

Abstract
The MUC1 glycoprotein is considered a tumor antigen due to its over-expression and aberrant glycosylation in cancer tissues. The latter results in appearance of new antigenic tumor-specific glycopeptides not found on normal glycoforms of the mucin. MUC1 glycopeptides can be presented by APCs on MHC class II molecules to activate glycopeptide-specific helper T-cells. No study has yet reported presentation of MUC1 glycopeptides on MHC class I molecules as stimulators of cytotoxic T-cells. In this study we show that human immuno- proteasomes and cathepsin-L can generate octa- to undecameric glycopeptides from the MUC1 repeat domain in vitro. We identified glycosylated fragments of which the decameric glycopeptide SAP10 [SAPDT(GalNAc)RPAPG] containing a single sugar binds with comparable strength to the MHC class I allele HLA-A*0201 as predicted high-score binding epitopes of the tandem repeat. The same sequence glycosylated with the disaccharide Gal-GalNAc does not bind. The glycan on SAP10 is predicted by molecular modeling to either protrude out or point into the MHC groove. SAPDTRPAPG peptide and the respective glycopeptide stimulated cytotoxic T-cells in vitro. Our findings suggest that MUC1 tandem repeat glycopeptides are capable of activating both helper and cytotoxic T-cells and thus represent good candidates for further development as vaccines.
Keywords: Cancer vaccine, MHC class I, MUC1, immunoproteasome, cathepsin-L
1. Introduction
The human mucin MUC1, a heavily O-glycosylated transmembrane glycoprotein, is a promising immune target in immunotherapeutic anti-cancer strategies. Strikingly, MUC1 glycopeptide-based vaccines that were tested in human MUC1 transgenic mice for their potential to break tolerance successfully induced B-cell responses to glycosylated MUC1 epitopes (Sorensen et al, 2006). Previous studies showed that the most immunogenic part of MUC1 is the highly O-glycosylated “variable number of tandem repeats” (VNTR) domain. Cancer cell-derived MUC1 VNTR peptides carry more densely clustered, but shorter glycans than VNTR peptides from normal cells (Müller et al., 1999; Müller et al. 1997). This structural difference could be recognized by glycopeptide-specific helper T cells (Vlad et al., 2002), but MUC1 glycopeptide-specific cytotoxic T cells (CTL) have not been reported. Yet, it appears possible for T cell receptors on CD8+ T cells to recognize glycopeptides. Xu et al. (Xu et al., 2004), showed that peptides with high affinity binding to MHC-class I and carrying the T- (Gal-GalNAc) and Tn- (GalNAc) antigens can elicit a CTL response directed to the combination of the sugar and the peptide sequence. Furthermore, Glithero et al. (Glithero et al., 1999) showed that MHC class I molecules bind viral glycopeptides with the glycans protruding out of the binding groove. Recently, Tn- glycopeptides of MUC1 outside of the VNTR domain were shown to elicit effective cytotoxic responses in HLA-A*0201 transgenic mice (Stepensky et al., 2006). However, the elicited CTLs did not induce lysis of human MUC1-expressing murine tumors, which was probably due to low abundance or differently glycosylated epitopes on the mouse cells.
We thus examined the MUC1 VNTR domain for 8- to11-meric glycopeptides that can result from processing in the class I pathway and would efficiently bind to MHC molecules, and stimulate glycopeptide-specific CTL. We analysed different MUC1 VNTR glycopeptides as substrates for APCs’ immunoproteasomes and cathepsin-L in vitro and compared the specificity of the in vitro system to a cellular assay. Processing products were analysed for their binding properties to MHC class I HLA-A*0201 and DC and stimulation of human cytotoxic T cells. The orientation of the glycan in the MHC groove of candidate peptides was analysed by molecular modelling.
2. Materials and Methods
2.1 Cells
The murine dendritic cell line DC2.4, obtained from the American Type Culture Collection, was grown in DMEM supplemented with 10% FCS, penicillin/streptomycin (200 IU/ml-200 μg/ml), 1% nonessential amino acids and 50 μM 2-mercaptoethanol at 37°C and 5% CO2. For propagation and analysis, cells were detached by 10 min incubation in 2mM EDTA/PBS, followed by centrifugation on 180×g/5min. In all experimental procedures followed by mass spectrometric analyses, the cultivation of cells was carried out in media without phenol red. The T2 cell line is a negative mutant for “Transporter associated with antigen processing” (TAP protein), expressing an empty HLA-A0201 allele of the MHC class I molecule on the cell surface. The cell line was a kind gift from Prof. Jonathan Howard and Dr. Michael Knittler, University of Cologne, and was grown at 37°C and 7.5% CO2 in IMDM media supplemented with 10% FCS and penicillin/streptomycin (200 IU/ml-200 μg/ml). For the MHC class I stabilization experiments, T2 cells were slowly adapted to reduced serum conditions (2% FCS). For T-cell assays T2 cell line was kindly provided by Prof. Walter J. Storkus, University of Pittsburgh (PA, USA) and grown in RPMI1640 medium supplemented with 10% FBS, 2mM L-glutamine, 1% nonessential amino acids, 100IU/mL penicillin and 100μg/mL streptomycin at 37°C and 6% CO2.
2.2 Peptides and glycopeptides
The MUC1 glycopeptides were chemically synthesized and kindly provided by Prof. Hans Paulsen (GP1-8, refer to table 1; SGGP1-5, refer to figure 1; and F1-F3). The P1 (GVT100) and P2 (ESR61) peptides were synthesized at the University of Pittsburgh Peptide Synthesis Facility and in vitro glycosylated with GalNAc using purified polypeptide GalNAc-transferases T1 and T2 (kindly provided by Dr. Henrik Clausen, School of Dentistry, University of Copenhagen, Denmark) and a previously published protocol (Hanisch et al., 2001). Other non-glycosylated peptides were ordered from Mimotopes/Perbio and glycosylated in vitro. TAP25 was synthesized in a local facility at the Institute of Biochemistry, Cologne, Germany.
Table 1.
