significantly (P < 0.05) increased intracellular IP3 levels of Mz-ChA-1 cells compared with their corresponding basal levels [0.98 ± 0.026 (basal) versus 1.82 ± 0.002 (RAMH) pmol/1 × 106 cells], but not cAMP levels [144.48 ± 5.6 (basal) versus 153.48 ± 5.7 (RAMH) fmol/1 × 105 cells]. These results strengthen the notion that in cholangiocarcinoma cells, the H3 histamine receptor signals through Gαo, resulting in activation of a PLC/IP3/Ca2+–dependent signaling pathway independent of cAMP activity.
Transduction Pathway Evaluation
RAMH (10 μmol/L) significantly increased the phosphorylation of PKCα (Fig. 3A), but not of PKCβI, PKCβII, and PKCγ (data not shown), in Mz-ChA-1 cells compared with their corresponding basal values. Total PKCα was similar in the two treatment groups (Fig. 3A). PKCα is a serine/threonine kinase and can be activated by numerous tyrosine residues (16). The serine 643 residue seems to be the autophosphorylation site, and several tyrosine sites compose the catalytic domain, which are crucial for activation (16). The antibody that we used detects PKCα phosphorylation on the serine 657 residue (17). By immunofluorescence, there was distinct positive staining for PKCα in bovine serum albumin (BSA)–stimulated (i.e., unstimulated) Mz-ChA-1 cells localized in the cytoplasm of these cells (Fig. 3B). After RAMH stimulation, there was translocation of PKCα from the cytosolic region into the membrane domain of the cells (Fig. 3B). This finding was further confirmed by immunoblotting for cytosolic and membrane fractions that were extracted (using the Qiagen Qproteome Cell Compartment kit) from Mz-ChA-1 cells stimulated in the absence or presence of RAMH (10 μmol/L for 15 minutes). Figure 3C clearly shows that PKCα expression is increased in the membrane fraction after RAMH treatment, compared with the higher basal expression seen in the cytosol fraction. To confirm that we were successful with the fractionation, we performed immunoblotting for GAPDH and Bcl2 (18). With the finding that RAMH induced the phosphorylation/translocation of PKCα, we sought to pinpoint the role of PKCα activity in Mz-ChA-1 growth using siRNA transfection. In PKCα siRNA–transfected cells (∼90% knockdown efficiency; Fig. 3D, top) treated with RAMH, there was a loss of the inhibitory effects of RAMH on Mz-ChA-1 growth compared with scrambled siRNA– or control-transfected cells (Fig. 3D, bottom). However, in mock-transfected cells, RAMH (10 μmol/L) decreased the proliferation (similar to levels seen in Fig. 2A) of Mz-ChA-1 cells compared with mock-transfected, basal cells (Fig. 3D, bottom).
Figure 3.
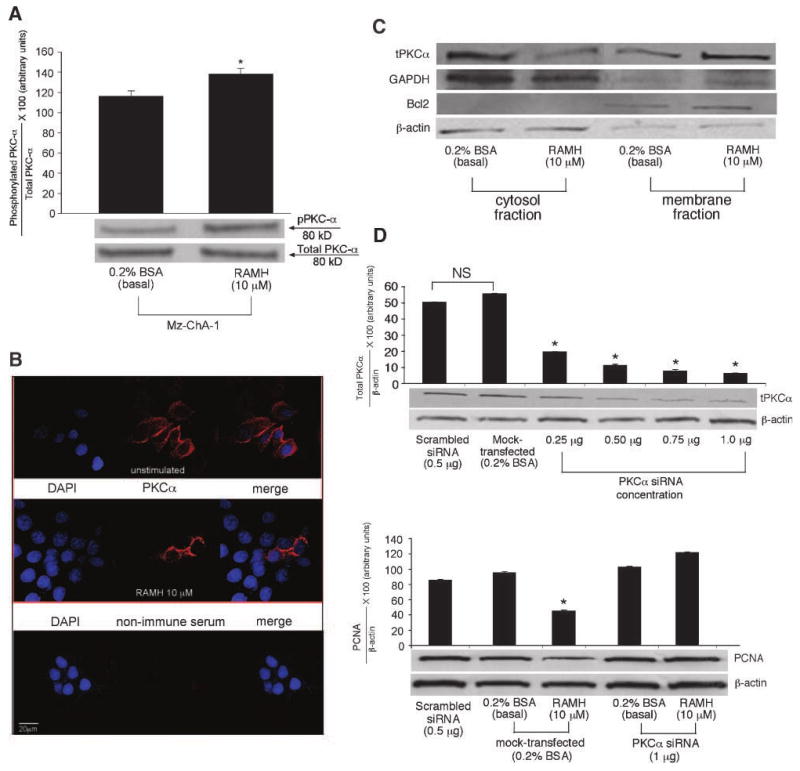
A. Phosphorylation of PKCα was significantly increased in Mz-ChA-1 cells treated with RAMH compared with the corresponding basal values. *, P < 0.05, versus the corresponding basal value. Columns, mean of six blots; bars, SEM. B. PKCα immunolocalization was shown using immunofluorescence. In unstimulated Mz-ChA-1 cells, there is an increased expression of PKCα in the cytosolic region; however, after treatment with RAMH (15 min), there is a shift in expression that seems to be stronger in the membrane region in Mz-ChA-1 cells, suggesting translocation of PKCα from the cytosol into the membrane region after RAMH stimulation. Red, PKCα expression; blue, nuclear counterstain [4′,6-diamidino-2-phenylindole (DAPI)]. Nonimmune serum was used as a negative control. Original magnification, ×60. C. Centrifugal fractionation for cytosol and membrane compartments shows that RAMH (10 μmol/L, 15 min) increases the expression of total PKCα in the membrane compartment compared with basal-treated cell lysates. A representative blot is shown for GAPDH (indicating the cytosolic fraction), Bcl2 (a control for the membrane fraction), total PKCα, and β-actin (for proper protein loading determination). D. Top, siRNA silencing for PKCα. Using increasing amounts of siRNA PKCα (0.25-1 μg), there was a significant reduction in PKCα expression as seen by PKCα immunoblotting compared with scrambled siRNA and control (0.2% BSA). A representative blot is shown normalized with β-actin. Bottom, knockdown of PKCα using siRNA (1 μg DNA) resulted in the loss of the inhibitory effects of RAMH (10 μmol/L) on Mz-ChA-1 cell growth. Mock-transfected cells stimulated with RAMH (10 μmol/L) inhibited the growth of Mz-ChA-1 cells compared with mock-transfected, BSA-treated cells. A representative blot is shown normalized with β-actin. *, P < 0.05, versus the corresponding basal value. Columns, mean of six experiments; bars, SEM.
