

Abstract
Background:
Progressive muscular atrophy (PMA) is clinically characterized by signs of lower motor neuron dysfunction and may evolve into amyotrophic lateral sclerosis (ALS). Whether PMA is actually a form of ALS has important consequences clinically and for therapeutic trials. We compared the survival of patients with PMA or ALS to analyze the clinical features that influence survival in PMA.
Methods:
We reviewed the medical records of patients with PMA (n = 91) or ALS (n = 871) from our ALS Center and verified survival by telephoning the families or using the National Death Index.
Results:
In PMA, patients were more likely to be male (p < 0.001), older (p = 0.007), and lived longer (p = 0.01) than in ALS. Cox model analysis suggested that the risk of death increased with age at onset in both patient groups (p < 0.005). Upper motor neuron (UMN) signs developed in 22% of patients with PMA within 61 months after diagnosis. Demographic and other clinical variables did not differ at diagnosis between those who did or did not develop UMN signs. In PMA, the factors present at diagnosis that predicted shorter survival were greater number of body regions affected, lower forced vital capacity, and lower ALS Functional Rating Scale–Revised score. Noninvasive ventilation and gastrostomy were used frequently in PMA.
Conclusion:
Although patients with progressive muscular atrophy (PMA) tended to live longer than those with amyotrophic lateral sclerosis (ALS), shorter survival in PMA is associated with the same risk factors that predict poor survival in ALS. Additionally, PMA is relentlessly progressive, and UMN involvement can occur, as also reported in imaging and postmortem studies. For these reasons, PMA should be considered a form of ALS.
GLOSSARY
- ALS
= amyotrophic lateral sclerosis;
- ALSFRS-R
= ALS Functional Rating Scale–Revised;
- CI
= confidence interval;
- FVC
= forced vital capacity;
- HR
= hazard ratio;
- LMN
= lower motor neuron;
- MND
= motor neuron disease;
- MRS
= magnetic resonance spectroscopy;
- NIV
= noninvasive ventilation;
- PEG
= percutaneous endoscopic gastrostomy;
- PMA
= progressive muscular atrophy;
- TMS
= transcranial magnetic stimulation;
- UMN
= upper motor neuron.
Progressive muscular atrophy (PMA) is clinically characterized solely by signs of lower motor neuron (LMN) dysfunction. Patients with LMN signs who at any time later in follow-up develop upper motor neuron (UMN) signs are then considered to have LMN-onset amyotrophic lateral sclerosis (ALS). PMA was identified in only 2.5% of all patients with adult-onset motor neuron disease (MND),1 and in another study with decades of observation, PMA was found in 11% of MND cases.2 In the World Federation of Neurology El Escorial ALS diagnostic criteria, PMA is not considered a form of ALS but is labeled as “suspected ALS,” and patients with that diagnosis are excluded from therapeutic trials.3 The revised Airlie House criteria discarded the “suspected ALS” category.4
The clinical limits of ALS and its subtypes remain to be more clearly defined. Histopathologic studies have shown pyramidal tract degeneration in more than half of all patients with purely LMN signs who were clinically diagnosed while alive with PMA.5–7 In most cases, ubiquitinated inclusions (characteristic of ALS) have been found in surviving motor neurons of patients with PMA.6 Additionally, magnetic resonance spectroscopy (MRS) has revealed UMN involvement in more than 60% of patients with PMA, and transcranial magnetic stimulation (TMS) identified UMN dysfunction in more than one-third of PMA cases,8–10 suggesting that PMA is a form of ALS. In familial ALS with SOD1 mutations, some patients have only LMN signs11,12—further evidence that PMA may be a form of ALS. Furthermore, the rate of progression and duration of PMA are comparable to those of ALS.13–15 All these reports prompted us to retrospectively examine our patients with PMA to compare survival in PMA with that in ALS and analyze the clinical features that influence survival in PMA.
