

Summary
Cardiolipin (CL), the signature lipid of mitochondria, plays a critical role in mitochondrial function and biogenesis. The availability of yeast mutants blocked in CL synthesis has facilitated studies of the biological role of this lipid. Perturbation of CL synthesis leads to growth defects not only during respiratory growth but also under conditions in which respiration is not essential. CL was shown to play a role in mitochondrial protein import, cell wall biogenesis, aging and apoptosis, ceramide synthesis, and translation of electron transport chain components. The genetic disorder Barth syndrome (BTHS) is caused by mutations in the tafazzin gene resulting in decreased total CL levels, accumulation of monolysocardiolipin (MLCL), and decreased unsaturated fatty acyl species of CL. The variation in clinical presentation of BTHS indicates that other physiological factors play a significant role in modifying the phenotype resulting from tafazzin deficiency. Elucidating the functions of CL is expected to shed light on the role of this important lipid in BTHS and other disorders of mitochondrial dysfunction.
Keywords: cardiolipin, Barth syndrome, protein import, aging, mitochondrial bioenergetics and biogenesis, cell wall
1. Introduction
Cardiolipin (CL) (1,3 diphosphatidyl-sn-glycerol) is a unique and ubiquitous anionic phospholipid that, in eukaryotes, is localized in the mitochondrial inner membrane. CL was first isolated from beef heart, hence its name [1]. While it is most abundant in the heart, CL can be found in all mammalian tissues. Unlike the other membrane phospholipids, it has a dimeric structure in which two phosphatidyl moieties are linked by a glycerol [2]. As a result, CL is hydrophobic by virtue of four fatty acyl groups and acidic due to two phosphates. CL interacts with a wide variety of mitochondrial proteins by both hydrophobic and electrostatic interactions [3, 4], and stabilizes proteins in the mitochondrial respiratory chain [5]. CL molecules can form lamellar or inverted hexagonal structures. The hexagonal phase is favored in the presence of divalent cations [6]. Although the biological relevance of these structures is not known, it is plausible that CL is involved in the formation of local non-bilayer structures within biological membranes. Such structures are believed to be involved in membrane fusion and in trans-bilayer movement of solutes [7]. The finding that mitochondrial biosynthesis of the non-bilayer forming phospholipid phosphatidylethanolamine is essential for the viability of yeast mutants lacking CL suggests a critical role of CL in the formation of these structures [8], although it should be noted that these structures have not been convincingly demonstrated in vivo.
The fatty acid composition of CL plays an important role in the function of the lipid, as aberrant CL remodeling (replacing one fatty acid with another) underlies the genetic disorder Barth syndrome (BTHS) [9]. However, no single species of fatty acid is required for function, as the acyl species of CL from different organisms vary considerably. Bacterial CL contains saturated and mono-unsaturated fatty acyl chains 14-19 carbons in length [10]. Mitochondrial CL is mainly composed of monounsaturated and diunsaturated fatty acyl chain of 16-18 carbons in length, resulting in a much higher unsaturation index than that of bacterial CL. In mammals and higher plants, the predominant species is linoleic acid, whereas oleic acid and palmitoleic acid species exist in yeast [11]. In humans, CL acyl species vary with tissue type, although the predominant species in heart is tetralinoleoyl CL. This species is absent from BTHS cells [12]. While specific CL acyl species vary among eukaryotic cardiolipins, the feature shared by different organisms is that the dominant species of CL contains only one or two types of fatty acid. This leads to symmetry and structural uniformity among cardiolipins [9].
This review focuses on the biosynthesis and function of CL in eukaryotes, primarily concentrating on yeast, the model in which null mutants of the genes for CL synthesis are available. The most exciting findings pertaining to CL in the last few years are that this phospholipid plays an important role not only in mitochondrial bioenergetics, which is not unexpected given the interaction of CL with mitochondrial proteins, but also in essential cellular functions not generally associated with respiratory function. Some of these include mitochondrial protein import, cell wall biogenesis, translational regulation of electron transport chain (ETC) components, aging, and apoptosis, and it is likely that this list will be longer in subsequent reviews. The importance of CL in these and other processes is underscored by the finding that mutations in tafazzin, the CL remodeling enzyme, leads to BTHS. In this paper, CL biosynthesis is summarized only briefly in order to provide a context in which to understand the defects in the mutants that have been used to elucidate function. For a more detailed discussion of the regulation of CL biosynthesis, we refer the reader to several recent reviews [13, 14]. We focus here on studies describing the role of CL in mitochondrial function and in the essential cellular functions that are not associated with mitochondrial bioenergetics. In addition, we discuss the role of CL in BTHS. Finally, we put forward several unanswered questions that can be exciting areas for further exploration in this field.
2. CL biosynthesis
All of the enzymes for de novo synthesis of CL are present in the mitochondria. As seen in Fig. 1, phosphatidylglycerolphosphate (PGP) synthase (Pgs1p) catalyzes the committed step, forming PGP from CDP-DG and glycerol-3-phosphate (G-3-P) [15]. PGP is then dephosphorylated to PG by PGP phosphatase [16]. CL synthase (Crd1p) catalyzes an irreversible condensation reaction in which CDP-DG is linked to PG via cleavage of a high energy anhydride bond to form CL [11, 17-22]. CL then undergoes remodeling - deacylation leads to the formation of monolysocardiolipin (MLCL), which is then reacylated with another fatty acid [23]. In rat liver, mitochondria associated phospholipase A2 was shown to catalyze the deacylation step to form MLCL [24]. The yeast enzyme that deacylates CL has not been identified. Reacylation is catalyzed by tafazzin [25]. Schlame et al. demonstrated that tafazzin is a CoA-independent transacylase that transfers acyl chains preferentially from phosphatidylcholine (PC) to CL [26]. In a study comparing CL species from a wide variety of organisms, Schlame and co-workers showed that the most abundant species of CL contained only one or two types of fatty acids, which results in a high degree of structural uniformity and molecular symmetry in cardiolipins [9]. In contrast, tafazzin-deficient cells were characterized by multiple species of CL.
Fig. 1.