Peptides and glycopeptides produced by proteasomal processing of VNTR sequences.
| Name | Sequence | Number of fragments | Average fragment length | MHC fitting fragments (%) | Glycosylated MHC fitting fragments (%) | ||
|---|---|---|---|---|---|---|---|
| PEPTIDE | P1 | (GVTSAPDTRPAPGSTAPPAH)x5 | 105 | 25,2 | 13 | 0 | |
| P2 | A(HGVTSAPESRPAPGSTAPPA)x3 | 105 | 17,5 | 26 | 0 | ||
| P3 | AHGVTSAPDTRPAPGSTAPPA | 29 | 10,8 | 52 | 0 | ||
| P4 | AHGVTSAPESRPAPGSTAPPA | 31 | 11,6 | 50 | 0 | ||
| GLYCOPEPTIDE | MONOSACCHARIDE | GP2 | AHGVTSAPDTRPAPGSTAPPA | 63 | 11,7 | 55 | 18 |
| GP3 | AHGVTSAPDTRPAPGSTAPPA | 33 | 10,9 | 15 | 98 | ||
| GP4 | AHGVTSAPDTRPAPGSTAPPA | 47 | 12,9 | 48 | 45 | ||
| GP5 | AHGVTSAPESRPAPGSTAPPA | 46 | 11,6 | 79 | 26 | ||
| GP6 | AHGVTSAPESRPAPGSTAPPA | 46 | 10,6 | 35 | 96 | ||
| GP7 | AHGVTSAPESRPAPGSTAPPA | 61 | 11,7 | 29 | 39 | ||
| GP8 | AHGVTSAPESRPAPGSTAPPA | 46 | 10,5 | 35 | 21 | ||
Non-glycosylated and glycosylated peptides with AHG starting motif were processed by immunoproteasomes, and analyzed by reverse-phase HPLC and MALDI mass spectrometry followed by identification and quantification of fragments [8]. Relative amounts of potentially MHC class I fitting octa-to undecapeptides were calculated and expressed relative to the total amount of digestion products generated by immunoproteasomal cleavage of the respective substrate. The percentage of glycosylated 8- to 11-meric fragments was calculated relative to the fraction of total 8- to 11-mers.
Fig. 1. In vitro generation of 10-/11-meric (glyco)peptides from MUC1 tandem repeats by human cathepsin-L.


A,The 100-meric P1 peptide was digested with human cathepsin-L for 24h and the proteolytic fragments were analyzed after HPLC by MALDI-TOF-MS. Major decapeptides STA10 and SAP10 were detected in the digest. B, Synthetic 21-meric sialoglycopeptides carrying one to three NeuAc2-3Gal1-3GalNAc moieties ( were digested with human cathepsin-L for 24h and the proteolytic fragments were analyzed after HPLC by MALDI-TOF-MS. Three cleavage sites (arrows) were identified within the repeat, which varied with the positions of the glycans.
were digested with human cathepsin-L for 24h and the proteolytic fragments were analyzed after HPLC by MALDI-TOF-MS. Three cleavage sites (arrows) were identified within the repeat, which varied with the positions of the glycans.
2.3 In vitro proteolysis of glycopeptides
A 1 mg/ml solution of immunoproteasomes prepared according to a published protocol (Tenzer et al., 2004) was purchased from Immatics Biotechnologies (Tübingen, Germany) and stored frozen at −80°C. Glycopeptides were incubated with 10 μg of human 20S immunoproteasomes (Immatics) for 48 h, at 37°C, in 10 μl of digestion buffer (20mM HEPES/KOH, pH 7.6 containing 2 mM Mg(CH3COO)2 and 1mM DTT) as described (Ninkovic and Hanisch, 2007). Digestion was stopped after 24 h by freezing the sample at −80°C. Human cathepsin-L was purchased from Sigma (Munich, Germany) and solubilized in 0.1M sodium acetate, pH 5.5, containing 1 mM EDTA and 1 mM dithiothreitol (Hanisch et al., 2003). About 5 mU of enzyme were added to sialoglycopeptide substrates SGGP1 to SGGP5 (75 – 100 μM) in a total volume of 20 μl. The reaction mixtures were incubated at 37°C for 24 h and 1 μl of trifluoroacetic acid was added to stop the reaction. The reaction products were cleaned by solid-phase extraction on ZipTipC18 pipette tips and 0.5 μl aliquots were applied onto the MALDI target for mass spectrometric analysis. Kinetic studies with the non-glycosylated 100-mer P1 were performed under the same conditions and the products were chromatographed on HPLC for purification and quantification.
2.4 Purification of glycopeptides by HPLC
Aliquots (50–100 μl) of the reaction mixtures or glycopeptides were run on a narrow-bore ODS Ultrasphere column (150 × 2 mm; Beckman Instruments, Munich, Germany) with flow rates of 0.3 ml/min (analytical scale) or on a Prevail C18 column (Alltech, Munich, Germany) with flow rates of 2 ml/min (preparative scale). Chromatography was performed on an HPLC system (System Gold Beckman Instruments, Munich, Germany) by gradient elution as previously described (Ninkovic and Hanisch, 2007). The photometrical detection was performed at 214 nm.
2.5 Structural analysis by mass spectrometry
Matrix-assisted laser desorption/ionization (MALDI)
MALDI analyses were performed on a Bruker–Reflex IV instrument (Brucker-Daltonics, Bremen, Germany) by positive ion detection in the reflectron mode (Ninkovic and Hanisch, 2007).
Liquid chromatography (LC)
MS/MS data were acquired on a Q-TOF2 quadruple-TOF mass spectrometer (Micromass, Manchester, United Kingdom) equipped with a Z spray source (Ninkovic and Hanisch, 2007).
2.6 Antigen processing and presentation assays
Peptide carrying poly-lactic acid based vesicles were generated by mixing two solutions: Solution A: 200 nmol of P2 peptide (1 mg) resolved in 3% poly-vinyl alcohol disolved in cold water; Solution B: 5% poly-lactic acid in a mixture of ethanol and acetone (9:1, v/v). Solution B (30 ml) was slowly added to solution A (150 ml) under constant mixing and stirring overnight at room temperature. The next day the mixture was centrifuged at 10.000×g for 10 min and washed three times with PBS (200 ml), followed by centrifugation steps as mentioned previously. The generated vesicles were resuspended in 330 μl of PBS and added to media of 4×108 DC2.4 mouse dendritic cells growing confluent in nine cell culture flasks (300cm2). The cells were grown as usual, but in bovine exosome-depleted medium for isolation of dendritic cell-derived exosomes. Exosomes are derived from endosomal invaginations of the plasmamembrane, which by a second invagination form multi-vesicular bodies. The nanovesicles thus expose MHC-complexes of the plasmamembrane, are known to elicit potent anti-tumor T cell responses and represent an easily separable source of MHC-bound peptides originating from DCs.
Cells were incubated with PLA-vesicles for 24 h at 37°C and 5% CO2. The cell culture supernatant was collected and exosomes were isolated by ultracentrifugation (see below). Isolated exosomes were resuspended in 100μl 0.1% TFA/H2O. Acidification of exosomes denatures tertiary MHC complex and releases bound peptide epitopes. The solution was shortly spun and injected on a HPLC narrow-bore ODS column for separation of eluted peptides and glycopeptides. Fractions were collected manually, dried and analyzed by MALDI-TOF-MS as described. Molecular masses of detected ions were analyzed by the FindPep software and by nanospray ESI-MS/MS analyses.
