

Abstract
Similar to CNG and HCN channels, EAG and ERG channels contain a cyclic nucleotide binding domain (CNBD) in their C terminus. While cyclic nucleotides have been shown to facilitate opening of CNG and HCN channels, their effect on EAG and ERG channels is less clear. Here we explored cyclic nucleotide binding and modulation of mEAG1 and hERG1 channels with fluorescence and electrophysiology. Binding of cyclic nucleotides to the isolated CNBD of mEAG1 and hERG1 channels was examined with two independent fluorescence-based methods: changes in tryptophan fluorescence and fluorescence of an analog of cAMP, 8-NBD-cAMP. As a positive control for cyclic nucleotide binding we used changes in the fluorescence of the isolated CNBD of mHCN2 channels. Our results indicated that cyclic nucleotides do not bind to the isolated CNBD domain of mEAG1 channels and bind with low affinity (Kd ≥ 51 μm) to the isolated CNBD of hERG1 channels. Consistent with the results on the isolated CNBD, application of cyclic nucleotides to inside-out patches did not affect currents recorded from mEAG1 channels. Surprisingly, despite its low affinity binding to the isolated CNBD, cAMP also had no effect on currents from hERG1 channels even at high concentrations. Our results indicate that cyclic nucleotides do not directly modulate mEAG1 and hERG1 channels. Further studies are necessary to determine if the CNBD in the EAG family of K+ channels might harbor a binding site for a ligand yet to be uncovered.
The EAG family of K+ channels comprises ether-à-go-go (EAG),2 EAG-related gene (ERG), and EAG-like (ELK) K+ channel subfamilies (1) with diverse tissue expression patterns and physiological functions (reviewed in Ref. 2). mEAG channels are overexpressed in tumor tissues (3, 4), where they are involved in regulation of tumor progression (5, 6). Inhibition of the EAG channel expression by RNAi interference (7), application of channel blockers (8, 9), and monoclonal antibody that selectively inhibits currents from EAG channels (10) decreased cell proliferation in tumor tissues.
ERG channels are best known for their function in the heart. Because of their unique physiological properties, fast inactivation, and slow deactivation, ERG channels are major contributors to the repolarization phase of the cardiac action potential (11–14). Mutations in the ERG channels and administration of ERG channel blockers, such as class III antiarrhythmic drugs, cause long QT syndrome, a potentially lethal cardiac arrhythmia characterized by a prolonged cardiac action potential (15–19). In addition to their role in cardiac excitability, ERG channels also regulate proliferation of tumor cells (20–22). The physiological role of ELK channels is not well understood, however, early reports suggest their possible involvement in the regulation of neuronal excitability (23).
K+ channels in the EAG family are structurally related to the cyclic nucleotide-gated (CNG) and hyperpolarization-activated cyclic nucleotide-modulated (HCN) K+ channels (1, 24). All of these channels contain a CNBD in their C-terminal region. Unlike HCN and CNG channels whose regulation by direct binding of cyclic nucleotides to the CNBD is well established (25–32), regulation of the EAG family of K+ channels by the direct binding of cyclic nucleotides is controversial. It has been reported that EAG channels in mouse (33), rat (34), and bovine retina (35) and ERG channels in humans (36) are not regulated by cyclic nucleotides. However, in similar studies other groups have shown that EAG channels in Drosophila (37, 38) and ERG channels in humans (39, 40) are regulated by cAMP. Most of the above mentioned studies were performed in a whole-cell or two-electrode voltage clamp configuration. In either of these configurations it is difficult if not impossible to control the concentration of the applied cyclic nucleotides and differentiate between direct effect of cyclic nucleotides on the EAG and ERG channels and secondary effects through signaling pathways regulated by cyclic nucleotides.