METHODS
We retrospectively reviewed the full medical records of all patients diagnosed with an MND between 2000 and 2007 at the Eleanor and Lou Gehrig MDA/ALS Research Center, Columbia University. For all cases, demographic and clinical data were collected at the first full evaluation and at follow-up through the last day we communicated with or examined the patient; survival data were also collected from families. For patients who were lost to follow-up, the duration of survival was determined using the National Death Index Interactive Search at http://ssdi.rootsweb.com/cgi-bin/ssdi.cgi.
The diagnosis of ALS was determined according to the El Escorial criteria3,4 using clinical and EMG findings. For purposes of this analysis, we excluded patients who had a limited number of affected nervous system segmental regions (cervical only or lumbosacral region only) or were diagnosed with flail arm or flail leg syndromes,16–18 monomelic amyotrophy, PLS, or other MNDs such as Kennedy disease or multifocal motor neuropathy.
In PMA, neurologic findings were also reviewed and recorded at all follow-up visits. The diagnosis of PMA was made based on the presence of pure LMN findings at the first full evaluation performed at our center. Specifically, from the record at the first full evaluation, we collected data about the date of symptom onset and the number of body regions (not nervous system segmental regions) involved (6 body regions: bulbar, right or left arm, right or left leg, and respiration symptoms). Neurologic examination data consisted of the presence of LMN signs (weakness and muscle atrophy, fasciculation, loss of or diminished stretch reflexes), presence of UMN signs (including probable UMN signs: preserved reflexes in limbs with atrophic muscles, pathologic hyperreflexia, spastic catch, Hoffmann or Babinski sign), score on the ALS Functional Rating Scale–Revised (ALSFRS-R), and forced vital capacity (FVC). In some patients, MRS and TMS were performed according to methods described in detail elsewhere.8 The use of noninvasive ventilation (NIV) and percutaneous endoscopic gastrostomy (PEG) was recorded.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Columbia University IRB.
Statistical analysis.
Descriptive statistics are presented as mean ± SD for continuous variables and proportions for categorical variables. χ2 or Fisher exact test and Wilcoxon rank sum test were used for 2-group comparisons of categorical or continuous variables for demographic and clinical features. Kaplan-Meier survival curves were estimated for each patient group, and the log-rank test was used to assess the difference between groups. Cox proportional hazards modeling was used to examine factors related to survival. All tests were 2-sided with a significance level of 0.05. Statistical analyses were performed using SAS software, version 9.1.3 (SAS Institute, Cary, NC).
RESULTS
Of 1,201 records with diagnosis of an MND, ALS was the initial diagnosis in 916 (76.3%) cases, PMA in 91 (7.6%), and PLS in 84 (7.0%). The remaining cases were flail arm or leg syndrome (n = 13), monomelic amyotrophy (n = 15), and other MNDs (n = 84). We excluded 45 ALS cases that were missing clinical or survival data, resulting in a total of 871 ALS cases for analysis. All 91 PMA cases had complete survival data; those that were missing clinical data were not excluded from the study, but were included for analysis only when they had data available for the specific variable being evaluated.
The median follow-up time after diagnosis (first clinic visit) in PMA patients was 23.4 months (range, 0.7 to 88.3 months), whereas in ALS, the median follow-up time was 13.8 months (range, 0.2 to 213.4 months). The overall mortality rate after diagnosis was 68.1% (62/91) in PMA and 68.4% (596/871) in ALS (p = 0.95).
Differences between PMA and ALS.
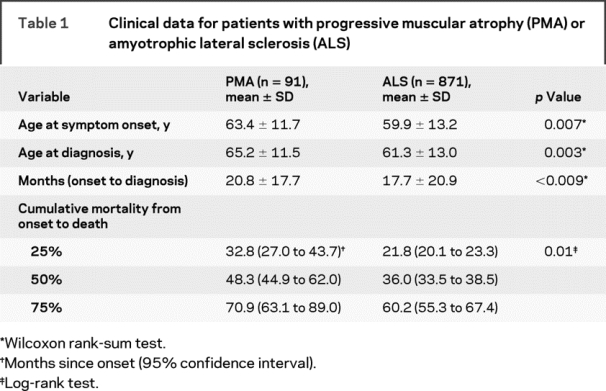
In PMA, the proportion of men was greater than in ALS (73.6% vs 54.9%, p = 0.0006). Table 1 shows differences in demographic and survival data between ALS and PMA.