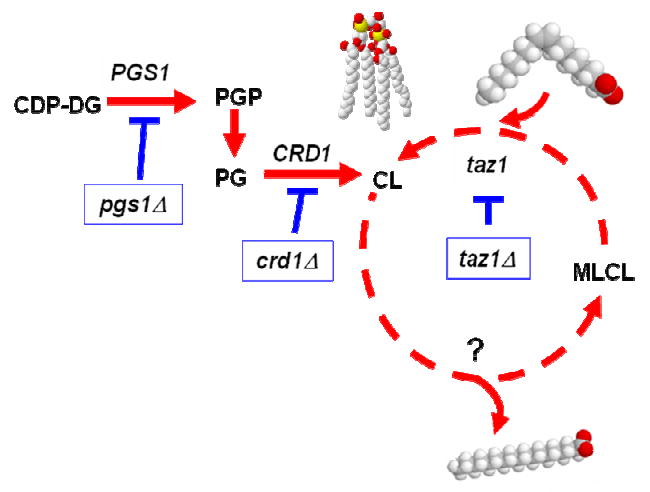
The cardiolipin (CL) biosynthetic pathway: PGS1 encodes phosphatidylglycerol phosphate (PGP) synthase, which converts glycerol-3-phosphate (G-3-P) and CDP-diacylglycerol (CDP-DAG) to PGP. PGP is dephosphorylated to phosphatidylglycerol (PG) by PGP phosphatase. CRD1 encodes CL synthase, which converts CDP-DAG and phosphatidylglycerol (PG) to CL. In the proposed remodeling pathway, CL is deacylated to monolysocardiolipin (MLCL) by a phospholipase that has not been identified. MLCL is then reacylated to mature CL by Taz1p.
The accumulation of MLCL in yeast and human tafazzin-deficient cells [25, 27-29] is consistent with the two step pathway of remodeling shown in Fig. 1. The importance of remodeling is underscored by its role in BTHS, a rare X-linked disorder caused by mutations in tafazzin. BTHS is characterized by cardiomyopathy, skeletal myopathy, neutropenia and growth retardation due to abnormal mitochondria and defective oxidative phosphorylation [12]. Patients also exhibit elevated urinary excretion of 3-methylglutaconic acid [30] and hypocholesterolemia [31]. BTHS patients display reduced CL, accumulation of MLCL, and aberrant CL species [24].
The power of the yeast system in elucidating the function of CL derives from the characterization of the yeast genes that encode Pgs1p, Crd1p, and Taz1p and the availability of null mutants of these genes [15, 20-22, 25, 27, 28]. These mutants are powerful molecular tools with which to elucidate the role of CL in vivo. The yeast taz1Δ null mutant exhibits biochemical defects similar to those observed in BTHS [14, 32-34]. These defects are complemented by expression of the human tafazzin gene in the taz1Δ mutant [25]. Many studies with the yeast mutants have shown that the CL pathway is required for optimal mitochondrial function, as discussed in section 3. The mutants exhibit growth defects with varying degrees of severity on non-fermentable carbon sources. The taz1Δ mutant grows poorly on ethanol at elevated temperature [25, 35], crd1Δ exhibits growth defects at elevated temperature on several carbon sources [35-37], and pgs1Δ cannot grow at all on non-fermentable carbon sources [15, 38]. Interestingly, pgs1Δ and crd1Δ exhibit growth defects even on glucose, suggesting that the CL pathway is required for essential cellular processes not directly associated with respiration. The crd1Δ mutant exhibits a strain dependent inability to form colonies at elevated temperature on glucose medium, and pgs1Δ cannot grow at all at 37°C on glucose unless supplemented with sorbitol [36, 37]. Moreover, pgs1Δ also loses mitochondrial DNA, which may account to some degree for the inability of the mutant to grow on non-fermentable carbon sources. The studies summarized in sections 4-7 describe cellular functions that are perturbed when CL synthesis is blocked.
3. CL and mitochondrial bioenergetics
CL is highly enriched in membranes designed to generate an electrochemical gradient for ATP synthesis, such as the bacterial plasma membrane [39] and the inner mitochondrial membrane [40]. It is also present in membranes of hydrogenosomes, a mitochondria-like organelle in protists [41]. This ubiquitous and intimate association between CL and energy transducing membranes suggests an important role for CL in bioenergetic reactions. It is not surprising that a number of biochemical studies carried out in the 80’s and 90’s have identified interactions between CL and several inner mitochondrial membrane proteins (Table 1). We refer readers to previous reviews [3, 4] for a more detailed discussion of this subject. CL modulates the catalytic activities of the interacting proteins, as seen in the ADP-ATP carrier [42, 43] and/or provides stability, as reported for complex III [44] and complex IV [45]. CL also binds specifically and irreversibly to cytochrome c [46], limiting the soluble pool of the protein. Therefore, it may play an important regulatory role in cytochrome c release, which triggers the downstream events in apoptosis.
Table 1.
CL interacting proteins in mitochondria
| Protein | Reference No. |
|---|---|
| a) Respiratory chain proteins | |
| 1) Complex I | 6 |
| 3) Complex III | 6,7,8 |
| 4) Complex IV | 9,10 |
| 5) Complex V | 11 |
| 6) Cytochrome c | 12 |
| b) Transporter/Carrier proteins | |
| 1) ADP-ATP carrier | 13,14 |
| 2) Phosphate carrier | 15 |
| 3) Pyruvate carrier | 16 |
| 4) Carnitine carrier | 17,18 |
| c) Others | |
| 1) Cardiolipin synthase | 19 |
| 2) Creatine kinase | 20 |
| 3) Glycerol 3-phosphate dehydrogenase | 21 |
| 4) Carbamoyl phosphate synthetase I | 22 |
| 5) Cytochrome P450scc | 23,24 |
In summary, in vitro studies suggest that CL is involved in site-directed structure-function relationships with mitochondrial respiratory chain complexes and transporters. However, it should be noted that these studies may not necessarily reflect in vivo interactions.
3.1 In vivo studies of the role of CL in mitochondrial respiration
The cloning of the CL synthase gene CRD1 [20-22] and the construction of crd1Δ yeast cells, which are completely devoid of CL, provided an excellent model to address the in vivo role of CL. Subsequent studies led to the surprising observation that crd1Δ cells can grow in non-fermentable carbon sources (which requires a functional respiratory chain for viability). This suggested that the interaction of CL with respiratory chain proteins is not essential for respiration, perhaps because other phospholipids such as PG and phosphatidylethanolamine (PE), which are elevated in crd1Δ cells, are able to compensate for the lack of CL [37, 47]. Although crd1Δ cells can grow in non-fermentable carbon sources, the growth rate of the mutant is reduced, suggesting the presence of mild defects in the mitochondrial energy transforming machinery. Indeed, bioenergetic measurements in isolated mitochondria from crd1Δ cells showed that CL is required for tight coupling (optimal ADP/O ratio and low state 4 respiration) of mitochondria at the maximal rate of respiration [48] and also for the maintenance of a mitochondrial membrane potential [35]. Furthermore, CL improved the efficiency of oxidative phosphorylation in unfavorable conditions, such as increased temperature and osmotic shock [48, 49]. The molecular basis underlying these physiological defects became clearer after the discovery of the supramolecular structural assembly of respiratory chain proteins [50].