RAMH had no effect on the phosphorylation of protein kinase A (PKA), p38, and c-jun NH2-terminal kinase (data not shown). However, RAMH decreased the phosphorylation of the mitogen-activated protein kinase (MAPK), ERK1/2, compared with its basal value (Fig. 4). Silencing of PKCα (by siRNA transfection; Fig. 3D, top) ablated the effects of RAMH on ERK1/2 phosphorylation (Fig. 4), further confirming the regulatory role of PKCα in RAMH-induced manipulation of cholangiocarcinoma. Total levels for ERK1/2 (Fig. 4) were unchanged between basal and RAMH-stimulated cells.
Figure 4.
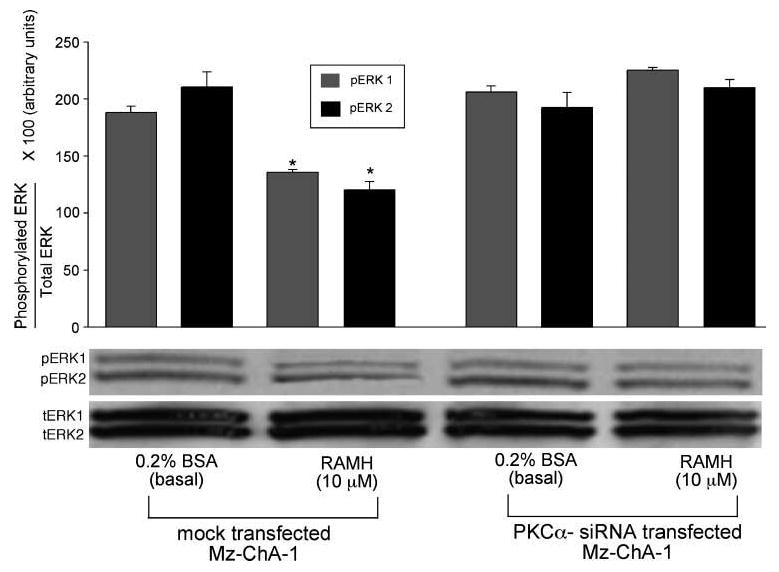
Phosphorylation of ERK1/2 was inhibited by RAMH (10 μmol/L) treatment in mock-transfected Mz-ChA-1 cells compared with basal, mock-transfected cells. After PKCα silencing, there was a loss of RAMH-induced ERK1/2 inhibition compared with basal, PKCα-transfected cells. A representative blot is shown. *, P < 0.05, versus the corresponding basal value. Columns, mean of six blots; bars, SEM.
Nude Mouse Evaluation
Chronic treatment with RAMH had a significant inhibitory effect on cholangiocarcinoma growth in vivo. After 44 days, tumor growth remained relatively unchanged in nude mice treated with RAMH compared with the vehicle group that increased ∼100% [151 ± 8.9 mm3 to 295.94 ± 15.36 mm3 (days 1-44, vehicle) versus 130.65 ± 12.5 mm3 to 135.06 ± 11.21 mm3 (days 1-44, RAMH)]. Representative pictures from treated mice and a summary graph of each data point are seen in Fig. 5A. There was no significant difference between final body weight, liver weight, and liver-to-body-weight ratio between vehicle- and RAMH-treated mice (data not shown). Histologically, no changes were seen in fibrosis or inflammation in tumors from either RAMH- or vehicle-treated mice (data not shown); however, necrosis was significantly upregulated in RAMH-treated tumors [20.17 ± 1.84 (vehicle) versus 36.88 ± 4.92 (RAMH)] compared with tumors from vehicle-treated mice. The number of proliferating cell nuclear antigen (PCNA)–positive cells was decreased in RAMH-treated animals compared with vehicle-treated mice (Fig. 5B, top), whereas apoptosis (evaluated by caspase-3 protein expression) increased in tumors from RAMH-treated compared with vehicle-treated mice (Fig. 5B, bottom). Compared with in vitro data for total PKCα that showed no significant difference in basal versus RAMH-stimulated cells (Fig. 3A), we found that total PKCα expression was significantly increased in tumors from RAMH-treated mice compared with vehicle (Fig. 5C). This result is presumably due to the chronic exposure of the tumor cells to RAMH compared with the acute effects seen in vitro. Expression of vascular endothelial growth factor (VEGF)-A, VEGF-C, VEGF receptor (VEGFR)-2, and VEGFR-3 was downregulated in tumor samples from RAMH-treated animals compared with vehicle-treated mice (Fig. 5D). Quantitative data for all immunohistochemistry are found in Table 1. Evaluation of other organs showed no histologic changes in RAMH-treated animals compared with vehicle (data not shown), indicating that the dosage of RAMH was not toxic to other organ systems.
Figure 5.