Exosomes were isolated from FCS or cell culture supernatant by subsequent centrifugation steps: 10 min/1200 ×g; 30 min/10000 ×g; 30 min/20.000 ×g; and 60 min/100.000 ×g. The exosomes were pelleted in the final centrifugation step, washed with PBS and used for further analyses. For the depletion of FCS from exosomes, exosomes from serum were pelleted by ultracentrifugation at 110.000 ×g for 1 h.
2.7 MHC stabilization assay
MHC stabilization assay is based on the increased affinity of HLA-A, -B, -C specific monoclonal W6/32 antibody for the stabilized conformation of MHC molecules after binding of a peptide-ligand. The T2 cells express empty MHC molecules with short half-life.
For MHC stabilization assay T2 cells, grown in IMDM medium supplemented with 2% FCS, were washed in PBS from the residual serum, and resuspended at 1×106/ml of IMDM supplemented with 0.1 % FCS and 25mM HEPES. Half of a million of cells were incubated in 48-well cell culture plate with 1 μg of β2-microglobulin and 100 μg of peptide for 16 h at 27°C and 8% CO2. Cells were washed with 5 ml PBS, centrifuged at 180×g for 5 min and the pellet resuspended in 10μl PBS containing 1μg of W6/32 antibody and incubated at 4°C for 30 minutes. After 3 washes in 1%BSA/PBS cells were incubated with 50μl of secondary anti-mouse Alexa 488 antibody (1:1000 in PBS with 0.1% BSA) for 30 minutes in the dark, washed with PBS (3×5 ml) and resuspended in 500μl of PBS. Binding of the W6/32 antibody to MHC class I molecules was analyzed by FACS on the Beckman Coulter ADC XL4C flow cytometer. As a positive control hepatitis B virus core antigen HBVc18–27 P6Y~C (FLPSDCFPSV) was used. As a negative control T2 cells were used, to which no peptide was added. Relative binding capacities of the peptides were expressed as the percent of the positive control signal (100%), normalized to the negative control (0%). A mean fluorescence that was at least 10% higher than the negative control level was considered as positive binding.
2.8 In vitro priming of peptide-specific CTL
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of HLA-A*0201 healthy donors by Ficoll-Hypaque (MP Biomedicals, Solon, OH) density gradient centrifugation. Dendritic cells (DCs) were generated by culturing adherent monocytes for 6 days in AIM-V medium (Invitrogen, Carlsbad, CA) supplemented with 500U/mL granulocyte macrophage colony-stimulating factor (GM-CSF, R&D, Minneapolis, MN) and 1000U/mL interleukin (IL)-4 (R&D), and then matured for 48 hours with a cocktail of cytokines consisting of 10ng/mL of each, tumor necrosis factor (TNF)-alpha, IL-6, IL-1beta (R&D), and 1μg/mL prostaglandine E2 (Sigma, St. Louis, MO). Maturity and activation of DCs was confirmed by flow cytometry (FACS LSRII, BD) using monoclonal antibodies (BD, San Diego, CA) against CD80, CD14 and CD11c. Matured DCs were pulsed for 4 hours in RPMI 1640 medium (Lonza BioWhittaker, Walkersville, MD) supplemented with 10% human AB serum (GemCell, Gemini BioProducts, Woodland, CA), 2mM L-glutamine (Mediatech cellgro, Herndon, VA, USA), 100 units/ml penicillin and 100 μg/ml streptomycin (Mediatech cellgro), and 1% nonessential amino acids (Gibco-Invitrogen), with the synthetic MUC1-derived peptides F1, F2 or F3 at the concentration of 30–50μg/106 DC. DCs pulsed with single peptides were combined, washed, resuspended and used as stimulator cells. They were added to the autologous lymphocyte fraction in 24-well Linbro plates (ICN, Aurora, OH) at a ratio of 1:10 at culture initiation and fed in RPMI 1640 media supplemented with 10% human AB serum, 10 ng/ml rhIL-6 (R&D) and 5 ng/ml rhIL-12 (PreproTech, Rocky Hill, NJ). On day 10 and thereafter every 9 days, T cells were restimulated at a ratio of 1 DC to 20 T cells, in RPMI 1640 media supplemented with 10% human AB serum, rhIL-2 and rhIL-7 (20U/ml each, R&D).
2.9 T cell assays
Antigen specificity of in vitro expanded T cells was assessed after 4 weekly stimulations with autologous DC loaded with peptides. Three days past each stimulation cell supernatant was sampled from the cultures and the amount of IFN-γ secreted measured using OptEIA human IFNγ ELISA kit (BD). CD8+ T-cells were purified using magnetic microbeads (human CD8+ T Cell Isolation Kit II from Miltenyi Biotech, Auburn, CA) according to the manufacture’s instructions and analyzed by flow cytometry for intracellular IFN-γ and TNF-α production after a final 5-hour restimulation with peptide-loaded DCs in the presence of monensin (Golgi Stop, BD) (Elson et al., 1995). CTL activity was determined 3 days after the last restimulation. T2 target cells were incubated over night at 37°C in the presence of F1 [SAPDTRPAPG], F2 [SAPDT(GalNAc)RPAPG] or F3 [SAPES(GalNAc)RPAG] peptides at 10 μg/ml concentration each. After washing, the cells were labeled with 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE) according to the manufacture’s instructions (Immunochemistry, Bloomington, MN) and distributed into a 96-well plate in duplicates at 5000 cells/well. CFSE labeled T2 cells not loaded with peptides were used as controls. Purified CD8+ T cells were added at various E:T ratios for a 6-hour incubation at 37°C. Target cell death was detected by adding 7-amino-actinomycin (7-AAD, Immunochemistry) to each well 10 min before data analysis by flow cytometry and analysis was performed according to the manufacture’s instructions (Lecoeur et al., 2001).
2.10 Molecular modelling
Molecular modelling was performed by the SYBYL 7.1 software package (Tripos, Inc., St. Louis, MO, USA). Two crystal structures of the HLA-A2/10-mer peptide complexes (PDB IDs: 1HHH and 1I4F) were used as corresponding templates for modelling of HLA/FLPSDCFPSV and HLA/SAPDTRPAPG (HLA/SAP10), either non-glycosylated or glycosylated at Thr5 with GalNAc (T5*) and Gal-GalNAc (T5**) residues. Two orientations of the sugar moieties, pointed into the groove and out of the groove, were considered for the glycosylated Thr5 of SAP10. Modelled peptides were energy-minimized within the binding groove of the HLA-A0201 molecule using the conjugate gradient algorithm (the Powell method implemented in SYBYL) to a maximum derivative of 0.05 kcal/(mol*Å).