Table 1 Clinical data for patients with progressive muscular atrophy (PMA) or amyotrophic lateral sclerosis (ALS)
Survival in PMA and ALS.
Kaplan-Meier estimates of survival for time from symptom onset to death differed in PMA and ALS (p = 0.01) (figure 1): patients with PMA had longer survival, up to 77 months after onset. At about 80 months, the curves crossed, with an estimated 14% survival rate; after that, the estimated survival in PMA was similar to or less than that of ALS. After symptom onset, the patients with PMA survived longer as a group (median, 48.3 months vs 36 months for ALS). Among the patients who died, the longest survival time was 123.3 months in the 62 decedents with PMA and 213.1 months in the 596 decedents with ALS. Among the patients who were still alive, the longest survival time was 130.7 months in PMA and 367.2 months in ALS. The patient with ALS with the longest survival began to have symptoms at age 26 and lived for 30.6 years.
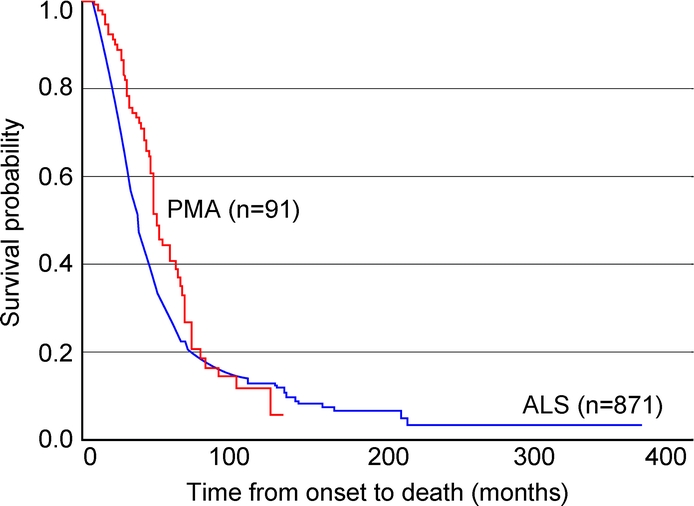
Figure 1 Kaplan-Meier survival curves for amyotrophic lateral sclerosis and progressive muscular atrophy
The 2 survival curves differed (log-rank test, p = 0.01).
Table 1 summarizes survival data in patients with PMA and ALS. To further examine the associations between the survival time from symptom onset and gender and age at onset, we applied a Cox proportional hazards model to each patient group. Gender had no effect on survival in PMA or ALS. However, the risk of death increased with age at onset (hazard ratio [HR] = 1.038, 95% confidence interval [CI] = 1.012 to 1.064, p = 0.003 in PMA; HR = 1.042, 95% CI = 1.035 to 1.049, p < 0.0001 in ALS). When we applied the model to all patients with PMA and patients with ALS combined (n = 962), and controlled for gender and age at onset, patients with ALS were at greater risk of death (HR = 1.737, 95% CI = 1.330 to 2.268; p < 0.0001).
The duration from symptom onset to diagnosis was longer in PMA (table 1). The Cox models for survival time after diagnosis revealed that when gender and age at diagnosis were controlled for, the duration from symptom onset to diagnosis was unrelated to the risk of death in PMA, whereas in ALS the risk of death decreased as the duration doubled (HR = 0.812, 95% CI = 0.759 to 0.867; p < 0.0001).
PMA with and without later appearance of UMN signs.