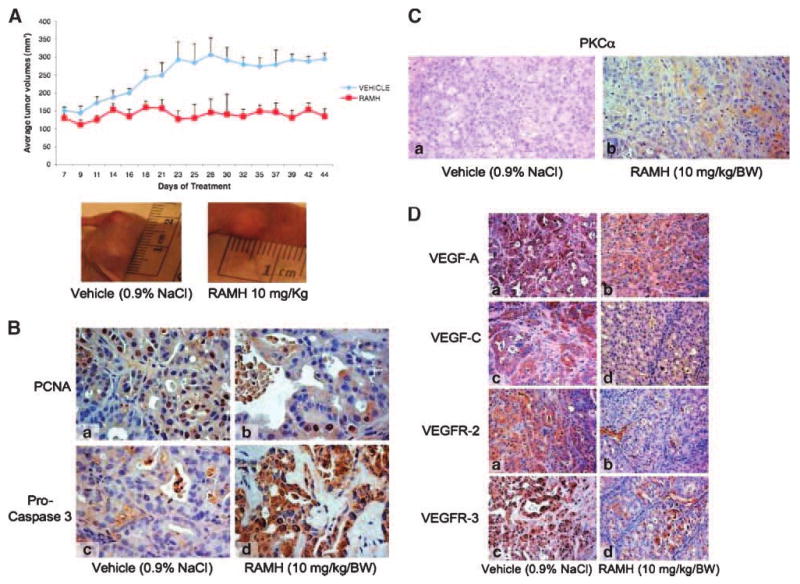
A. RAMH (10 mg/kg/bw) stunts tumor growth compared with vehicle (0.9% NaCl)–treated mice. Data shown is average tumor values from four mice (eight tumors in total) for each treatment group. Representative images for vehicle- and RAMH-treated tumors are shown. Points, mean tumor size; bars, SEM. B. RAMH induces a decrease in the number of PCNA-positive cells coupled with an increase in the number of apoptotic cells compared with vehicle treatment. Original magnification, ×40. a and c, vehicle; b and d, RAMH. C. PKCα expression increased in tumors from RAMH-treated mice compared with tumors from vehicle-treated mice. Original magnification, ×20. a, vehicle (0.9% NaCl); b, RAMH (10 mg/kg/bw). D. RAMH decreased the expression of VEGF-A, VEGF-C, VEGFR-2, and VEGFR-3 compared with vehicle-treated mice. Original magnification, ×20. a and c vehicle; b and d, RAMH. See Table 1 for quantitative data.
Table 1. Quantitative Data for the Expression of PCNA, Cleaved Caspase-3, PKCα, VEGF-A, VEGF-C, VEGFR-2, and VEGFR-3 in Nude Mice Treated with Vehicle (0.9% NaCl) or RAMH (10 mg/kg/bw).
| Parameter | Vehicle (0.9% NaCl) | RAMH (10 mg/kg/bw) |
|---|---|---|
| PCNA | 33.11 ± 3.29 | 13.14 ± 1.49* |
| Cleaved caspase-3 | 21.80 ± 2.6 | 41.4 ± 2.71* |
| PKCα | 2.60 ± 0.98 | 26.80 ± 2.52* |
| VEGF-A | 74.2 ± 2.92 | 63.4 ± 3.25* |
| VEGF-C | 53.00 ± 3.44 | 9.83 ± 1.40* |
| VEGFR-2 | 66.33 ± 1.67 | 11.00 ± 3.30* |
| VEGFR-3 | 77.60 ± 1.50 | 30.40 ± 3.26* |
NOTE: Values are mean ± SEM of four mice per group.
P < 0.05 versus vehicle.
Discussion
In this study, we have shown that activation of HRH3 by RAMH decreases both in vitro and in vivo cholangiocarcinoma growth via IP3/Ca2+/PKCα–dependent dephosphorylation of ERK1/2. In vitro, we showed that (a) HRH3 are upregulated in cholangiocarcinoma cells and in human tissue arrays compared with normal cells and tissues; (b) RAMH inhibits the growth of cholangiocarcinoma growth by increased levels of IP3; further, RAMH induces translocation and enhanced phosphorylation of PKCα, which leads to a dephosphorylation of ERK1/2; and (c) PKCα knockdown prevents RAMH inhibition of cholangiocarcinoma growth and ERK1/2 phosphorylation. In vivo, we showed that (a) RAMH inhibits the growth of Mz-ChA-1 cells implanted in nude mice coupled with enhanced apoptosis, and (b) RAMH inhibition of cholangiocarcinoma growth was associated with enhanced expression of PKCα and reduced expression of VEGF-A, VEGF-C, VEGFR-2, and VEGFR-3.
Our present studies correlate with our previous study showing that RAMH decreases the growth of hyperplastic cholangiocytes (6). However, whereas RAMH (acting via Gαi) inhibited hyperplastic biliary growth by inhibition of cAMP-dependent PKA/ERK1/2/Elk-1 (6), in the present study, RAMH (acting through Gαo) decreased cholangiocarcinoma growth by PLC/IP3/Ca2+/PKCα–dependent inhibition of ERK1/2 phosphorylation, which is independent of cAMP activation. It is known that histamine exerts its effects on cells via activation of the four histamine receptors that couple to diverse G-proteins (4, 5). Our studies involving hyperplastic (6) along with the current study show the ability of HRH3 to preferentially couple to different G-proteins. In cardiac sympathetic nerves, Levi et al. (19) reported a novel, signaling pathway for the H3 histamine receptor. In this study, they found that HRH3 exerted its actions via both the GαI-mediated inhibition of adenylyl cyclase-cAMP-PKA and the Gβγ-mediated activation of a MAPK-dependent pathway, specifically the MAPK-phospholipase A2-cyclooxygenase-prostaglandin E2-prostaglandin EP3 receptor pathway (19). These studies offer evidence that the histamine receptors, including the H3 receptor, are able to diversify their signaling effects in different cell systems.