Tripos force-field with Kollman-all-atom partial charges for the MHC molecule and Gasteiger-Marcili charges for (glyco)peptide was used in structure optimization and in subsequent FlexiDock runs. The distance-dependent dielectric of 4r was used to mimic aqueous solvent environment. The energy-minimized structures were analyzed for atomic fluctuations and hydrogen bonding pattern and were used for docking with the FlexiDock module of SYBYL. All single bonds of the (glyco)peptide ligand were considered as rotatable in FlexiDock runs, except amide and ring bonds..
3. Results
3.1 Immunoproteasomes generate glycosylated class I epitopes
Immunoproteasomes can process O-glycosylated MUC1 VNTR, if the glycans are short, neutral (non-sialylated) and located at permissive sites of the tandem repeat (Ninkovich and Hanisch, 2007). Here we addressed whether immunoproteasomes can process longer peptides from the O-glycosylated MUC1 VNTR domain into short glycopeptides that potentially bind to MHC class I.
Analysis of in vitro digests of non-glycosylated MUC1 tandem repeat substrates revealed that: 1) longer substrates (100-mer and 61-mer) on average yielded peptides of 20 amino acids with about 20% having an appropriate size to fit into the MHC class I groove (8- to 11-mers); and 2) shorter peptides (21-mers) yielded higher numbers of 8- to 11-meric peptides (50%) (Tab. 1).
O-Glycosylation at specific positions in the sequence did not change the average peptide fragment length, but did influence the quantity of 8- to 11-mers (Tab. 1). The 8- to 11-mers resulted from two predominant cleavages within the processing regions SAP and GST (Tab. 2):
AHGVTSAPD(E)T(S)RPAPGSTAPP (sequence of MUC1 repeat peptide with glycosylation sites given in bold face; the preferred cleavage sites of immunoproteasomes are underlined [8])
The position of the glycosylated residue significantly influenced processing and therefore the relative amount of glycopeptides within the group of 8- to 11-mers. Substrates of the AHG21 series carrying glycans at VTSA or GSTA (GP2 and GP5) led to abundant yet mostly unglycosylated 8- to 11-mers. In contrast to this, processing of substrates glycosylated at the central DTR (GP3 and GP6) delivered fewer 8- to 11-mers, but the amount of glycosylated peptides generated was higher. Thus, substrates glycosylated at the DTR motif were the best source of glycopeptides that would potentially bind to MHC.
3.2 Cathepsin-L proteolysis products resemble the patterns of immuno- proteasomal cleavage
Cross-presentation of extracellular MUC1domains in the class I pathway of APCs can occur as a result of endocytosis and processing by endosomal cathepsins (Chen and Jondal, 2004; Shen et al., 2004). Our previous work on MHC class II restricted MUC1 peptides revealed cathepsin-L as one of the major processing enzymes in human and mouse DCs (Hanisch et al., 2003). Here we wanted to address whether cathepsin-L is able to cleave VNTR peptides into potential class I epitopes and whether peptides glycosylated at permissive sites (DTR or GST) with sialyltrisaccharides (as found on cancer-associated mucin), would be effective substrates and yield reaction products in the appropriate size range for MHC class I binding.
The in vitro digestion of nonglycosylated VNTR 100-mer revealed 1) quantitative digestion of the substrate within 24 h (Fig. 1A); 2) formation of a complex pattern of fragments ranging in size from 7-mers to 33-mers (Fig. 1A) that compares to a more restricted pattern in the cellular processing (Hanisch et al., 2003); 3) three major and one minor cleavage sites within the repeat peptide; and 4) the SAP10 and STA10 fragments as the dominant products. Similar results were obtained with the nonglycosylated 61-mer comprised of three MUC1 repeats with variant sequences (DT > ES).
While short glycans (neutral mono- and disaccharides) are tolerated by cathepsin-L at permissive sites, we wanted to learn whether the tumor-associated sialyltrisacchraide sialyl-T would affect processing. A series of sialoglycopeptides carrying the trisaccharide NeuAc2-3Gal1-3GalNAc on various residues are listed in Fig. 1B. As we showed previously for glycopeptides with GalNAc substitution, the sialoglycopeptides with glycosylation at Thr or Ser in the VTSA motif of the tandem repeat (SGGP1 to SGGP3 and SGGP5) were not cleaved within this motif, whereas the tri-glycosylated peptide SGGP4 lacking glycans at either of the two positions was an effective substrate (Fig. 1B). SGGP1 and SGGP2 instead showed cleavage of the Gly-Ser bond in the GSTA motif. In SGGP4 the Gly-Ser linkage in the GST motif was resistant to cleavage, probably because of steric hindrance by the adjacent glycans. However, a new cleavage site was used by the enzyme in SGGP4: the Thr-Arg linkage in the DTR motif (see SGGP4 in Fig. 1B) yielding the RPA11 glycopeptide, a fragment with potential MHC class I binding capacity.
The structural identity of the proteolytic products was confirmed by ESI-MS/MS of the peptides and glycopeptides. In particular, the major cathepsin-L cleavage product with a molecular mass of m/z 1171 was demonstrated in collision-induced dissociation (CID) MS/MS experiments to be identical with SAPDT(GalNAc) RPAPG (Fig. 2).
Fig. 2. Identification of major cathepsin-L processing product as SAP10(Thr5-GalNAc) by tandem mass spectrometry.

The MS/MS (CID) spectrum of the doubly charged ion at m/z 586 (corresponding to the singly charged ion at m/z 1171) generated by in vitro cathepsin-L proteolysis of AHG21(T10-GalNAc) is shown. The molecular mass in conjunction with the oxonium ion at m/z 204 (indicating loss of a HexNAc residue), and the major y- and b-series ions registered after dissociation of the sugar confirm the identity of the cathepsin-L processing product as SAPDT(GalNAc) RPAPG.
3.3 Mouse dendritic cells simulate in vitro cleavage patterns
To validate our in vitro method, we analyzed the processing of the MUC1 VNTR peptides on the cellular level. Cross-presentation of MUC1 61-mer (P2 in Tab. 1) was tested in the mouse dendritic cell line DC2.4. The loading of poly-lactic vesicles with this VNTR peptide resulted in the generation of high amounts of MUC1-derived peptides in MHC complexes that could be identified on the peptidomic level by mass spectrometry. After incubation of mouse dendritic cells DC2.4 with poly-lactic acid vesicles (PLA) carrying the VNTR 61-mer, exosomes produced by the cells were collected by ultracentrifugation, acidified and the liberated peptides were fractionated by reverse phase HPLC. Collected fractions were dried by vacuum centrifugation and analyzed by MALDI/MS. The molecular masses of detected ions were analyzed by the FindPept software application (http://expasy.org/tools/findpept.html). The mass spectra revealed the presence of at least 18 distinct peptide species in the size range from 8- to 25-mers (Tab. 3). Eight peptides were in the class I size range (8- to 11-mers), while ten fell into the class II size range (12- to 25-mers). Among the peptides with potential class I binding activity the major species were identical to those found in the cell-free in vitro studies (APE8, APE9, APE10, STA10, SAP10, SAP11, PES9, and PES10). Their structural identity with fragments generated by immunoproteasomes in vitro clearly validates the results of cell-free processing studies.