3.2 The role of CL in supercomplex assembly
The respiratory chain supercomplexes, first reported by Schagger and Pfeiffer, refer to the organization of electron transport chain complexes into supramolecular structures [50]. In mammalian mitochondria, complex I is associated with dimeric complex III and multiple copies of complex IV (up to 4) resulting in multiple supercomplexes. In S. cerevisiae mitochondria, which lack complex I, complex III exists as a free dimer and in two supercomplexes comprising an additional one or two complex IV monomers, which are referred to as the small and large supercomplexes, respectively [50]. Blue-native PAGE (BN-PAGE) analysis of crd1Δ mitochondria showed a complete absence of these supercomplexes [51]. However, analysis by milder colorless-native PAGE revealed the presence of large supercomplexes, suggesting that the anionic dye used in BN-PAGE induced dissociation of the loosely bound complexes III and IV in the absence of CL. This implies that supercomplexes exist in the mutant mitochondria, but the absence of CL significantly reduces their stability [51]. An alternate explanation for the detection of supercomplexes in crd1Δ by CN-PAGE is that highly hydrophobic membrane proteins aggregate under the conditions of CN-PAGE in which membrane complexes migrate through the native gel by virtue of their intrinsic charges. The experiments by Zhang et al. [52] support the latter explanation and suggest that CL may be required for the assembly of supercomplexes. Additional evidence for the role of CL in respiratory chain supercomplex assembly and stability came from studies with yeast taz1Δ cells. BN-PAGE analysis of taz1Δ mitochondria showed a decrease in the amount of large supercomplex and an increase in free complex IV monomer. Additionally, the assembly of complex IV into supercomplexes is severely affected in taz1Δ mutant mitochondria [53]. This indicates that CL is critical for the biogenesis of respiratory chain supercomplexes, and that the role of CL in supercomplex assembly may not be limited to a stabilizing effect. However, the possibility that Taz1p is a cofactor for the assembly of respiratory supercomplexes cannot be ruled out. Similar results were seen in mitochondria from BTHS patients [53]. Furthermore, in BTHS patient mitochondria, the interaction between complexes I and III was also less stable, with decreased levels of the complex I/III2 supercomplex. Thus, the decreased CL and/or loss of mature CL species in BTHS results in unstable respiratory chain supercomplexes, which may contribute to subsequent pathology. What are the bioenergetic consequences of the absence of supercomplexes and how do they contribute to the pathology in BTHS? It has been proposed that supercomplexes allow substrate channeling and thus prevent leakage of electrons from the respiratory chain. The leaked electrons can easily react with oxygen molecules to generate reactive oxygen species (ROS). Thus, one possible consequence of unstable supercomplexes is increased oxidative stress, and this may be a contributing factor in the pathogenesis of BTHS. Consistent with this, Chen et al. observed an increase in protein carbonylation in yeast taz1Δ cells during respiratory growth (see section 6) [54].
4. CL and mitochondrial protein import
More than 98% of mitochondrial proteins are encoded in the nucleus and synthesized as precursors in the cytosol. These preproteins are imported into the mitochondria via translocases present in the outer and inner mitochondrial membrane [55]. The preproteins are targeted to receptor proteins on the outer mitochondrial membrane and to general import proteins of the TOM complex [56]. Transport across the inner membrane is mediated by the TIM complex [56]. A role for CL in protein import was first suggested by the finding that protein import was blocked when yeast cells were treated with the CL-binding compound doxorubicin [57]. Subsequent studies with the crd1Δ mutant indicated that the absence of CL leads to a reduced membrane potential and decreased protein import [35]. More recently a reconstitution study indicated that the membrane integration of mitochondrial preproteins is most efficient when a presequence translocase is reconstituted in CL-containing membranes [58].
4.1 Defective protein import in the absence of CL
Jiang and co-workers showed that crd1Δ cells exhibit a decreased mitochondrial membrane potential as well as defective import of proteins into mitochondria [35]. The import defect was greater for a preprotein that required a membrane potential. More recently, a link between BTHS and protein import was suggested by the finding that a BTHS-like illness known as dilated cardiomyopathy with ataxia (DCMA) syndrome is caused by mutations in the protein import gene DNAJC19/TIM14 [59]. Like BTHS, DCMA syndrome is characterized by cardiomyopathy, neutropenia and elevated 3-methylglutaconic acid. The DNAJC19 protein shares sequence similarity with Tim14p, a protein that is associated with the inner mitochondrial membrane motor complex of Tim23p. Because the clinical presentation of DCMA is very similar to that of BTHS, it is interesting to speculate that the defect in BTHS may be caused or exacerbated by defective mitochondrial protein import.
5. Mitochondrial anionic phospholipids and cell wall biogenesis
Experiments to isolate suppressors of the pgs1Δ temperature sensitivity phenotype led to the identification of a loss of function mutant of KRE5, a gene involved in cell wall biogenesis [60]. The cell wall is an essential organelle that determines cell shape and integrity. It is a dynamic structure that undergoes numerous modifications in response to changes in growth phase and environment [61, 62]. The yeast cell wall is made up of two layers, including a highly glycosylated mannoprotein outer layer, and an inner layer enriched in chitin and glucan [63]. β-1,3-glucan and chitin together provide mechanical strength to the cell wall. β-1,3-glucan, the product of the plasma membrane protein β-1,3-glucan synthase, interconnects cell wall components [63, 64]. Glucan synthase is composed of a catalytic subunit encoded by homologous genes FKS1 and FKS2 [65, 66], and a regulatory subunit, Rho1p [64]. Chitin is enriched in and around bud scars and is increased in number or size upon weakening of the cell wall [63, 67]. Defects in the assembly of cell wall components affect the cellular response to heat and osmotic stress.
Suppression of pgs1Δ temperature sensitivity by kre5Δ suggested a connection between the CL pathway and cell wall biogenesis [60], consistent with the finding of Lussier et al. that disruption of the PGS1 promoter leads to hypersensitivity to cell wall perturbing agents such as zymolyase, calcofluor white, papulacandin and caffeine [68]. Biochemical analysis of the cell wall of pgs1Δ indicated that the mutant has reduced levels of β-1,3-glucan [60]. Consistent with this, cytological studies revealed that pgs1Δ cells exhibited the enlarged cell phenotype characteristic of cell wall mutants. Levels of β-1,3-glucan were increased in the kre5 suppressor mutant, and the phenotypes of temperature sensitivity and enlarged spherical morphology were suppressed by kre5.
What are the mechanisms linking mitochondrial anionic phospholipids to the cell wall, where these lipids are not present? A clue to this question comes from the finding that cell wall defects in the pgs1Δ mutant are associated with perturbation of the cell integrity pathway [69]. In this pathway, activation of Pkc1p in response to cell wall stress results in activation of a cascade of proteins in the Mpk1/Slt2 mitogen activated protein kinase (MAPK) pathway, culminating in the activation/dual phosphorylation of Slt2. The dual phosphorylated Slt2p activates transcription factors that up-regulate genes involved in cell wall synthesis [70, 71]. Interestingly, the pgs1Δ mutant exhibits defective Slt2p activation, which is restored by the kre5 suppressor [69]. The mechanism linking PG/CL to the Slt2 pathway is not known.