To dissect the role of HRH3 in the regulation of cholangiocarcinoma growth, we evaluated the expression of this receptor in malignant and normal cell lines. Expression and overexpression of histamine and histamine receptors have been seen in other tissues and cell types (12, 20). HRH3 and HRH4 are upregulated in human mammary tissue and carcinomas compared with normal samples (12). Further, in colon carcinoma cells, HRH1, HRH2, and HRH4 were found to be present (20). We have previously shown that (a) HRH3 is found in cholangiocytes from normal and bile duct–ligated rats (6); and (b) small and large mouse cholangiocytes express HRH1, HRH2, HRH3, and HRH4 (7). However, in small cholangiocytes, the HRH1 agonist was the only receptor agonist to induce changes in proliferation (7). These studies are important in understanding the differential effects of histamine (via the four receptors) in the proliferative capacity of different cell types, including cholangiocarcinoma. Here, the HRH1 and HRH2 agonists elicit stimulatory effects in our cholangiocarcinoma cells, whereas the HRH4 agonist clobenpropit reduced cell growth (data not shown), showing an innate ability for histamine to regulate its effects by activating specific receptors when warranted.
Differential regulation of histamine receptors is found in many cell types including Leydig cells where there is an inhibitory (via HRH1) and a stimulatory (via HRH3) effect on steroidogenesis (21). We have found both a stimulatory (by HRH1; ref. 7) and an inhibitory (by HRH3; ref. 6) response in cholangiocyte growth.
The PKC isozymes are important regulators of proliferation, differentiation, angiogenesis, and apoptosis in a number of cells including cholangiocytes (22-25). In our study, we have shown that PKCα plays a key role in RAMH inhibition of cholangiocarcinoma growth because (a) this HRH3 agonist induces activation of the PLC/Ca2+/IP3/PKCα signaling pathway; (b) RAMH inhibitory action is dependent on this signaling pathway; (c) RAMH induces activation and translocation of PKCα; and (d) gene silencing of PKCα prevents the inhibitory effects induced by RAMH including phosphorylation of ERK1/2. PKCα is important in the regulatory mechanisms of the growth of many cancers including cholangiocarcinoma (14, 22, 24). A recent study has shown that activation of PKCα plays an important role in the cytotoxicity and mutagenicity of human lung cancer cell lines by changes in Raf-1-MAPK kinase 1/2-ERK1/2 signaling (15). Inhibition of the human hepatoma cells was also found to be dependent on PKCα activation, which was found to signal via the ERK pathway (26). In addition, tauroursodeoxycholic acid inhibits human cholangiocarcinoma growth via Ca2+-PKCα–dependent dephosphorylation of ERK1/2 (22). These results are consistent with other studies showing that PKCα is a key player in cancer growth regulation whether by activation of PKCα or by deactivation of this important protein (25).
Our next experiments were aimed to evaluate the chronic effects of RAMH on tumor growth. Using an established protocol (27), we showed that, in comparison with vehicle-treated mice, chronic treatment with RAMH (a) decreased tumor growth and increased apoptosis, (b) significantly increased PKCα protein expression, and (c) decreased the expression of the angiogenic factors (VEGF-A and VEGF-C; ref. 28) and their receptors (VEGFR-2 and VEGFR-3). VEGF proteins and their receptors are notorious partners in crime with regard to regulation of cancer growth (29, 30). Upregulation of these factors has been seen in numerous human cancers including breast (29) and colorectal (30) cancers. We propose that inhibition of tumor growth by RAMH is also due to decreased expression of the trophic factors, VEGF-A and VEGF-C and their receptors, which are important for the vascularization, development, and growth of neoplasias (31). Compounds that are able to inhibit the expression of VEGF (and its receptors) are important in decreasing the growth of cancer (27). The growth of human breast cancer cells was inhibited by treatment with epigallocatechin-3-gallate, which was coupled with decreased VEGF expression (32). In hepatocellular carcinoma, rapamycin decreases tumor growth and angiogenesis via downregulation of both hypoxia-inducible factor 1α and VEGF (33). These studies are among just a few that have come to light in recent years showing the importance of VEGF in cancer growth and the implications of identifying factors that have the ability to downregulate VEGF expression.
In summary, we have shown that the specific HRH3 agonist RAMH inhibits the growth of cholangiocarcinoma cell lines and tumors implanted into nude mice. This decrease in cholangiocarcinoma growth by RAMH is associated with increased levels of IP3, translocation of PKCα, and IP3/Ca2+–dependent dephosphorylation of ERK1/2. Further, RAMH increased PKCα expression and significantly decreased the expression of VEGF (and its receptors) in vivo. These studies further emphasize the importance of histamine in cholangiocyte regulation via activation of the four receptors, whether it is hyperplastic or neoplastic cholangiocyte growth.
Whereas novel treatments for cholangiocarcinoma are actively being sought out, our study shows that curative therapy may begin with the prevention of tumor growth and by blocking the components that aid in the maintenance of the blood supply that feeds the tumor. A compound such as the H3 histamine receptor agonist may offer new hope as a therapeutic strategy for patients facing a dismal prognosis.
Materials and Methods
Materials
High-quality reagents were obtained from Sigma unless indicated otherwise. Antibodies for siRNA, immunoblotting, immunohistochemistry, and immunofluorescence were purchased from Santa Cruz Biotechnology unless indicated otherwise. The RIA kits for the measurement of intracellular cAMP (125I Biotrak Assay System, RPA509) and IP3 (3H Biotrak Assay System, TRK1000) levels were purchased from GE Healthcare. Primers and other reagents for real-time PCR were obtained from SABiosciences.