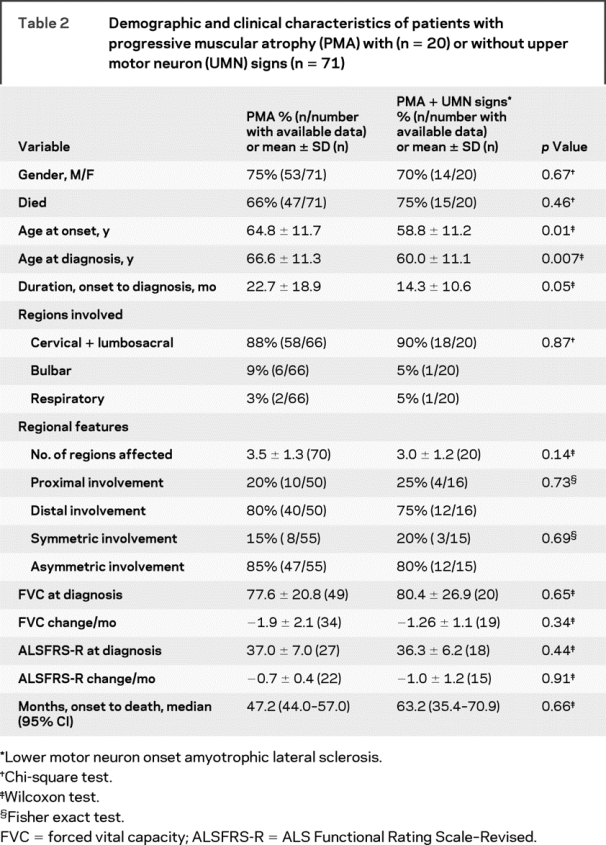
During follow-up, 20 of the 91 patients with PMA developed UMN signs; thus, by definition, these patients had ALS with LMN onset; 14 of the 20 died. Among those with delayed UMN signs, the time to appearance of the first UMN signs ranged from 0.5 to 61.3 months. The median was 8.5 months and the upper quartile 25.4 months, indicating that, if UMN signs developed, they tended to appear within 2 years of diagnosis.
As table 2 shows, patients who developed UMN signs had PMA symptom onset at a younger age (p = 0.01). The same patients had a shorter duration between onset and diagnosis, so the age at diagnosis also was younger (p = 0.007). However, there was no difference in gender or other clinical characteristics. In more than half of the PMA patients with MRS studies (19 of 32) and in one third of patients with TMS studies (9 of 28), the changes suggested UMN involvement based on reduced N-acetylaspartate on MRS and prolonged central motor conduction time on TMS.8–10
Table 2 Demographic and clinical characteristics of patients with progressive muscular atrophy (PMA) with (n = 20) or without upper motor neuron (UMN) signs (n = 71)
Predictors of survival in PMA.
We examined the impact on survival in PMA of UMN signs and the factors associated with survival in ALS (table 2). The Cox model revealed that the presence of UMN signs was unrelated to survival time after diagnosis when controlling for gender and age at diagnosis (HR = 1.129, 95% CI = 0.579 to 2.202; p = 0.72).
The mortality rates did not differ significantly, regardless of the number of body regions affected at diagnosis (table 2). However, the survival curves for time from diagnosis to death differed according to the number of regions involved (log-rank test, p = 0.003), with the worst survival in the group with 5 to 6 affected regions. The Cox model revealed that the risk of death grew as the number of affected regions increased (p = 0.009; table 3, model 1). The FVC was evaluated in 69 PMA patients and correlated with the number of regions affected (r = −0.40, p = 0.0008) and with the ALSFRS-R score at diagnosis (n = 45, r = 0.33, p = 0.03), but was not related to age at diagnosis or time from symptom onset to diagnosis. When we analyzed the FVC (expressed as the percent of expected FVC) using the median value of 79% as a cutpoint, those with an FVC >79% had longer survival (p = 0.0005; figure 2). In the Cox model, patients had a lower risk of death for every unit of increase in the FVC (p = 0.002; table 3, model 2). Only 45 of the 91 patients with PMA had an ALSFRS-R score available at diagnosis (median = 38; range, 11 to 47) (table 2). Among patients with a score above the median of 38, 14 of 21 died (66.7%), whereas of those with a score of 38 or less, 20 of 24 (83.3%) died (Fisher exact test, p = 0.30). The Cox model revealed that an ALSFRS-R score ≤38 tended to be associated with a greater risk of death (p = 0.047; table 3, model 3).