6. CL is associated with aging and apoptosis
A role for CL has been implicated in the inter-related processes of aging and apoptosis [72-74], and cells lacking CL exhibit accelerated entry into apoptosis and an increased level of cell death in response to stimuli that induce apoptosis [75]. Decreased CL and a loss of mitochondrial function have been observed in the early stages of aging in rats and humans [76, 77]. An increased content of CL species containing highly unsaturated acyl groups is also observed in old cells [78]. CL is required for the loose binding of cytochrome c to the mitochondrial inner membrane by electrostatic interactions in normal cells, and PG can only partly compensate for this function [73]. CL is also required for the function of the adenine nucleotide translocase, which is a component of the mitochondrial permeability transition pore [79-81]. It has been proposed that the permeability transition pore controls a mitochondrial amplification cycle in which cytochrome c and other apoptogenic proteins are released from the mitochondrial intermembrane space into the cytosol [81]. During apoptosis, CL peroxidation and a decrease in total CL levels result in a significant increase in cytochrome c release into the cytoplasm and an accumulation of free cytochrome c in the intermembrane space [73, 75, 82-84]. The release of cytochrome c represents a central step in apoptotic signaling [72, 83, 85]. Interestingly, in addition to loose binding between cytochrome c and CL, tight binding through hydrophobic interactions in the inner membrane induces partial unfolding of cytochrome c and the activation of its peroxidase catalytic activity [86]. Recent studies showed that cytochrome c is a catalyst for CL peroxidation, which contributes to permeation of the mitochondrial outer membrane and release of cytochrome c [87]. In vitro studies showed that cytochrome c has a lower affinity for peroxidized CL than normal CL [88]. These findings suggest that during apoptosis, CL peroxidation caused by peroxidase activity of cytochrome c may facilitate the release of cytochrome c from both loose and tight binding with CL.
As discussed above, CL is required for maintaining the stability and function of electron transport chain supercomplexes [72, 89-91]. As a result, the loss of CL results in respiration defects that may contribute to the generation of ROS. Increased ROS, which is linked to mitochondrial dysfunction, triggers both apoptosis and aging [73, 92, 93], and is associated with a shortened life span in many species [94]. CL is particularly susceptible to ROS attack because of its location in the mitochondrial inner membrane close to the site of ROS generation, and due to its high content of unsaturated fatty acids that are highly vulnerable to oxidative damage [84, 95]. In this manner, increased ROS in CL-deficient cells may contribute to a further decrease in CL.
Chen and co-workers have shown that, in response to ethanol, CL mutants taz1Δ and crd1Δ exhibit increased protein carbonylation, an indicator of ROS [54]. As the presence of paraquat, menadione or hydrogen peroxide (H2O2) did not lead to severe growth defects in the CL mutants as compared to wild type, the increase in ROS was most likely not due to defective oxidant defense systems. Ethanol sensitivity and increased protein carbonylation in the taz1Δ mutant but not in crd1Δ were rescued by supplementation with oleic acid, suggesting that oleoyl-CL and/or oleoyl-MLCL enables growth of taz1Δ in ethanol by decreasing oxidative stress. The finding of increased oxidative stress in the taz1Δ mutant during respiratory growth may have important implications for understanding the pathogenesis of BTHS [54].
The observations of Chen et al. that growth of crd1 was not severely affected by H2O2 differed somewhat from a previous report in which a phenotypic screen of yeast deletion mutants identified crd1Δ as sensitive to H2O2 [96]. The most likely reason for the disparity is that sensitivity was determined differently in the two studies. Chen et al. compared growth of crd1Δ to mutants in SOD1 and SOD2, which are extremely sensitive to H2O2, under conditions that would mask mild sensitivity. In contrast, the experiments in Higgins et al. compared sensitivity of mutants to wild type cells in short term incubations, conditions under which mild sensitivity would easily be detected. It is thus likely that, while loss of CL does not impair SOD1 or SOD2 mediated antioxidant defense, mild sensitivity of crd1Δ cells to H2O2 is most likely mediated via a compromised respiratory chain function, as has been observed in a number of respiratory chain deletion mutants [97].
7. CL, inositolsphingolipid phospholipase C (Isc1p), and translational regulation of ETC components
Dowhan and co-workers made the surprising observation that pgs1Δ exhibits defective translational regulation of several mitochondria-encoded ETC components and of Cox4p, a nuclear-encoded component of the ETC [98]. The translational defect resulted from the lack of PG/CL in the mitochondrial membrane, as re-introduction of PGS1 on a high copy plasmid restored expression of Cox4p. RNA levels were not affected, and in vitro studies indicated that the defect was not due to decreased protein import but rather to a failure of translation [99]. Deletion analysis of the upstream non-coding region of COX4 suggested that a cis-acting sequence with two stem-loops in the 5’ UTR appeared to be responsible for inhibition of COX4 translation. Trans-acting factors that bind to this region have not been identified; however, binding of a protein factor(s) to this sequence was observed with cytoplasm from pgs1Δ but not wild type PGS1 cells, and loss of function mutants that allowed expression of reporter constructs under control of the COX4 promoter were isolated. These findings identify a novel cross talk pathway between mitochondria and the nucleus, in which translation of nuclear-encoded proteins destined for the mitochondrial membrane respond to a deficiency of mitochondrial anionic lipids PG and/or CL [99].
Defective translation of Cox4p was also seen with the loss of inositol phosphosphingolipid phospholipase C (Isc1p), a member of the family of neutral sphingomyelinases that regulate ceramide synthesis [100]. Isc1p activity is impaired in the pgs1Δ mutant, suggesting that PG/CL is required for activation of this enzyme [100]. Interestingly, the phenotypic defects of isc1Δ and pgs1Δ are similar, which suggests that these genes may have overlapping functions. These findings led to the speculation that PG regulates translation of the ETC proteins indirectly by activation of Isc1p.
8. CL and BTHS
The studies cited above indicate that the CL pathway is crucial for mitochondrial bioenergetics and for essential cellular functions not generally associated with respiration. It is, therefore, not surprising that perturbation of this pathway in humans leads to deleterious consequences. As discussed earlier, BTHS is a disorder resulting from loss of tafazzin, a CL remodeling enzyme. About ten years after the first description of BTHS, the locus was mapped to Xq28 [101] and mutations were identified in G4.5, the tafazzin gene [102]. The link between CL and BTHS was first reported by Peter Vreken and colleagues, who demonstrated that fibroblast cultures from BTHS patients contain less CL than control cultures [33]. Furthermore, BTHS cells were defective in acylation of CL and PG with unsaturated fatty acids. Subsequent analysis showed that BTHS cells contained a decrease in total CL content [33] and an accumulation of MLCL [29]. The predominant acyl species, tetralinoleoyl-CL is absent from BTHS cells [12].