Cell Culture
We used a number of intrahepatic and extrahepatic cholangiocarcinoma cell lines along with nonmalignant, immortalized cholangiocytes (H69). Mz-ChA-1 cells from human gallbladder (24, 34) were a gift of Dr. G. Fitz (University of Texas Southwestern Medical Center, Dallas, TX). HuH-28 cells (Cancer Cell Repository, Tohoku University) from human intrahepatic bile duct (35) were maintained as described (24). CCLP-1 (ref. 36; from intrahepatic bile ducts; from Dr. A.J. Demetris, University of Pittsburg, Pittsburg, PA) were cultured as described (36-38). The nonmalignant cholangiocyte cell line, H69 (from Dr. G.J. Gores, Mayo Clinic, Rochester, MN), was cultured as described (39).
Receptor Expression
We evaluated HRH3 receptor expression in all cell lines by (a) immunofluorescence (7, 40), (b) real-time PCR in total RNA (1 μg; refs. 7, 40), and (c) immunohistochemistry (41) in commercially available Accumax tissue arrays. We also evaluated the expression of HRH1, HRH2, and HRH4 by real-time PCR (7, 40) in total RNA from the selected cell lines.
Immunofluorescence
For immunofluorescence, cells were seeded on coverslips in a six-well plate (500,000 per well) and allowed to adhere overnight. Immunofluorescence was done as described (40) using the HRH3 receptor antibody (diluted 1:50 in 1% BSA/PBST or nonimmune serum). Images were visualized using an Olympus IX-71 confocal microscope.
Real-time PCR
To evaluate the expression of HRH1, HRH2, HRH3, and HRH4 mRNA in total RNA (1 μg) from cholangiocarcinoma cell lines and H69 cells, we used the RT2 Real-time assay from SABiosciences (6, 7). Human primers (SABiosciences) were designed for the histamine receptor subtypes and GAPDH, the housekeeping gene (7, 40). A ΔΔCT (delta threshold cycle) analysis was done using the H69 cell line as the control sample.
Human Tissue Array
Immunoreactivity for HRH3 was evaluated in Accumax tissue arrays by immunohistochemistry (41) using a specific antibody for HRH3 (6). These tissue arrays contain 48 well-characterized human cholangiocarcinoma biopsy samples from a variety of tumor differentiation grades as well as 4 control liver biopsy samples. Semiquantitative analysis was done as described (41).
Evaluation of Cholangiocarcinoma Growth
MTS Assays
Cells were seeded into 96-well plates and stimulated for 24 and 48 h with the HRH3 agonist, RAMH (ref. 6; 5-200 μmol/L). In separate experiments, we evaluated the effect of histamine trifluoromethyl toluidide (HTMT dimaleate; HRH1 agonist, 10 μmol/L; ref. 7), amthamine dihydrobromide (HRH2 agonist, 10 μmol/L; ref. 42), or clobenpropit (HRH4 agonist, 10 μmol/L; ref. 43) on cholangiocarcinoma growth for 48 h. Cholangiocarcinoma growth was evaluated by the CellTiter 96 AQueous One Solution Cell Proliferation Assay (6, 27, 40). Absorbance was measured at 490 nm on a micro-plate spectrophotometer (Versamax, Molecular Devices). Data were expressed as the fold change of treated cells as compared with vehicle-treated controls.
To evaluate the intracellular mechanisms by which RAMH regulates cholangiocarcinoma growth, Mz-ChA-1 cells were stimulated with 0.1% BSA (basal) or RAMH (10 μmol/L with 0.1% BSA, 24 h; ref. 6) in the absence or presence of the following inhibitors: BAPTA/AM (an intracellular Ca2+ chelator, 5 μmol/L; ref. 24), Gö6976 (a PKCα inhibitor, 1 μmol/L; ref. 28), and U73122 (a PLC inhibitor; 1 μmol/L; ref. 6). Growth of Mz-ChA-1 cells was measured by MTS assay (6, 27, 40).
Effect of RAMH on Intracellular IP3 and cAMP Levels
To evaluate the transduction pathway activated by the HRH3 agonist, we measured intracellular IP3 (7) and cAMP (6, 44) levels in Mz-ChA-1 cells stimulated with 0.1% BSA or RAMH (10 μmol/L with 0.1% BSA). Following trypsinization, Mz-ChA-1 cells were incubated at 37°C for 1 h (22, 45) and subsequently stimulated with 0.1% BSA (basal) or RAMH (10 μmol/L with 0.1% BSA) for 5 min before determining intracellular cAMP and IP3 levels by RIA (22, 23, 27, 45).
Effect of RAMH on the Phosphorylation of PKA, PKC, and MAPK Isoforms
Mz-ChA-1 cells were stimulated with 0.1% BSA or RAMH (10 μmol/L with 0.1% BSA) for 2 h at 37°C. Following stimulation, we evaluated the expression of the phosphorylated form (expressed as ratio to the corresponding total protein expression) of PKA; Ca2+-dependent PKCα, PKCβI, PKCβII, and PKCγ; and the MAPKs, ERK1/2, c-jun NH2-terminal kinase, and p38, by immunoblots (7). The amount of protein loaded was normalized by immunoblots for α-tubulin (6). Band intensity was determined by scanning video densitometry using the phospho-imager Storm 860 (GE Healthcare) and the Image-Quant TL software, version 2003.02 (GE Healthcare).