3.4 In silico prediction of peptide binding to MHC class I allele HLA-A2
In the digests of all analyzed 21-meric glycopeptides, 117 fragments in the size range of 8 to 11 amino acid long peptides and glycopeptides were generated. To identify MHC binding glycopeptides, we performed a pre-selection based on the predicted binding affinity of the peptide. The ligation strength of non-glycosylated 8- to 11-meric sequences to MHC class I was analyzed by the SYFPEITHI MHC-epitopes predicting software (http://www.syfpeithi.de/). Only 55 peptide sequences were analyzed, however the variation due to glycosylation had to be ignored (the influence of glycans on MHC binding is unknown) as well as 11-meric sequences. Analyses were restricted to the HLA-A0201 as the most frequent MHC class I allele (Tab. 4). STA9 and STA10 were predicted as high-score binders (>20), whereas SAP10 was revealed as a low- to medium-score binder (<10), which is in conflict with results from MHC stabilization assays.
3.5 SAPDT(GalNAc)RPAPG binds HLA-A0201 while the respective disaccharide peptide does not
The predicted good binders with a potential glycosylation site were synthesized, glycosylated in vitro and tested for binding to empty HLA-A0201 molecules on T2 cells by MHC stabilization assay. Ligation strength measured by flow cytometry was compared to the positive control HLA-A2 restricted peptide FLPSDCFPSV and to non-loaded T2 cells as the negative control. Results for glycosylated peptides were compared with affinities of their non-glycosylated forms. Fig. 3 presents the relative ligation strengths of tested peptides compared with a negative control considered as 0% and positive control considered as 100%. As predicted by the SYFPEITHI software tool, the strongest binders in the MHC class I stabilization assays were predicted high-score binders STA9 and STA10. Contrasting to the predicted binding strength, SAP10 bound comparably strongly to the A2 allele as the high-score binder STA10. A strong reduction of ligation strength was caused by the introduction of GalNAc into peptides (STA9, STA10), reducing it to the level of the negative control. However, an exception represents the glycopeptide SAPDT(-GalNAc)RPAPG (SAP10-T5*), which was revealed as the only MUC1 VNTR glycopeptide binding comparably strongly in the glycosylated and non-glycosylated form. All other peptides lost their binding abilities after glycosylation or were already inactive in the non-glycosylated form.
Fig. 3. Ligation strength of MUC1 repeat peptides and glycopeptides to HLA-A0201.

Binding of peptides and glycopeptides was measured by MHC stabilization assay and expressed as percent (%) of the positive control (100%). The negative control was considered as 0%. Strongly binding peptides STA9 and STA10 lost their binding activity on glycosylation, while SAP10 glycopeptide retained it.
3.6 Monosaccharide glycosylated peptide shows mixed orientation into and out of the groove
Each model of the MHC/(glyco)peptide complex included a 10-residue (glyco)- peptide and 180 residues of the MHC heavy chain that formed the binding groove (Saper et al., 1991; Bjorkman et al., 1987). The bound peptides were mostly buried into the MHC groove and demonstrated extended conformations with slightly bulged central parts P4-P6 (not shown). The modelled complexes maintained very similar patterns of specific interactions between the side-chain atoms of MHC molecule and the main-chain atoms of the peptide near N- and C-termini that resulted in a conserved set of hydrogen bonds. All models demonstrated high similarity to the template crystal structures with root-mean-square deviations of the modelled atomic coordinates from the corresponding template that were less than 0.15 Å for the heavy atoms of the peptide backbone. The crystal structure of the HLA-A2/FLPSDFFPSV complex (1HHH) was used as a template to model the reference structure, HLA/FLPSDCFPSV, which served as a positive control in the MHC stabilization assays. The lowest binding energy after FlexiDock runs was equal to -241 kcal/mol that was used as a reference for comparison to binding of non-glycosylated or glycosylated SAP10 peptides. A crystal structure of the HLA-A2/GVYDGREHTV complex (1I4F) with native D4 and R6 residues at corresponding positions was used for the molecular modelling of HLA/SAPDTRPAPG binding. After FlexiDock runs, all generated bound peptides demonstrated very similar structural features: S1, A2, and P3 residues (refer to the one-letter code for amino acids) were located at the binding pockets A, B, and D; while P7 and G10 residues were located at the E and F binding pockets, respectively. The non-glycosylated T5 residue was positioned at the pocket C; whereas D4 and R6 were mostly exposed out of the MHC binding groove. The significant conformational variations were only observed for side-chains of the D4 and R6 residues of the docked peptide. The lowest binding energy for the HLA/SAP10 complex without sugar was higher by +3 kcal/mol than for the reference structure HLA/FLPSDCFPSV.
For modelling of the glycosylated peptides bound to MHC, the T5 residue of the HLA/SAP10 complex was converted to Thr-GalNac (T5*) or Thr-Gal-GalNAc (T5**) with torsion angles corresponding to the lowest energy conformation of the glycosylated threonine (Rubinstein et al., 2004). The only allowed orientation of Gal-GalNAc (T5**) was pointed out of the MHC groove for all docked conformers (not shown). The binding energy calculated by FlexiDock was significantly higher (+28 kcal/mol) than for the reference structure. The T5** direction into the groove was prohibited due to intermolecular clashes of the sugar moiety with the amino acid residues A69, H70, T73, and H74 of the C pocket of the HLA-A0201 molecule that resulted in significant structural perturbations of the peptide backbone and disruption of the hydrogen binding.
Both orientations of GalNAc, directed either into the groove (Fig. 4) or out of the groove, were allowed for the glycosylated T5*. The binding energy of the SAP10 with T5* directed into the groove was slightly better (-3 kcal/mol); whereas, for T5* oriented out of the groove, the lowest binding energy was higher (+8 kcal/mol) than for the reference strucure. Although the calculated relative binding energies demonstrated good correlation with the litigation strength measured by MHC stabilization assays (Fig. 3), these binding energies provide rather qualitative estimates of the (glyco)peptide binding that could serve as the relative scoring binding propensities of the potential (glyco)peptide epitopes.