Table 3 Hazards ratio (HR) for 3 clinical predictors of survival after diagnosis derived from Cox proportional hazards models controlling for age at diagnosis
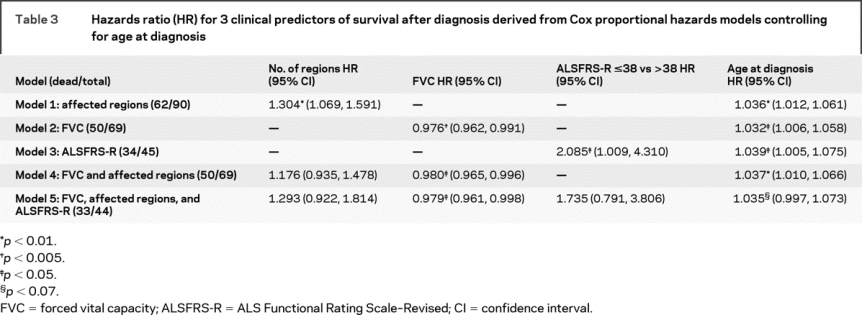

Figure 2 Survival according to forced vital capacity (FVC) at diagnosis as dichotomized at the median of 79%
Patients with an FVC of 79% or less (low FVC) had shorter survival than those with an FVC greater than 79% (high FVC; log-rank test, p = 0.0005).
To examine the simultaneous effect of multiple risk factors, we first included the FVC and number of affected regions in the model while controlling for age at diagnosis (n = 69). Model 4 (table 3) showed that greater FVC was still related to survival time (p = 0.01), whereas the impact of the number of regions became insignificant. The Cox model including the 3 predictors of FVC, number of regions, and ALSFRS-R showed the effect of FVC remained significant (table 3, model 5).
Medical management in PMA and ALS.
In PMA patients without UMN signs, 10% (7/71) received PEG, not significantly different from those with UMN signs (15%, 3/20). In contrast, NIV was used in 30% (21/71) of PMA patients without UMN signs, but less than 60% (12/20) of patients with later UMN signs required it (p = 0.03). Neither PEG nor NIV predicted survival time after diagnosis. In comparison, PEG was used in 7.5% (65/871) of patients with ALS, which did not differ from use in all patients with PMA (11%, 10/91; p = 0.32); however, fewer patients with ALS were treated with NIV (15.8% in ALS vs 36.3% in PMA; p < 0.0001), probably because a large number of patients with ALS were in the early stages of ALS.
DISCUSSION
Survival time was the main difference we found between PMA and ALS. Median survival in patients with PMA was 12 months longer than in those with ALS for up to 77 months after onset. Few studies have directly compared survival in PMA and ALS.1,19 Other differences included older age at onset and marked male predominance in PMA. Sex differences are also seen in other MND disorders, including ALS, flail arm syndrome,16,17,18 respiratory- onset ALS,20 and benign monomelic amyotrophy (Hirayama disease),21,22 being seen much more often in men, whereas bulbar-onset ALS is much more common in women.23 One of the most consistent epidemiologic findings in ALS is that patients diagnosed at an older age have shorter survival.19,24,25 Our data reaffirmed this in ALS, and we also found it to be true in PMA.