Changes in PKCα Translocation Induced by RAMH
To detect membrane translocation, Mz-ChA-1 cells were plated, stimulated with 0.1% BSA (basal) or RAMH (10 μmol/L with 0.1% BSA) for 15 min, and processed for immunofluorescence (7, 40) as described above using the antibody for total PKCα. Negative controls were done with the use of preimmune serum instead of the respective primary antibody. Slides were visualized using an Olympus IX-71 inverted confocal microscope. To further evaluate the localization of PKCα in Mz-ChA-1 cytosol and membrane fractions, we performed an extraction for these compartments using the Qiagen Qproteome Cell Compartment kit followed by immunoblotting for total PKCα (as described above). Mz-ChA-1 cells were plated and brought to confluency; 5 × 106 cells were used per treatment [0.2% BSA (basal) or RAMH (10 μmol/L with 0.1% BSA)] for 15 min. The extraction was done using these cellular lysates according to the instructions provided by the vendor. Specifically, sequential addition of different buffers to the cell pellets was followed by incubation and centrifugation (at 4°C) at varying speeds allowing for the isolation of different cellular compartments. The first buffer breaks down the plasma membrane without solubilizing it, resulting in cytosolic protein isolation. The remaining compartmentalized organelles and plasma membrane are left intact and pelleted by centrifugation. After the addition of the second buffer that solubilizes the plasma membrane, centrifugation allows for the collection of membrane proteins (18). After fractionation, we performed immunoblotting for GAPDH (localized in the cytosol of liver) and Bcl2 (localized in the membrane region) to confirm separation (18). Immunoblotting (6) was then done for total PKCα; blots were stripped and normalized to β-actin to validate proper protein loading. Blots were visualized as described above.
Effect of PKCα Silencing on RAMH Modulation of Mz-ChA-1 Cell Growth
To evaluate the effects of PKCα on cholangiocarcinoma growth, we used siRNA to knock down the expression of PKCα and evaluated cholangiocarcinoma growth by PCNA protein expression and the phosphorylation of ERK1/2 by immunoblotting analysis (6, 23). Mz-ChA-1 cells were plated into six-well plates and allowed to adhere overnight. siRNA transfection (0.25-1 μg of PKCα siRNA was used) was done according to the instructions provided by Santa Cruz Biotechnology. Diluted siRNA duplexes were added to the cells and allowed to incubate for 5 h at 37°C in a CO2 incubator. For controls, a scrambled siRNA duplex (obtained from Santa Cruz Biotechnology) was added to corresponding wells. The extent of PKCα silencing was evaluated by measuring the expression of total PKCα in transfected versus control Mz-ChA-1 cells by immunoblotting for PKCα (6, 7). PCNA and phosphorylated ERK1/2 protein expression was evaluated in Mz-ChA-1 cells stimulated with 0.1% BSA or RAMH (10 μmol/L with 0.1% BSA) for 24 h in the absence or presence of PKCα siRNA (1 μg).
Effect of RAMH on Cholangiocarcinoma Tumor Implanted in Nude Mice
Treatment Schedule
Eight-week-old male BALB/c nude (nu/nu) mice (∼30 grams in weight; Taconic Farms) were kept in a temperature-controlled environment (20-22°C) with a 12-h light-dark cycle with access to drinking water and standard chow. Mz-ChA-1 cells (3 × 106) were suspended in 0.5 mL of extracellular matrix gel and injected s.c. in the flanks of these animals (27). The first group of mice (n = 4; control) received 0.9% NaCl injections i.p. (150 μL), whereas the second group (n = 4) received RAMH (10 mg/kg body weight in NaCl; 150 μL; ref. 46), injected i.p., for 44 d. The injections were done every other day starting from “day 0,” when the tumors were implanted, for 44 d. Tumor parameters were measured twice a week by an electronic caliper, and volume was determined as follows: tumor volume (mm3) = 0.5 × [length (mm) × width (mm) × height (mm)]. The measurements started from the 3rd week, day 23, when the tumor mass was well established. After 44 d, mice were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and tissues harvested. All experiments were conducted under the guidelines of the Scott & White and Texas A&M Health Science Center Institutional Animal Care and Use Committee. Along with tumor excision, samples for the heart, liver, and kidney were removed for evaluation of organ damage.
Morphologic Analysis of Tumor Tissues
Tumor samples were excised from the flanks of the mice, fixed in 10% buffered formalin for 2 to 4 h, and embedded in low-temperature fusion paraffin, and 4-μm sections were stained with H&E (for evaluation of necrosis and inflammation) and Masson's trichrome (for evaluation of fibrosis). Immunohistochemistry was done as described (28) using primary antibodies for cytokeratin-7, PCNA, cleaved caspase-3, total PKCα, and VEGF-A and VEGF-C (two trophic factors regulating cholangiocyte growth; ref. 28) and their receptors, VEGFR-2 and VEGFR-3. For all immunoreactions, negative controls were also included. Light microscopy and immunohistochemistry observations were taken using a BX-51 light microscope (Olympus) with a video-cam (Spot Insight, Diagnostic Instrument, Inc.) and evaluated with an Image Analysis System (Delta Sistemi). Quantitation was assessed in six slides for each group. Positive cells were counted in six nonoverlapping fields for each slide, and the data expressed as percentage of positive cells.
Statistical Analysis
All data are expressed as mean ± SEM. Differences between groups were analyzed by the Student unpaired t test when two groups were analyzed and by ANOVA when more than two groups were analyzed, followed by an appropriate post hoc test. P value of <0.05 was used to indicate statistical significance.
Acknowledgments
We thank the Texas A&M Health Science Center Microscopy Imaging Center for their assistance with the confocal imaging, and Taylor Francis for her technical assistance.