Fig. 4. Model of the SAP10(T5*) glycopeptide bound into the HLA-A2 groove.

The crystal structure of the HLA-A2/GVYDGREHTV complex (1I4F) was used as a template. The GalNAc moiety of the glycosylated T5* residue is pointed into the groove. Line ribbon represents the peptide-binding MHC groove. The glycosylated peptide is shown from a side view through the α2 α-helix toward the α1 α-helix of the groove. The peptide is shown as capped sticks; the GalNAc moiety is shown as balls and sticks.
3.7 T cells are activated by SAPDT(GalNAc)RPAP
Lymphocytes from a HLA-A*0201 healthy donor were repeatedly stimulated in vitro with autologous DCs pulsed with SAPDTRPAPG (F1), SAPDT(GalNAc)RPAPG (F2) and SAPES(GalNAc)RPAPG (F3) for a period of 4 weeks. CD8+ T-cells were then enriched by negative selection using magnetic microbeads and specific immunoreactivity of the induced CTLs was tested with T2 target cells loaded with the relevant peptides or no peptides. Stimulated T-cells secreted large amounts of IFN-γ, but not TNF-α into the cell supernatant during the 4-week culture period. A peak in IFN-γ secretion (>23 ng/ml) was reached after the first restimulation with autologous peptide-loaded DCs (not shown). After the last cycle of restimulation (4 weeks of in vitro culture) 28.8% of F1-stimulated CD8+ cells, 22% of F2-induced CD8+ T-cells and 18.1% of F3-induced CD8+ T-cells produced IFN-γ, but not TNF-α (not shown). Three days after the last restimulation the induced CD8+ T-cells secreted >17 ng/ml IFN-γ, indicating an immunological response and activity. The responder cells were tested for cytotoxic activity and as shown in Fig. 5, CD8+ T-cells efficiently lysed T2 target cells pulsed with F1 or F2 peptide, whereas T2 cells loaded with F3 peptide showed only minimal increase in cell lysis above background. These results demonstrate that the induced CTL recognized F1 and F2 antigen-peptides specifically and were able to target cells presenting those peptides.
Fig. 5. MUC1 peptides F1 and F2, but not F3 can elicit specific CD8+ CTL and serve as their targets.

Bars represent different target cells and the efficiency of target cell lysis at an E:T ratio of 50:1. T2, target cells not loaded with peptides; F1, target cells loaded with peptide F1 [SAPDTRPAPG]; F2, target cells loaded with glycopeptide F2 [SAPDT(GalNAc)RPAPG]; F3, target cells loaded with glycopeptide F3 [SAPES(GalNAc)RPAPG]. Data represent the average of two experiments performed with PBMC of two different HLA-A*0201 healthy donors, in duplicate cell cultures. Error bars: SD.
4. Discussion
The glycoprotein MUC1 is of special interest for cancer immunobiology as cancer-specific glycoforms of the over-expressed mucin are found on carcinoma cells and on cancer stem cells (Engelmann et al., 2008). Many MUC1 peptide-based cancer vaccines have been designed and tested in animal models and in clinical trials. All of them were based on unglycosylated instead of glycosylated MUC1 peptide sequences. The major difference between normal MUC1 and tumor MUC1 in addition to their expression levels is that tumor MUC1 carries tumor-specific shorter glycan chains with a much higher portion of negatively charged sialic acids and thus could yield more immunogenic epitopes. In this study we addressed the question whether glycopeptides derived from human MUC1 by processing can be efficient MHC class I-binders and therefore good candidate antigens for eliciting a tumor-specific CTL response. We show for the first time for an exogenous antigen that irrespective of the cross-presentation route the antigen processing machinery can produce high amounts of short glycopeptides from MUC1 glycopeptide substrates that fit the MHC binding groove. The 21-meric MUC1 glycopeptides used in our experiments correspond to a single repeat plus one residue of the highly immunogenic MUC1 VNTR domain. MUC1 has five potential glycosylation sites per repeat, and in previous work we have shown that glycosylation density and location within the repeats influence the cleavage specificity of proteasomes (Ninkovic and Hanisch, 2007). This knowledge can be used to specifically design glycopeptides of MUC1 with permissive glycosylation sites as potential immunogens and eventual vaccine. Based on our results we propose the monoglycosylated 21-mer AHGVTSAPDTRPAPGSTAPPA carrying a GalNAc at the central Thr (DTR) for further development as a vaccine candidate. Processing of this glycopeptide by immunoproteasomes or by cathepsin-L released the 10-meric glycopeptide SAPDTRPAPG (T5-GalNAc) as a major product that strongly bound to empty MHC class I HLA-A0201 molecules, the most frequent MHC class I allele in Caucasians. Importantly, the same sequence carrying a Gal-GalNAc disaccharide at the central Thr shows only weak binding affinity in the MHC stabilisation assay. This is consistent with the idea that MHC binding is dependent on glycan structure and size. Molecular modelling of SAPDTRPAPG glycopeptide in the MHC groove using two templates (1HHH and 1I4F) has shown that for the disaccharide Gal-GalNAc the only allowed orientation of the sugar moiety was pointed out-of-groove, which is in full agreement with the published crystal structure (Glithero et al., 1999). According to the model, the high binding energy of Gal-GalNAc glycosylated peptides leads to low ligation strength to MHC class I molecules, which was confirmed in the MHC stabilisation assays. In contrast, modelling the GalNAc glycosylated MUC1 peptide showed that the sugar can point towards or out of the MHC binding groove. A mixture of the stronger (pointing towards the groove) and the weaker bound conformers (pointing out of the groove) might explain the slightly weaker binding of the GalNAc glycosylated relative to the unglycosylated peptide SAP10. Therefore, for an efficient vaccine design it is important that the antigen carries glycans on positions permissive for cleavage and MHC binding. Similar results have been reported by others working in mouse models. Haurum et al. showed that positioning of the glycan at the peptide anchor residues causes a dramatic decrease of a glycopeptide binding affinity (Haurum et al., 1995). Apostolopoulos et al. demonstrated that the octameric MUC1 glycopeptide SAPDT(GalNAc)RPA strongly binds the mouse H2-Kb molecule, while the introduction of the Gal-GalNAc disaccharide at the same Thr [SAPDT(GalNAc-Gal)RPA completely abolished its binding (Apostolopoulos et al., 2003). In our study, SAP10 peptide modified with Gal-GalNAc disaccharide at T5 also failed to bind human class I MHC molecule (HLA-A2).