Lymphoblast mitochondria from patients with BTHS exhibit hyperproliferation and abnormalities in energy metabolism [103]. These abnormalities led to impaired coupling, consistent with the studies in tafazzin deficient yeast [104]. Due to the adhesion of opposing membranes, the intracrista space in BTHS mitochondria appears to be deformed [105], which may explain the decrease in mitochondrial membrane potential in BTHS [103] and in yeast taz1Δ [104].
Approximately 28 different mutations resulting in single amino acid changes in tafazzin have been identified in BTHS patients[26]. The mutations result in a complete loss of tafazzin or in expression of a severely truncated protein [28]. Interestingly, the clinical presentation of BTHS varies a great deal, from those who have severe incapacitating disease to those who are nearly asymptomatic, even among patients with identical mutations. This variation indicates that physiological modifiers may play a significant role in the BTHS phenotype. Thus, while it is clear that tafazzin is a CL transacylase, the cellular consequences of defective tafazzin and the molecular basis underlying the pathologies observed in BTHS patients are not understood. Homologues of human tafazzin are present throughout eukaryotic species from yeast to mammals, and yeast [25, 27, 28], Drosophila [106] and zebrafish [107] models of BTHS have been characterized. Genetic studies in these model systems may help to elucidate the mechanisms linking tafazzin to the cellular defects in BTHS, and to identify the physiological modifiers of the BTHS phenotype.
9. Future directions
While a great deal has been learned about the biochemical role of CL from in vitro studies, the availability of yeast CL mutants has opened the door to in vivo studies of CL function. The studies cited above indicate that CL plays a role not only in mitochondrial bioenergetics, but also in cell wall integrity, mitochondrial protein import, aging and apoptosis, ceramide synthesis, and translational regulation (Fig. 2). While these studies comprise a promising beginning to understanding CL function, numerous questions remain unanswered.
Fig. 2.
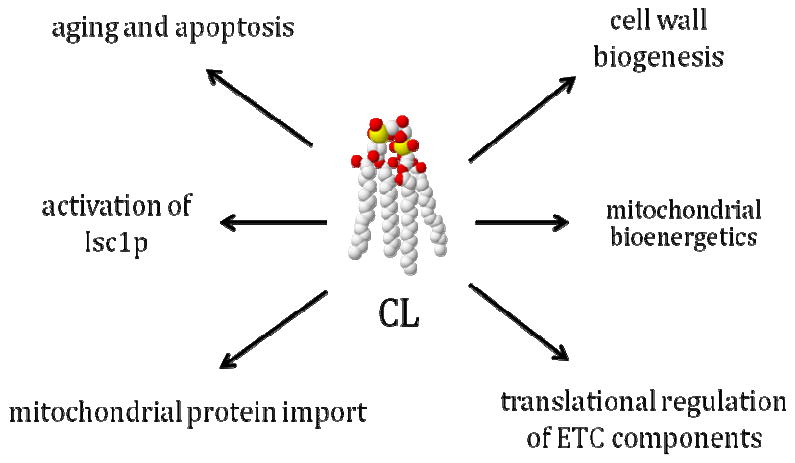
Cellular functions of the CL pathway in yeast
The regulation of CL metabolism is not well-characterized. Genes that mediate the breakdown and turnover of CL and the regulatory networks that control CL synthesis have not been identified.
Molecular mechanisms underlying the role of CL in mitochondrial bioenergetics are only beginning to be understood, and the degree to which PG can compensate for CL varies with specific functions. What are the consequences of defective supercomplex formation associated with CL deficiency? How does the loss of CL lead to increased generation of ROS? What is the nature of CL interactions with the proteins of the ETC? What are the trans-acting factors that regulate translation of ETC proteins in response to PG/CL? These questions remain to be addressed.
The mechanisms linking PG/CL to the essential cellular functions described above are not known. What role does PG/CL play in mitochondrial protein import? Are import defects the indirect results of perturbation of the membrane potential in CL mutants, or does CL play a more direct role by interacting with the protein import machinery? Does DCMA syndrome identify an import defect that is downstream from perturbation of CL synthesis? The answers to these questions may help to elucidate the direct or indirect role of CL in mitochondrial import.
The link between PG/CL and cell wall biogenesis is intriguing and may shed light on the role of CL in aging. In yeast, cell integrity and the stress response are inter-related,and both pathways affect longevity [108-113]. As discussed above, the Pkc1-activated Mpk1/Slt2 MAPK pathway is defective in pgs1Δ cells in some genetic backgrounds. This pathway is interconnected with the high osmolarity glycerol (HOG) MAPK pathway that is activated in response to osmotic and other stresses [114]. The central kinase HOG1/p38 affects longevity and replicative capacity [115, 116]. There are also several cell wall associated pathways that affect aging independent of the stress pathways [110, 111]. Because several of these interconnected pathways are defective in CL mutants, it is tantalizing to think that they identify possible routes through which CL regulates longevity. Further studies are required to elucidate the mechanisms connecting PG/CL to cell wall biogenesis, to the stress response pathways, and to aging.
Finally, how does loss of tafazzin lead to the devastating consequences in BTHS? What are the physiological modifiers that affect the phenotype associated with tafazzin deficiency? While it is clear that tafazzin remodels CL, the possibility cannot be ruled out that defective functions in tafazzin deficiency that are unrelated to CL may underlie the pathology in BTHS. The answers to these questions may shed light on the mechanisms underlying BTHS, and may point the way to new therapies for this life-threatening illness.