Our survival estimate for PMA was much shorter than the mean survival of 200 (SE = 30) months previously reported, but that study included only 17 cases of PMA.1 Comparable to our survival rates of 73.3% (SE = 4.8%) at 3 years and 40.7% (SE = 5.7%) at 5 years after symptom onset, survival rates of 61.3% at 3 years and 56.4% at 5 years have been recorded in 155 patients with PMA; however, the differences imply that our patients had a shorter duration of disease.26
More than 20% of patients with PMA develop UMN signs at some time, and among those who do, 50% develop UMN signs within 1 year after LMN symptom onset. Applying the El Escorial Diagnostic Criteria, the diagnosis of these patients with PMA who later develop UMN signs could simply be reclassified as ALS or LMN-onset ALS.3,4 We found no difference in the survival time between patients with PMA who did or did not develop UMN signs. Ince et al.,6 however, found that patients originally diagnosed with PMA but who had UMN pathology at autopsy had an aggressive, rapid course.
The El Escorial Criteria for diagnosis of ALS are based on the number of nervous system segmental regions (bulbar, cervical, thoracic, and lumbosacral) involved. For example, when at least 3 of these segmental regions show both UMN and LMN signs, the diagnostic certainty is definite ALS; when 2 regions are involved, it is probable ALS; and for 1 region it is possible ALS.3,4 Survival is shorter in definite or probable ALS than in possible ALS, indicating that as more segmental regions are involved, the shorter the survival is in ALS.27,28 Impaired FVC or a lower ALSFRS-R score also predicts shorter survival in ALS.29,30 We found that each of these factors also was independently associated with shorter survival in PMA. However, when the simultaneous effect of multiple risk factors was examined in patients in whom all 3 factors were available, lower FVC was most likely to predict shorter survival (table 3). Others have reported similar trends.13 When PMA onset occurs in the axial muscles, the disease is more aggressive and more rapidly leads to the need for respiratory care.15 Other investigators have also emphasized that low vital capacity at baseline and a sharp decline in FVC during the first 6 months are associated with shorter survival in PMA.13
Based on the current ALS diagnostic criteria,3,4 patients with PMA are excluded from current clinical trials because of slow progression, long survival, and absence of UMN signs. Based on our large sample size, we can conclude that patients with PMA indeed lived longer than those with ALS. However, few differences were found between PMA with or without later UMN signs, suggesting that ALS and PMA share the clinical spectrum of the same disease, as suggested almost 5 decades ago.2 Further, it is not possible to distinguish the 2 conditions based on individual prognosis or need for multidisciplinary care.
Postmortem pathology, neuroimaging, and TMS studies suggest that UMNs and the corticospinal tracts are affected in more than half of patients with PMA.5–10 Our neuroimaging and electrophysiologic findings also support these findings. Therefore we suggest modifying the current El Escorial diagnostic criteria so that patients with PMA can participate in clinical trials.
We also recommend incorporating the number of affected body regions into the definition of PMA. PMA with one affected body region, as seen in the flail arm and leg syndromes16,17,18 or monomelic amyotrophy,21,22 should be considered separately because progression in these syndromes is much slower than in other MNDs.31,32 The distribution and number of regions involved in LMN disease affect prognosis in PMA,13,26 as confirmed in this study. Secondly, the disease should become progressively more severe, as documented clinically and also by FVC or ALSFRS-R changes.13
Whether PMA is indeed ALS in a particular patient cannot be resolved until autopsy proves that it is ALS or, alternatively, a reliable biomarker is found that can distinguish ALS from PMA. Currently, patients are frustrated, and clinical trialists often feel compelled to find “soft” UMN signs. Clinical criteria for trial eligibility that would include more patients and a wider range of ALS forms might help identify drugs that have a broader effectiveness in ALS.
Because this study was retrospective and based on one ALS Center, generalization of the results might be limited. Nevertheless, we included all cases, except for the 45 ALS cases missing survival data, and believe the study results are valuable because of the large number of patients analyzed.
ACKNOWLEDGMENT
Cassandra Talerico-Kaplin, PhD, Neurological Institute, Cleveland Clinic, provided editorial suggestions. The authors thank the patients and their families who came to see us at the Eleanor and Lou Gehrig MDA/ALS Research Center for giving us the opportunity to conduct a retrospective study.