Activation of vaccine antigen-specific cytotoxic T cells that can kill tumor cells presenting the same antigen is considered to be a requirement for a successful cancer vaccine. The ability to elicit a strong CTL response depends on many different aspects of the vaccine design. We have focused on a selection of potentially good antigens. From the many peptides we analyzed in this study we selected two MUC1 glycopeptides and one non-glycosylated MUC1 peptide that showed strong MHC class I binding capacity to test CTL stimulatory ability. We were able to show generation of IFN-γ secreting CD8+ T cells specific for F1 (SAP10) peptide and F2 (SAP10) glycopeptide, but not F3 glycopeptide. These T cells also lysed F2 presenting cells. F3 glycopeptide contains the naturally occurring variant ESR motif instead of the immuno-dominant DTR motif in the tandem repeat and has recently been shown to induce IgG responses in healthy people (von Mensdorff-Pouilly et al., 2005). To the contrary, antibodies against MUC1 from cancer patients preferentially bind to DTR motif containing repeats, such as F1 or F2. This is in agreement with the observed weak cytotoxicity towards F3 glycopeptide present on tumor cells. The higher background lysis is based on residual NK cell activity due to lack of MHC molecules on T2 target cells.
Based on our current knowledge, we propose that MUC1 vaccines of the future should include both peptides and glycopeptides to stimulate multiple populations of T cells recognizing cancer specific structural changes in the MUC1 molecule, such as those induced by altered glycosylation.
Table 2.
Relative frequencies of 8- to 11-meric proteolytic fragments grouped according to their N-terminus.
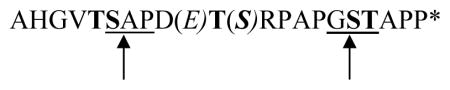 | |||
|---|---|---|---|
| AHG 8 to -11 | SAP8 to -11 | PAP8 to -11 | |
| GP2 | 18% | 42% | 40% |
| GP3 | 20% | 78% | 2% |
| GP4 | 3% | 60% | 37% |
| GP5 | 26% | 27% | 47% |
| GP6 | 55% | 42% | 3% |
| GP7 | 8% | 61% | 31% |
Schematic presentation of VNTR sequence and major proteasomal processing sites (underlined). The 8- to 11-meric fragments in digests of MUC1 repeat glycopeptides were grouped according to their N-terminal tripeptide motif into three groups and the relative amounts in each group were calculated (expressed as % of the total amount of 8- to –11-mers). Dark squares mark the position of glycosylated threonine within VTS (GP2, GP5), DTR/ESR (GP3/GP6) or STA motifs (GP4/GP7).
Table 3.
Peptides eluted from exosomal MHC molecules after cross-presentation of MUC1 61-mer P2 in a murine dendritic cell line. Peptides found also in cell-free in vitro proteasomal processing are written in bold face.
| MW (m/z) | Potential MHC class I binding 8- to 11-mers | MW (m/z) | Potential MHC class II binding12- to 25-mers |
|---|---|---|---|
| 823 | APESRPAP | 1169 | VTSAPESRPAPG VTSAPESRPAPG |
| 844 | TSAPESRP | 1264 | PESRPAPGSTAPP |
| 881 | APESRPAPG | 1523 | TSAPESRPAPGSTAPP |
| 896 | PESRPAPGS | 1549 | HGVTSAPESRPAPGST |
| 937 |
STAPPAHGVT TAPPAHGVTS |
1629 | SAPESRPAPGSTAPPAH |
| 968 |
SAPESRPAPG APESRPAPGS |
1692 | AHGVTSAPESRPAPGSTA VTSAPESRPAGSTAPPA |
| 999 | PESRPAPGST | 1786 | SAPESRPAPGSTAPPAHGV |
| 1056 | SAPESRPAPGS | 1986 | VTSAPDTRPAPGSTAPPAHGV |
| 2323 | TAPPAHGVTSAPESRPAPGSTAPPA | ||
| 2459 | TSAPESRPAPGSTAPPAHGTSAPES |
Table 4.
HLA-A0201 allele binding prediction of proteasomal products.
| FRAGMENT | SCORE | FRAGMENT | SCORE | FRAGMENT | SCORE |
|---|---|---|---|---|---|
| SAPESRPAP | 10 | RPAPGSTAPP | 6 | HGVTSAPD | 0 |
| SAPESRPAPG | 10 | AHGVTSAPES | 5 | PAPGSTAP | 0 |
| TSAPDTRPA | 10 | ESRPAPGST | 5 | PDTRPAPG | 0 |
| PGSTAPPA | 9 | ESRPAPGSTA | 5 | PDTRPAPGS | 0 |
| RPAPGSTA | 9 | AHGVTSAPE | 4 | PESRPAPG | 0 |
| SAPDTRPA | 9 | APDTRPAPG | 4 | PESRPAPGS | 0 |
| SAPDTRPAP | 9 | GVTSAPDTRP | 4 | SRPAPGST | 0 |
| SAPDTRPAPG | 9 | GVTSAPESRP | 4 | TRPAPGST | 0 |
| SAPESRPA | 9 | APESRPAPG | 3 | TSAPDTRP | 0 |
| SRPAPGSTA | 9 | APESRPAPGS | 3 | TSAPESRP | 0 |
| VTSAPESRPA | 9 | HGVTSAPES | 3 | VTSAPDTR | 0 |
| PAPGSTAPPA | 8 | PESRPAPGST | 3 | APGSTAPP | 0 |
| TSAPESRPA | 8 | SRPAPGSTAP | 3 | APDTRPAP | 0 |
| APGSTAPPA | 7 | HGVTSAPDTR | 2 | APESRPAP | 0 |
| PAPGSTAPP | 7 | HGVTSAPESR | 2 | DTRPAPGS | 0 |
| RPAPGSTAP | 7 | PDTRPAPGST | 2 | ESRPAPGS | 0 |
| TRPAPGSTA | 7 | TRPAPGSTAP | 1 | VTSAPESR | 0 |
| GVTSAPDTR | 6 | GVTSAPDT | 0 | ||
| GVTSAPESR | 6 | GVTSAPES | 0 |
Altogether 55 identified 8- to 10-meric fragments from in vitro processing were analyzed by the SYFPEITHI software tool for their ligation strength to the human HLA-A0201 allele. The information about glycosylation position was not included in these analyses due to software restrictions.
Acknowledgments
The work was supported by NIH grant 1RO1 CA84106 and the Köln-Fortune Programme (to F.G.H.) and by NIH grant 2PO CA73743 (to O.J.F.).