Acknowledgments
The authors acknowledge support from the National Institutes of Health (Grants HL 62263 and HL084218) and The Barth Syndrome Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pangborn MC. Method of recovering and refining cardiolipin. No. 2,456,836. J Pat Off Soc. 1948;108:UNKNOWN. [PubMed] [Google Scholar]
- 2.Lecocq J, Ballou CE. On the Structure of Cardiolipin. Biochemistry. 1964;3:976–80. doi: 10.1021/bi00895a023. [DOI] [PubMed] [Google Scholar]
- 3.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39(3):257–88. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 4.Hoch FL. Cardiolipins and biomembrane function. Biochim Biophys Acta. 1992;1113(1):71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- 5.Fry M, Green DE. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem. 1981;256(4):1874–80. [PubMed] [Google Scholar]
- 6.Vasilenko I, De Kruijff B, Verkleij AJ. Polymorphic phase behaviour of cardiolipin from bovine heart and from Bacillus subtilis as detected by 31P-NMR and freeze-fracture techniques. Effects of Ca2+, Mg2+, Ba2+ and temperature. Biochim Biophys Acta. 1982;684(2):282–6. doi: 10.1016/0005-2736(82)90018-9. [DOI] [PubMed] [Google Scholar]
- 7.de Kruijff B, et al. Molecular aspects of the bilayer stabilization induced by poly(L-lysines) of varying size in cardiolipin liposomes. Biochim Biophys Acta. 1985;820(2):295–304. doi: 10.1016/0005-2736(85)90124-5. [DOI] [PubMed] [Google Scholar]
- 8.Gohil VM, Thompson MN, Greenberg ML. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J Biol Chem. 2005;280(42):35410–6. doi: 10.1074/jbc.M505478200. [DOI] [PubMed] [Google Scholar]
- 9.Schlame M, et al. Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids. 2005;138(12):38–49. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Kito M, et al. Differences in fatty acid composition among phosphatidylethanolamine, phosphatidylglycerol and cardiolipin of Escherichia coli. Biochim Biophys Acta. 1972;260(3):475–8. doi: 10.1016/0005-2760(72)90062-8. [DOI] [PubMed] [Google Scholar]
- 11.Schlame M, Brody S, Hostetler KY. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur J Biochem. 1993;212(3):727–35. doi: 10.1111/j.1432-1033.1993.tb17711.x. [DOI] [PubMed] [Google Scholar]
- 12.Barth PG, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060) J Inherit Metab Dis. 1999;22(4):555–67. doi: 10.1023/a:1005568609936. [DOI] [PubMed] [Google Scholar]
- 13.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res. 2007 doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li G, et al. New insights into the regulation of cardiolipin biosynthesis in yeast: implications for Barth syndrome. Biochim Biophys Acta. 2007;1771(3):432–41. doi: 10.1016/j.bbalip.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Chang SC, et al. The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J Biol Chem. 1998;273(16):9829–36. doi: 10.1074/jbc.273.16.9829. [DOI] [PubMed] [Google Scholar]
- 16.Kelly BL, Greenberg ML. Characterization and regulation of phosphatidylglycerolphosphate phosphatase in Saccharomyces cerevisiae. Biochim Biophys Acta. 1990;1046(2):144–50. doi: 10.1016/0005-2760(90)90181-v. [DOI] [PubMed] [Google Scholar]
- 17.Tamai KT, Greenberg ML. Biochemical characterization and regulation of cardiolipin synthase in Saccharomyces cerevisiae. Biochim Biophys Acta. 1990;1046(2):214–22. doi: 10.1016/0005-2760(90)90192-z. [DOI] [PubMed] [Google Scholar]
- 18.Hostetler KY, van den Bosch H, van Deenen LL. The mechanism of cardiolipin biosynthesis in liver mitochondria. Biochim Biophys Acta. 1972;260(3):507–13. doi: 10.1016/0005-2760(72)90065-3. [DOI] [PubMed] [Google Scholar]
- 19.Hostetler KY, Van den Bosch H, Van Deenen LL. Biosynthesis of cardiolipin in liver mitochondria. Biochim Biophys Acta. 1971;239(1):113–9. doi: 10.1016/0005-2760(71)90201-3. [DOI] [PubMed] [Google Scholar]
- 20.Tuller G, et al. YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae. FEBS Lett. 1998;421(1):15–8. doi: 10.1016/s0014-5793(97)01525-1. [DOI] [PubMed] [Google Scholar]
- 21.Jiang F, Rizavi HS, Greenberg ML. Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol Microbiol. 1997;26(3):481–91. doi: 10.1046/j.1365-2958.1997.5841950.x. [DOI] [PubMed] [Google Scholar]
- 22.Chang SC, et al. Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J Biol Chem. 1998;273(24):14933–41. doi: 10.1074/jbc.273.24.14933. [DOI] [PubMed] [Google Scholar]
- 23.Schlame M, Rustow B. Lysocardiolipin formation and reacylation in isolated rat liver mitochondria. Biochem J. 1990;272(3):589–95. doi: 10.1042/bj2720589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hauff KD, Hatch GM. Cardiolipin metabolism and Barth Syndrome. Prog Lipid Res. 2006;45(2):91–101. doi: 10.1016/j.plipres.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Gu Z, et al. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol. 2004;51(1):149–58. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 26.Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580(23):5450–5. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 27.Vaz FM, et al. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J Biol Chem. 2003;278(44):43089–94. doi: 10.1074/jbc.M305956200. [DOI] [PubMed] [Google Scholar]
- 28.Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol. 2006;174(3):379–90. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valianpour F, et al. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J Lipid Res. 2005;46(6):1182–95. doi: 10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Cardonick EH, et al. Prenatal clinical expression of 3-methylglutaconic aciduria: Barth syndrome. Prenat Diagn. 1997;17(10):983–8. doi: 10.1002/(sici)1097-0223(199710)17:10<983::aid-pd174>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 31.Mazzocco MM, Henry AE, Kelly RI. Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr. 2007;28(1):22–30. doi: 10.1097/01.DBP.0000257519.79803.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valianpour F, et al. Linoleic acid supplementation of Barth syndrome fibroblasts restores cardiolipin levels: implications for treatment. J Lipid Res. 2003;44(3):560–6. doi: 10.1194/jlr.M200217-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Vreken P, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279(2):378–82. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 34.Schlame M, et al. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51(5):634–7. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 35.Jiang F, et al. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem. 2000;275(29):22387–94. doi: 10.1074/jbc.M909868199. [DOI] [PubMed] [Google Scholar]
- 36.Jiang F, et al. Cardiolipin synthase expression is essential for growth at elevated temperature and is regulated by factors affecting mitochondrial development. Mol Microbiol. 1999;31(1):373–9. doi: 10.1046/j.1365-2958.1999.01181.x. [DOI] [PubMed] [Google Scholar]
- 37.Zhong Q, et al. Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J Biol Chem. 2004;279(31):32294–300. doi: 10.1074/jbc.M403275200. [DOI] [PubMed] [Google Scholar]
- 38.Dzugasova V, et al. Phosphatidylglycerolphosphate synthase encoded by the PEL1/PGS1 gene in Saccharomyces cerevisiae is localized in mitochondria and its expression is regulated by phospholipid precursors. Curr Genet. 1998;34(4):297–302. doi: 10.1007/s002940050399. [DOI] [PubMed] [Google Scholar]