DISCLOSURE
Dr. Liu receives partial support from the NIEHS Center [P30 ES09089]. J. Sandner, M. Pasmantier, and Dr. Andrews report no disclosures. Dr. Rowland serves as Ombudsman for Neurology®; receives royalties for Merritt’s Neurology and Principles of Neural Science (Lippincott Williams & Wilkins, 2005); serves as Editor-in-Chief of the AAN publication Neurology Today; and has received travel expenses or speaker’s honoraria for lectures not funded by industry. Dr. Mitsumoto has served on scientific advisory boards for and/or received speaker honoraria from Avanir Pharmaceuticals, Knopp Neurosciences Inc., Neuralstem, Inc., and Sanofi-Aventis, and Otsuka Pharmaceutical Co., Ltd.; and has received research support from Avanir Pharmaceuticals, Teva Pharmaceutical Industries Ltd., Knopp Neurosciences Inc., the NIH [NINDS R01NS04812 (Co-I) and NIEHS 1R01ES016348-01A2 (PI)], the Muscular Dystrophy Association, the ALS Association, and the MDA Wings Over Wall Street. Dr. Kim is supported by a fellowship from the Hallym University College of Medicine, Seoul, Korea.
Address correspondence and reprint requests to Dr. Hiroshi Mitsumoto, The Eleanor and Lou Gehrig MDA/ALS Research Center, The Neurological Institute of New York, 710 West 168th Street, New York, NY 10032 hm264@columbia.edu
Disclosure: Author disclosures are provided at the end of the article.
Received March 23, 2009. Accepted in final form August 19, 2009.
REFERENCES
- 1.Norris F. Adult progressive muscular atrophy and hereditary spinal muscular atrophies. Handbook Clin Neurol 1991;59:13–34. [Google Scholar]
- 2.Mackay R. Course and prognosis in amyotrophic lateral sclerosis. Arch Neurol 1963;8:17–27. [Google Scholar]
- 3.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis: Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci 1994;124(suppl):96–107. [DOI] [PubMed] [Google Scholar]
- 4.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 5.Leung D, Hays AP, Karlikaya G, Del Bene ML, Rowland LP. Diagnosis of ALS: clinicopathologic analysis of 76 autopsies. Neurology 1999;52(suppl 2):A164. [Google Scholar]
- 6.Ince PG, Evans J, Knopp M, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology 2003;60:1252–1258. [DOI] [PubMed] [Google Scholar]
- 7.Rowland L. Clinical aspect of sporadic ALS and MND. In: Shaw P, Strong MJ, eds. Motor Neuron Disorder. Philadelphia: Butterworth-Heinemann; 2003:111–144. [Google Scholar]
- 8.Kaufmann P, Pullman SL, Shungu DC, et al. Objective tests for upper motor neuron involvement in amyotrophic lateral sclerosis (ALS). Neurology 2004;62:1753–1757. [DOI] [PubMed] [Google Scholar]
- 9.Mitsumoto H, Ulug AM, Pullman SL, et al. Quantitative objective markers for upper and lower neuron dysfunction in amyotrophic lateral sclerosis. Neurology 2007;17:1402–1410. [DOI] [PubMed] [Google Scholar]
- 10.Floyd AG, Yu QP, Piboolnurak P, et al. Transcranial magnetic stimulation in ALS: utility of central motor conduction tests. Neurology 2009;72:498– 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cudkowicz ME, McKenna-Yasek D, Chen C, Hedley-Whyte ET, Brown RH, Jr. Limited corticospinal tract involvement in amyotrophic lateral sclerosis subjects with the A4V mutation in the copper/zinc superoxide dismutase gene. Ann Neurol 1998;43:703–710. [DOI] [PubMed] [Google Scholar]
- 12.Cervenakova L, Protas II, Hirano A, et al. Progressive muscular atrophy variant of familial amyotrophic lateral sclerosis (PMA/ALS). J Neurol Sci 2000;177:124– 130. [DOI] [PubMed] [Google Scholar]