Abbreviations
- ESI-MS/MS
electrospray tandem mass spectrometry
- HPLC
high-pressure liquid chromatography
- MALDI-MS
matrix-assisted laser desorption mass spectrometry
- VNTR
variable number of tandem repeats
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sorensen AL, Reis CA, Tarp MA, Mandel U, Ramachandran K, Sankaranarayanan V, Schwientek T, Graham R, Taylor-Papadimitriou J, Hollingsworth MA, Burchell J, Clausen H. Chemoenzymatically synthesized multimeric Tn/STn MUC1 glycopeptides elicit cancer-specific anti-MUC1 antibody responses and override tolerance. Glycobiology. 2006;16:96–107. doi: 10.1093/glycob/cwj044. [DOI] [PubMed] [Google Scholar]
- 2.Müller S, Alving K, Peter-Katalinic J, Zachara N, Gooley AA, Hanisch FG. High density O-glycosylation on tandem repeat peptide from secretory MUC1 of T47D breast cancer cells. J Biol Chem. 1999;274:18165–18172. doi: 10.1074/jbc.274.26.18165. [DOI] [PubMed] [Google Scholar]
- 3.Müller S, Goletz S, Packer N, Gooley A, Lawson AM, Hanisch FG. Localization of O-glycosylation sites on glycopeptide fragments from lactation-associated MUC1. J Biol Chem. 1997;272:24780–24793. doi: 10.1074/jbc.272.40.24780. [DOI] [PubMed] [Google Scholar]
- 4.Vlad AM, Muller S, Cudic M, Paulsen H, Otvos L, Jr, Hanisch FG, Finn OJ. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J Exp Med. 2002;196:1435–1446. doi: 10.1084/jem.20020493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Y, Gendler SJ, Franco A. Designer glycopeptides for cytotoxic T cell-based elimination of carcinomas. J Exp Med. 2004;199:707–716. doi: 10.1084/jem.20031865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glithero A, Tormo J, Haurum JS, Arsequell G, Valencia G, Edwards J, Springer S, Townsend A, Pao YL, Wormald M, Dwek RA, Jones EY, Elliott T. Crystal structures of two H-2Db/glycopeptide complexes suggest a molecular basis for CTL cross-reactivity. Immunity. 1999;10:63–74. doi: 10.1016/s1074-7613(00)80007-2. [DOI] [PubMed] [Google Scholar]
- 7.Stepensky D, Tzehoval E, Vadai E, Eisenbach L. O-glycosylated versus non-glycosylated MUC1-derived peptides as potential targets for cytotoxic immunotherapy of carcinoma. Clin Exp Immunol. 2006;143:139–149. doi: 10.1111/j.1365-2249.2005.02965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ninkovic T, Hanisch FG. O-Glycosylated human MUC1 repeats are processed in vitro by immunoproteasomes. J Immunol. 2007;179:2380–2388. doi: 10.4049/jimmunol.179.4.2380. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Jondal M. Endolysosomal processing of exogenous antigen into major histocompatibility complex class I-binding peptides. Scand J Immunol. 2004;59:545–552. doi: 10.1111/j.1365-3083.2004.01426.x. [DOI] [PubMed] [Google Scholar]
- 10.Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity. 2004;21:155–165. doi: 10.1016/j.immuni.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Hanisch FG, Schwientek T, Von Bergwelt-Baildon MS, Schultze JL, Finn OJ. O-Linked glycans control glycoprotein processing by antigen-presenting cells: a biochemical approach to the molecular aspects of MUC1 processing by dendritic cells. Eur J Immunol. 2003;33:3242–3254. doi: 10.1002/eji.200324189. [DOI] [PubMed] [Google Scholar]
- 12.Saper MA, Bjorkman PJ, Wiley DC. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J Mol Biol. 1991;219:277–319. doi: 10.1016/0022-2836(91)90567-p. [DOI] [PubMed] [Google Scholar]
- 13.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 14.Rubinstein A, Kinarsky L, Sherman S. Molecular dynamics simulations of the O-glycosylated 21-residue MUC1 peptides. Int J Mol Sci. 2004;5:119–128. [Google Scholar]
- 15.Engelmann K, Shen H, Finn OJ. MCF-7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008;68:2419–2426. doi: 10.1158/0008-5472.CAN-07-2249. [DOI] [PubMed] [Google Scholar]
- 16.Haurum JS, Tan L, Arsequell G, Frodsham P, Lellouch AC, Moss PA, Dwek RA, McMichael AJ, Elliott T. Peptide anchor residue glycosylation: effect on class I major histocompatibility complex binding and cytotoxic T lymphocyte recognition. Eur J Immunol. 1995;25:3270–3276. doi: 10.1002/eji.1830251211. [DOI] [PubMed] [Google Scholar]
- 17.Apostolopoulos V, Yuriev E, Ramsland PA, Halton J, Osinski C, Li W, Plebanski M, Paulsen H, McKenzie IF. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc Natl Acad Sci U S A. 2003;100:15029–15034. doi: 10.1073/pnas.2432220100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Mensdorff-Pouilly S, Kinarsky L, Engelmann K, Baldus SE, Verheijen RH, Hollingsworth MA, Pisarev V, Sherman S, Hanisch FG. Sequence-variant repeats of MUC1 show higher conformational flexibility, are less densely O-glycosylated and induce differential B-lymphocyte responses. Glycobiology. 2005;15:735–746. doi: 10.1093/glycob/cwi058. [DOI] [PubMed] [Google Scholar]
- 19.Hanisch FG, Reis CA, Clausen H, Paulsen H. Evidence for glycosylation-dependent activities of polypeptide N-acetylgalactosaminyl- transferases rGalNAc-T2 and -T4 on mucin glycopeptides. Glycobiology. 2001;11:731–740. doi: 10.1093/glycob/11.9.731. [DOI] [PubMed] [Google Scholar]
- 20.Tenzer S, Stotze L, Schönfisch B, Dengjel J, Müller M, Stefanovic S, Rammensee HG, Schild H. Quantitative analysis of prion-protein degradation by constitutive and immuno-20S proteasomes indicates differences correlated with disease susceptibility. J Immunol. 2004;172:1083–1091. doi: 10.4049/jimmunol.172.2.1083. [DOI] [PubMed] [Google Scholar]
- 21.Elson LH, Nutman TB, Metcalfe DD, Prussin C. Flow cytometric analysis for cytokine production identifies T helper 1, T helper 2, and T helper 0 cells within the human CD4+CD27- lymphocyte subpopulation. J Immunol. 1995;154:4294–4301. [PubMed] [Google Scholar]
- 22.Lecoeur H, Fevrier M, Garcia S, Riviere Y, Gougeon ML. A novel flow cytometric assay for quantitation and multiparametric characterization of cell-mediated cytotoxicity. J Immunol Meth. 2001;253:177–187. doi: 10.1016/s0022-1759(01)00359-3. [DOI] [PubMed] [Google Scholar]