- 39.Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem. 1997;66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- 40.Daum G. Lipids of mitochondria. Biochim Biophys Acta. 1985;822(1):1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 41.de Andrade Rosa I, et al. Cardiolipin in hydrogenosomes: evidence of symbiotic origin. Eukaryot Cell. 2006;5(4):784–7. doi: 10.1128/EC.5.4.784-787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beyer K, Klingenberg M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry. 1985;24(15):3821–6. doi: 10.1021/bi00336a001. [DOI] [PubMed] [Google Scholar]
- 43.Beyer K, Nuscher B. Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry. 1996;35(49):15784–90. doi: 10.1021/bi9610055. [DOI] [PubMed] [Google Scholar]
- 44.Fry M, Green DE. Cardiolipin requirement by cytochrome oxidase and the catalytic role of phospholipid. Biochem Biophys Res Commun. 1980;93(4):1238–46. doi: 10.1016/0006-291x(80)90622-1. [DOI] [PubMed] [Google Scholar]
- 45.Sedlak E, Robinson NC. Phospholipase A(2) digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure. Biochemistry. 1999;38(45):14966–72. doi: 10.1021/bi9914053. [DOI] [PubMed] [Google Scholar]
- 46.Rytomaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem. 1994;269(3):1770–4. [PubMed] [Google Scholar]
- 47.Pfeiffer K, et al. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278(52):52873–80. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 48.Koshkin V, Greenberg ML. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem J. 2002;364(Pt 1):317–22. doi: 10.1042/bj3640317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koshkin V, Greenberg ML. Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem J. 2000;347(Pt 3):687–91. [PMC free article] [PubMed] [Google Scholar]
- 50.Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19(8):1777–83. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280(33):29403–8. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, et al. The enzymatic function of tafazzin. J Biol Chem. 2006;281(51):39217–24. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 53.McKenzie M, et al. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361(3):462–9. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 54.Chen S, He Q, Greenberg ML. Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol Microbiol. 2008;68(4):1061–72. doi: 10.1111/j.1365-2958.2008.06216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pfanner N, Craig EA, Meijer M. The protein import machinery of the mitochondrial inner membrane. Trends Biochem Sci. 1994;19(9):368–72. doi: 10.1016/0968-0004(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 56.Lill R, Neupert W. Mechanisms of protein import across the mitochondrial outer membrane. Trends Cell Biol. 1996;6(2):56–61. doi: 10.1016/0962-8924(96)81015-4. [DOI] [PubMed] [Google Scholar]
- 57.Eilers M, Endo T, Schatz G. Adriamycin, a drug interacting with acidic phospholipids, blocks import of precursor proteins by isolated yeast mitochondria. J Biol Chem. 1989;264(5):2945–50. [PubMed] [Google Scholar]
- 58.van der Laan M, et al. Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat Cell Biol. 2007;9(10):1152–9. doi: 10.1038/ncb1635. [DOI] [PubMed] [Google Scholar]
- 59.Davey KM, et al. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43(5):385–93. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong Q, et al. Loss of function of KRE5 suppresses temperature sensitivity of mutants lacking mitochondrial anionic lipids. Mol Biol Cell. 2005;16(2):665–75. doi: 10.1091/mbc.E04-09-0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smits GJ, van den Ende H, Klis FM. Differential regulation of cell wall biogenesis during growth and development in yeast. Microbiology. 2001;147(Pt 4):781–94. doi: 10.1099/00221287-147-4-781. [DOI] [PubMed] [Google Scholar]
- 62.Cabib E, et al. The yeast cell wall and septum as paradigms of cell growth and morphogenesis. J Biol Chem. 2001;276(23):19679–82. doi: 10.1074/jbc.R000031200. [DOI] [PubMed] [Google Scholar]
- 63.Klis FM, et al. Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2002;26(3):239–56. doi: 10.1111/j.1574-6976.2002.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 64.Qadota H, et al. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science. 1996;272(5259):279–81. doi: 10.1126/science.272.5259.279. [DOI] [PubMed] [Google Scholar]
- 65.Mazur P, et al. Differential expression and function of two homologous subunits of yeast 1,3-beta-D-glucan synthase. Mol Cell Biol. 1995;15(10):5671–81. doi: 10.1128/mcb.15.10.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inoue SB, et al. Signaling toward yeast 1,3-beta-glucan synthesis. Cell Struct Funct. 1996;21(5):395–402. doi: 10.1247/csf.21.395. [DOI] [PubMed] [Google Scholar]
- 67.Popolo L, Gualtieri T, Ragni E. The yeast cell-wall salvage pathway. Med Mycol. 2001;39 Suppl 1:111–21. [PubMed] [Google Scholar]
- 68.Lussier M, et al. Completion of the Saccharomyces cerevisiae genome sequence allows identification of KTR5, KTR6 and KTR7 and definition of the nine-membered KRE2/MNT1 mannosyltransferase gene family in this organism. Yeast. 1997;13(3):267–74. doi: 10.1002/(SICI)1097-0061(19970315)13:3<267::AID-YEA72>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 69.Zhong Q, et al. Up-regulation of the cell integrity pathway in saccharomyces cerevisiae suppresses temperature sensitivity of the pgs1Delta mutant. J Biol Chem. 2007;282(22):15946–53. doi: 10.1074/jbc.M701055200. [DOI] [PubMed] [Google Scholar]
- 70.Terashima H, et al. Up-regulation of genes encoding glycosylphosphatidylinositol (GPI)-attached proteins in response to cell wall damage caused by disruption of FKS1 in Saccharomyces cerevisiae. Mol Gen Genet. 2000;264(12):64–74. doi: 10.1007/s004380000285. [DOI] [PubMed] [Google Scholar]
- 71.Jung US, Levin DE. Genome-wide analysis of gene expression regulated by the yeast cell wall integrity signalling pathway. Mol Microbiol. 1999;34(5):1049–57. doi: 10.1046/j.1365-2958.1999.01667.x. [DOI] [PubMed] [Google Scholar]
- 72.McMillin JB, D W. Cardiolipin and apoptosis. Biochim Biophys Acta. 2002;1585:97–107. doi: 10.1016/s1388-1981(02)00329-3. [DOI] [PubMed] [Google Scholar]
- 73.Iverson SL, O S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423:37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 74.Pollack M, L C. Apoptosis and aging: role of the mitochondria. J Gerontol A Biol Sci Med Sci. 2001;56(11):B475–82. doi: 10.1093/gerona/56.11.b475. [DOI] [PubMed] [Google Scholar]
- 75.Choi SY, G F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007;14(3):597–606. doi: 10.1038/sj.cdd.4402020. [DOI] [PubMed] [Google Scholar]
- 76.Paradies G, R F. Age-related changes in the activity of the pyruvate carrier and in the lipid composition in rat-heart mitochondria. Biochim Biophys Acta. 1990;1016:207–12. doi: 10.1016/0005-2728(90)90060-h. [DOI] [PubMed] [Google Scholar]
- 77.Maftah A, R M, Dumas M, Bonte F, Meybeck A, Julien R. Human epidermal cells progressively lose their cardiolipins during ageing without change in mitochondrial transmembrane potential. Mech Ageing Dev. 1994;77:83–96. doi: 10.1016/0047-6374(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 78.Lee HJ, M J, Rapoport SI, Bazinet RP. Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis. 2006;5:2. doi: 10.1186/1476-511X-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoffmann B, S A, Schlame M, Beyer K, Klingenberg M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J Biol Chem. 1994;6:1940–4. [PubMed] [Google Scholar]