Grant support: Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White; a VA Research Scholar Award; a VA Merit Award; NIH grants DK58411, DK062975, and DK076898 (G. Alpini); University funds (P. Onori); PRIN 2007 and Federate Athenaeum funds from University of Rome “La Sapienza” (E. Gaudio); and NIH K01 grant award DK078532 (S. DeMorrow).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 2.Blechacz BR, Gores GJ. Cholangiocarcinoma. Clin Liver Dis. 2008;12:131–50. doi: 10.1016/j.cld.2007.11.003. ix. [DOI] [PubMed] [Google Scholar]
- 3.Khan SA, Toledano MB, Taylor-Robinson SD. Epidemiology, risk factors, and pathogenesis of cholangiocarcinoma. HPB (Oxford) 2008;10:77–82. doi: 10.1080/13651820801992641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen T, Shapiro DA, George SR, et al. Discovery of a novel member of the histamine receptor family. Mol Pharmacol. 2001;59:427–33. doi: 10.1124/mol.59.3.427. [DOI] [PubMed] [Google Scholar]
- 5.Repka-Ramirez MS. New concepts of histamine receptors and actions. Curr Allergy Asthma Rep. 2003;3:227–31. doi: 10.1007/s11882-003-0044-3. [DOI] [PubMed] [Google Scholar]
- 6.Francis H, Franchitto A, Ueno Y, et al. H3 histamine receptor agonist inhibits biliary growth of BDL rats by downregulation of the cAMP-dependent PKA/ERK1/2/ELK-1 pathway. Lab Invest. 2007;87:473–87. doi: 10.1038/labinvest.3700533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francis H, Glaser S, DeMorrow S, et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CAMK I/CREB pathway. Am J Physiol Cell Physiol. 2008;295:C499–513. doi: 10.1152/ajpcell.00369.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hill SJ, Ganellin CR, Timmerman H, et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–78. [PubMed] [Google Scholar]
- 9.Liu C, Ma X, Jiang X, et al. Cloning and pharmacological characterization of a fourth histamine receptor (H(4)) expressed in bone marrow. Mol Pharmacol. 2001;59:420–6. doi: 10.1124/mol.59.3.420. [DOI] [PubMed] [Google Scholar]
- 10.Lovenberg TW, Roland BL, Wilson SJ, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol. 1999;55:1101–7. [PubMed] [Google Scholar]
- 11.Endou M, Poli E, Levi R. Histamine H3-receptor signaling in the heart: possible involvement of Gi/Go proteins and N-type Ca2+ channels. J Pharmacol Exp Ther. 1994;269:221–9. [PubMed] [Google Scholar]
- 12.Medina V, Croci M, Crescenti E, et al. The role of histamine in human mammary carcinogenesis: H3 and H4 receptors as potential therapeutic targets for breast cancer treatment. Cancer Biol Ther. 2008;7:28–35. doi: 10.4161/cbt.7.1.5123. [DOI] [PubMed] [Google Scholar]
- 13.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Rayes BF, Ali S, Philip PA, Sarkar FH. Protein kinase C: a target for therapy in pancreatic cancer. Pancreas. 2008;36:346–52. doi: 10.1097/MPA.0b013e31815ceaf7. [DOI] [PubMed] [Google Scholar]
- 15.Wang CY, Lin YW, Yang JL. Activation of protein kinase Cα signaling prevents cytotoxicity and mutagenicity following lead acetate in CL3 human lung cancer cells. Toxicology. 2008;250:55–61. doi: 10.1016/j.tox.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Takai Y, Kishimoto A, Iwasa Y, Kawahara Y, Mori T, Nishizuka Y. Calcium-dependent activation of a multifunctional protein kinase by membrane phospholipids. J Biol Chem. 1979;254:3692–5. [PubMed] [Google Scholar]
- 17.Kikkawa U, Takai Y, Tanaka Y, Miyake R, Nishizuka Y. Protein kinase C as a possible receptor protein of tumor-promoting phorbol esters. J Biol Chem. 1983;258:11442–5. [PubMed] [Google Scholar]
- 18.Lerner M, Corcoran M, Cepeda D, et al. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol Biol Cell. 2007;18:1670–82. doi: 10.1091/mbc.E06-03-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levi R, Seyedi N, Schaefer U, et al. Histamine H3-receptor signaling in cardiac sympathetic nerves: identification of a novel MAPK-PLA2-COX-PGE2-3R pathway. Biochem Pharmacol. 2007;73:1146–56. doi: 10.1016/j.bcp.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boer K, Helinger E, Helinger A, et al. Decreased expression of histamine H1 and H4 receptors suggests disturbance of local regulation in human colorectal tumours by histamine. Eur J Cell Biol. 2008;87:227–36. doi: 10.1016/j.ejcb.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Khan UW, Rai U. Differential effects of histamine on Leydig cell and testicular macrophage activities in wall lizards: precise role of H1/H2 receptor subtypes. J Endocrinol. 2007;194:441–8. doi: 10.1677/JOE-06-0225. [DOI] [PubMed] [Google Scholar]
- 22.Alpini G, Kanno N, Phinizy JL, et al. Tauroursodeoxycholate inhibits human cholangiocarcinoma growth via Ca2+-, PKC–, and MAPK-dependent pathways. Am J Physiol Gastrointest Liver Physiol. 2004;286:G973–82. doi: 10.1152/ajpgi.00270.2003. [DOI] [PubMed] [Google Scholar]
- 23.Glaser S, Benedetti A, Marucci L, et al. Gastrin inhibits cholangiocyte growth in bile duct-ligated rats by interaction with cholecystokinin-B/gastrin receptors via d-myo-inositol 1,4,5-triphosphate-, Ca2+-, and protein kinase Cα-dependent mechanisms. Hepatology. 2000;32:17–25. doi: 10.1053/jhep.2000.8265. [DOI] [PubMed] [Google Scholar]