- 13.Visser J, van den Berg-Vos RM, Franssen H, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol 2007;64:522–528. [DOI] [PubMed] [Google Scholar]
- 14.Meyer T, Munch C, van Landeghem FK, Borisow N, Dullinger J, Linke P. [Progressive muscle atrophy: a rarely diagnosed variant of amyotrophic lateral sclerosis.] Nervenarzt 2007;78:1383–1388. [DOI] [PubMed] [Google Scholar]
- 15.de Carvalho M, Scotto M, Swash M. Clinical patterns in progressive muscular atrophy (PMA): a prospective study. Amyotroph Lateral Scler 2007;8:296–299. [DOI] [PubMed] [Google Scholar]
- 16.Katz JS, Wolfe GI, Andersson PB, et al. Brachial amyotrophic diplegia: a slowly progressive motor neuron disorder. Neurology 1999;53:1071–1076. [DOI] [PubMed] [Google Scholar]
- 17.Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78:849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wijesekera LC, Mathers S, Talman P, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology 2009;72:1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray B. Natural history and prognosis in amyotrophic lateral sclerosis. In: Mitsumoto HPS, Gordon PH, ed. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis; 2006:227–248. [Google Scholar]
- 20.Andrews J, Gordon PH, Basner R, Mitsumoto H. Clinical characteristics of respiratory onset ALS and respiratory symptoms at diagnosis in patients with ALS: 17th International Symposium on ALS/MND. Amyotroph Lateral Scler 2006;7(suppl 1):63–64. Abstract.
- 21.Tashiro K, Kikuchi S, Itoyama Y, et al. Nationwide survey of juvenile muscular atrophy of distal upper extremity (Hirayama disease) in Japan. Amyotroph Lateral Scler 2006;7:38–45. [DOI] [PubMed] [Google Scholar]
- 22.Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease). Intern Med 2000;39:283–290. [DOI] [PubMed] [Google Scholar]
- 23.Christensen PB, Hojer-Pedersen E, Jensen NB. Survival of patients with amyotrophic lateral sclerosis in 2 Danish counties. Neurology 1990;40:600–604. [DOI] [PubMed] [Google Scholar]
- 24.Eisen A, Schulzer M, MacNeil M, Pant B, Mak E. Duration of amyotrophic lateral sclerosis is age dependent. Muscle Nerve 1993;16:27–32. [DOI] [PubMed] [Google Scholar]
- 25.Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology 1993;43:1316–1322. [DOI] [PubMed] [Google Scholar]
- 26.Chio A, Brignolio F, Leone M, et al. A survival analysis of 155 cases of progressive muscular atrophy. Acta Neurol Scand 1985;72:407–413. [DOI] [PubMed] [Google Scholar]
- 27.Chio A, Mora G, Leone M, et al. Early symptom progression rate is related to ALS outcome: a prospective population-based study. Neurology 2002;59:99–103. [DOI] [PubMed] [Google Scholar]
- 28.Turner MR, Parton MJ, Shaw CE, Leigh PN, Al-Chalabi A. Prolonged survival in motor neuron disease: a descriptive study of the King’s database 1990–2002. J Neurol Neurosurg Psychiatry 2003;74:995–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brooks BR, Sanjak M. Disease-modifying drug therapies. Amyotroph Lateral Scler Other Motor Neuron Disord 2004;5(suppl 1):68–75. [DOI] [PubMed] [Google Scholar]
- 30.Kaufmann P, Levy G, Thompson JL, et al. The ALSFRSr predicts survival time in an ALS clinic population. Neurology 2005;64:38–43. [DOI] [PubMed] [Google Scholar]
- 31.van den Berg-Vos RM, Visser J, Franssen H, et al. Sporadic lower motor neuron disease with adult onset: classification of subtypes. Brain 2003;126:1036– 1047. [DOI] [PubMed] [Google Scholar]
- 32.Rowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve (in press 2009). [DOI] [PubMed]