- 80.Halestrap AP. The mitochondrial permeability transition: its molecular mechanism and role in reperfusion injury. Biochem Soc Symp. 1999;66:181–203. doi: 10.1042/bss0660181. [DOI] [PubMed] [Google Scholar]
- 81.Crompton M. Mitochondrial intermembrane junctional complexes and their role in cell death. 2000;529(1121) doi: 10.1111/j.1469-7793.2000.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ostrander DB, S G, Amoscato AA, McMillin JB, Dowhan W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem. 2001;276:38061–7. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- 83.Ott M, R J, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–63. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shidoji Y, H K, Komura S, Ohishi N, Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun. 1999;264(2):343–7. doi: 10.1006/bbrc.1999.1410. [DOI] [PubMed] [Google Scholar]
- 85.Goldstein JC, M-P C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12(5):453–62. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- 86.Belikova NA, et al. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry. 2006;45(15):4998–5009. doi: 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kagan VE, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1(4):223–32. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 88.Gonzalvez F, Gottlieb E. Cardiolipin: Setting the beat of apoptosis. Apoptosis. 2007;12(5):877–85. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- 89.Robinson NC. Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr. 1993;25:153–63. doi: 10.1007/BF00762857. [DOI] [PubMed] [Google Scholar]
- 90.Ostrander DB, Z M, Mileykovskaya E, Rho M, Dowhan W. Lack of mitochondrial anionic phospholipids causes an inhibition of translation of protein components of the electron transport chain. A yeast genetic model system for the study of anionic phospholipid function in mitochondria. J Biol Chem. 2001;276:25262–72. doi: 10.1074/jbc.M103689200. [DOI] [PubMed] [Google Scholar]
- 91.Pfeiffer K, G V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–80. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 92.Osiewacz HD, S C. Impact of ROS on aging of two fungal model systems: Saccharomyces cerevisiae and Podospora anserina. Free Radic Res. 2006;40(12):1350–8. doi: 10.1080/10715760600921153. [DOI] [PubMed] [Google Scholar]
- 93.Buttner S, E T, Herker E, Carmona-Gutierrez D, Kroemer G, Madeo F. Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol. 2006;175(4):521–5. doi: 10.1083/jcb.200608098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chan DC. Mitchondria: dynamic organelles in disease, aging, and developement. Cell. 2006;125(7):121–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 95.Petrosillo G, R F, Pistolese M, Paradies G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001;509:435–8. doi: 10.1016/s0014-5793(01)03206-9. [DOI] [PubMed] [Google Scholar]
- 96.Higgins VJ, et al. Phenotypic analysis of gene deletant strains for sensitivity to oxidative stress. Yeast. 2002;19(3):203–14. doi: 10.1002/yea.811. [DOI] [PubMed] [Google Scholar]
- 97.Thorpe GW, et al. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc Natl Acad Sci U S A. 2004;101(17):6564–9. doi: 10.1073/pnas.0305888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ostrander DB, et al. Lack of mitochondrial anionic phospholipids causes an inhibition of translation of protein components of the electron transport chain. A yeast genetic model system for the study of anionic phospholipid function in mitochondria. J Biol Chem. 2001;276(27):25262–72. doi: 10.1074/jbc.M103689200. [DOI] [PubMed] [Google Scholar]
- 99.Su X, Dowhan W. Translational regulation of nuclear gene COX4 expression by mitochondrial content of phosphatidylglycerol and cardiolipin in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26(3):743–53. doi: 10.1128/MCB.26.3.743-753.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vaena de Avalos S, et al. The phosphatidylglycerol/cardiolipin biosynthetic pathway is required for the activation of inositol phosphosphingolipid phospholipase C, Isc1p, during growth of Saccharomyces cerevisiae. J Biol Chem. 2005;280(8):7170–7. doi: 10.1074/jbc.M411058200. [DOI] [PubMed] [Google Scholar]
- 101.Bolhuis PA, et al. Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28. Am J Hum Genet. 1991;48(3):481–5. [PMC free article] [PubMed] [Google Scholar]
- 102.Bione S, et al. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12(4):385–9. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 103.Xu Y, et al. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest. 2005;85(6):823–30. doi: 10.1038/labinvest.3700274. [DOI] [PubMed] [Google Scholar]
- 104.Ma L, et al. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem. 2004;279(43):44394–9. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- 105.Acehan D, et al. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87(1):40–8. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu Y, et al. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A. 2006;103(31):11584–8. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Khuchua Z, et al. A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ Res. 2006;99(2):201–8. doi: 10.1161/01.RES.0000233378.95325.ce. [DOI] [PubMed] [Google Scholar]
- 108.Barros MH, et al. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J Biol Chem. 2004;279(48):49883–8. doi: 10.1074/jbc.M408918200. [DOI] [PubMed] [Google Scholar]
- 109.Kaeberlein M, et al. High osmolarity extends life span in Saccharomyces cerevisiae by a mechanism related to calorie restriction. Mol Cell Biol. 2002;22(22):8056–66. doi: 10.1128/MCB.22.22.8056-8066.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kaeberlein M, et al. Saccharomyces cerevisiae SSD1-V confers longevity by a Sir2p-independent mechanism. Genetics. 2004;166(4):1661–72. doi: 10.1534/genetics.166.4.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaeberlein M, Guarente L. Saccharomyces cerevisiae MPT5 and SSD1 function in parallel pathways to promote cell wall integrity. Genetics. 2002;160(1):83–95. doi: 10.1093/genetics/160.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vilella F, et al. Pkc1 and the upstream elements of the cell integrity pathway in Saccharomyces cerevisiae, Rom2 and Mtl1, are required for cellular responses to oxidative stress. J Biol Chem. 2005;280(10):9149–59. doi: 10.1074/jbc.M411062200. [DOI] [PubMed] [Google Scholar]
- 113.Wojda I, et al. Response to high osmotic conditions and elevated temperature in Saccharomyces cerevisiae is controlled by intracellular glycerol and involves coordinate activity of MAP kinase pathways. Microbiology. 2003;149(Pt 5):1193–204. doi: 10.1099/mic.0.26110-0. [DOI] [PubMed] [Google Scholar]
- 114.Hohmann S, Krantz M, Nordlander B. Yeast osmoregulation. Methods Enzymol. 2007;428:29–45. doi: 10.1016/S0076-6879(07)28002-4. [DOI] [PubMed] [Google Scholar]
- 115.Haq R, et al. Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 2002;62(17):5076–82. [PubMed] [Google Scholar]
- 116.Reiser V, et al. The stress-activated mitogen-activated protein kinase signaling cascade promotes exit from mitosis. Mol Biol Cell. 2006;17(7):3136–46. doi: 10.1091/mbc.E05-12-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]