- 24.Kanno N, Glaser S, Chowdhury U, et al. Gastrin inhibits cholangiocarcinoma growth through increased apoptosis by activation of Ca2+-dependent protein kinase C-α. J Hepatol. 2001:284–91. doi: 10.1016/s0168-8278(00)00025-8. [DOI] [PubMed] [Google Scholar]
- 25.Ali AS, Ali S, El-Rayes BF, Philip PA, Sarkar FH. Exploitation of protein kinase C: a useful target for cancer therapy. Cancer Treat Rev. 2009;35:1–8. doi: 10.1016/j.ctrv.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Wen-Sheng W, Jun-Ming H. Activation of protein kinase C α is required for TPA-triggered ERK (MAPK) signaling and growth inhibition of human hepatoma cell HepG2. J Biomed Sci. 2005;12:289–96. doi: 10.1007/s11373-005-1210-5. [DOI] [PubMed] [Google Scholar]
- 27.Fava G, Marucci L, Glaser S, et al. γ-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005;65:11437–46. doi: 10.1158/0008-5472.CAN-05-1470. [DOI] [PubMed] [Google Scholar]
- 28.Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–82. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 29.Timoshenko AV, Chakraborty C, Wagner GF, Lala PK. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br J Cancer. 2006;94:1154–63. doi: 10.1038/sj.bjc.6603067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawinkels LJ, Zuidwijk K, Verspaget HW, et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur J Cancer. 2008;44:1904–13. doi: 10.1016/j.ejca.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 31.Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–9. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sen T, Moulik S, Dutta A, et al. Multifunctional effect of epigallocatechin-3-gallate (EGCG) in downregulation of gelatinase-A (MMP-2) in human breast cancer cell line MCF-7. Life Sci. 2009;84:194–204. doi: 10.1016/j.lfs.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Jia WD, Xu GL, et al. Antitumoral activity of rapamycin mediated through inhibition of HIF-1α and VEGF in hepatocellular carcinoma. Dig Dis Sci. 2009;54:2128–36. doi: 10.1007/s10620-008-0605-3. [DOI] [PubMed] [Google Scholar]
- 34.Knuth A, Gabbert H, Dippold W, et al. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol. 1985;1:579–96. doi: 10.1016/s0168-8278(85)80002-7. [DOI] [PubMed] [Google Scholar]
- 35.Kusaka Y, Muraoka A, Tokiwa T, Sato J. Establishment and characterization of a human cholangiocellular carcinoma cell line. Hum Cell. 1988;1:92–4. [PubMed] [Google Scholar]
- 36.Shimizu Y, Demetris AJ, Gollin SM, et al. Two new human cholangiocarcinoma cell lines and their cytogenetics and responses to growth factors, hormones, cytokines or immunologic effector cells. Int J Cancer. 1992;52:252–60. doi: 10.1002/ijc.2910520217. [DOI] [PubMed] [Google Scholar]
- 37.Storto PD, Saidman SL, Demetris AJ, Letessier E, Whiteside TL, Gollin SM. Chromosomal breakpoints in cholangiocarcinoma cell lines. Genes Chromosomes Cancer. 1990;2:300–10. doi: 10.1002/gcc.2870020408. [DOI] [PubMed] [Google Scholar]
- 38.Miyagiwa M, Ichida T, Tokiwa T, Sato J, Sasaki H. A new human cholangiocellular carcinoma cell line (HuCC-T1) producing carbohydrate antigen 19/9 in serum-free medium. In Vitro Cell Dev Biol. 1989;25:503–10. doi: 10.1007/BF02623562. [DOI] [PubMed] [Google Scholar]
- 39.Grubman SA, Perrone RD, Lee DW, et al. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol Gastrointest Liver Physiol. 1994;266:G1060–70. doi: 10.1152/ajpgi.1994.266.6.G1060. [DOI] [PubMed] [Google Scholar]
- 40.DeMorrow S, Glaser S, Francis H, et al. Opposing actions of endocannabinoids on cholangiocarcinoma growth: recruitment of Fas and Fas ligand to lipid rafts. J Biol Chem. 2007;282:13098–113. doi: 10.1074/jbc.M608238200. [DOI] [PubMed] [Google Scholar]
- 41.Alpini G, Invernizzi P, Gaudio E, et al. Serotonin metabolism is dysregulated in cholangiocarcinoma, which has implications for tumor growth. Cancer Res. 2008:9184–93. doi: 10.1158/0008-5472.CAN-08-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shayo C, Fernandez N, Legnazzi BL, et al. Histamine H2 receptor desensitization: involvement of a select array of G protein-coupled receptor kinases. Mol Pharmacol. 2001;60:1049–56. doi: 10.1124/mol.60.5.1049. [DOI] [PubMed] [Google Scholar]
- 43.Baumer W, Wendorff S, Gutzmer R, et al. Histamine H4 receptors modulate dendritic cell migration through skin-immunomodulatory role of histamine. Allergy. 2008;63:1387–94. doi: 10.1111/j.1398-9995.2008.01720.x. [DOI] [PubMed] [Google Scholar]
- 44.Kato A, Gores GJ, LaRusso NF. Secretin stimulates exocytosis in isolated bile duct epithelial cells by a cyclic AMP-mediated mechanism. J Biol Chem. 1992;267:15523–9. [PubMed] [Google Scholar]
- 45.Kanno N, LeSage G, Phinizy JL, Glaser S, Francis H, Alpini G. Stimulation of α2-adrenergic receptor inhibits cholangiocarcinoma growth through modulation of Raf-1 and B-Raf activities. Hepatology. 2002;35:1329–40. doi: 10.1053/jhep.2002.33330. [DOI] [PubMed] [Google Scholar]
- 46.Girard P, Pansart Y, Coppe MC, Verniers D, Gillardin JM. Role of the histamine system in nefopam-induced antinociception in mice. Eur J Pharmacol. 2004;503:63–9. doi: 10.1016/j.ejphar.2004.09.030. [DOI] [PubMed] [Google Scholar]