To resolve this controversy we took a direct approach by applying cyclic nucleotides directly to the isolated CNBD and membrane patches expressing channels in the inside-out configuration. The direct binding of cAMP and cGMP to the isolated CNBD of the mEAG1 (also known as KCNH1 and Kv10.1) and hERG1 (also known as KCNH2 and Kv11.1) channels was examined with fluorescence-based methods. To demonstrate the validity of our approach, the fluorescence methods were also applied to the isolated CNBD of mHCN2 channels. The effect of cAMP and cGMP on full-length channels was examined by direct application of cyclic nucleotides to inside-out patches expressing mEAG1 and hERG1 channels. The fluorescent-based experiments indicated no binding of the cyclic nucleotides to the CNBD of mEAG1 and only low affinity binding (Kd ≥ 51 μm) of cAMP to the CNBD of hERG1 channels. Direct application of cAMP and cGMP had no effect on the currents recorded from mEAG1 and hERG1 channels. Our results indicate that cAMP and cGMP do not regulate mEAG1 and hERG1 channels by direct binding to the CNBD.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
To express portions of the C-terminal regions containing the CNBD, the DNA fragments encoding residues 505–702 of the mEAG1 channel (mEAG1-(505–702)), 666–872 of the hERG1 channel (hERG1-(666–872)), and 443–645 of the wild-type and mutant mHCN2 channels with Trp substituted for Leu at the position 586 (mHCN2J and mHCN2J-L586W, respectively) were subcloned into a pETGQ vector (41). The constructs were grown in BL21 (DE3) cells at 37 °C. At OD 0.6–0.8, the cell cultures were cooled on ice and induced with 1 m isopropyl-1-thio-β-d-galactopyranoside. After growing overnight at 18 °C, the cells were harvested by centrifugation at 5000 × rpm for 15 min at 4 °C, and the cell pellets were frozen at −80 °C. The cells were then resuspended in a lysis buffer (150 mm KCl, 10% glycerol, 1 mm TCEP, 30 mm HEPES, 1 mm phenylmethylsulfonyl fluoride, and 2.5 mg/ml DNase; pH 7.5) and lysed in an Emulsiflex-C5 (Avestin). Insoluble protein was separated by centrifugation for 45 min at 40,000 × rpm at 4 °C. The protein of interest was then purified from the supernatant by Ni2+-nitrilotriacetic acid chromatography and eluted on a linear gradient to 500 mm imidazole. The 6× His tag was cleaved with thrombin protease (Calbiochem) and separated with size exclusion chromatography. The protein was purified on a Superdex 200 column (Amersham Biosciences) equilibrated with the buffer used for the subsequent experiments (150 mm KCl, 10% glycerol, 1 mm TCEP, 30 mm HEPES; pH 7.5). The purified protein was stored at −80 °C in small aliquots and thawed immediately before the experiments. The molecular weight of the constructs used was verified on Coomassie Blue-stained gels and with mass spectrometry. The protein concentration was determined by absorbance at 280 nm and with RC DC protein assay (Bio-Rad) based on the Lowry method (42). Both methods gave similar values for the protein concentration.
hERG1-(666–872) was eluted at the void volume of the column when expressed in pETGQ vector. In an attempt to increase monodispersity hERG1-(666–872) was subcloned into pHMalc2T vector, a modified version of pMalc2T (New England Biolabs), with maltose-binding protein (MBP) as a tag. The purification steps were carried out as described above except the protein was purified on amylose affinity column (New England Biolabs), instead of the Ni2+ affinity column, and eluted on a linear gradient to 50 mm maltose. hERG1-(666–872) MBP fusion protein (hERG1-(666–872)/MBP) was monodisperse and used for the fluorescence-based assays.
Fluorescence Measurements
Fluorescence intensity was recorded with a Fluorolog 3 spectrophotometer (HORIBA Jobin Yvon) using FluorEssence software. For the experiments with cAMP and cGMP, the sample was excited at 295 nm, and the emission spectrum was recorded from 300 to 500 nm. To account for the decrease in the excitation and emission intensities due to the absorbance, observed fluorescence intensities of the sample and buffer were corrected for the inner filter effect according to Equation 1 (43),
where Fci and Foi are the corrected and observed fluorescence intensities at the i nm wavelength, A295 and Ai are the absorbance recorded at 295 nm and i nm wavelength, respectively. To calculate the final fluorescence intensity, the corrected buffer intensity was subtracted from the corrected sample intensity. Each of the experiments was repeated at least three times. The error bars on the figures correspond to the S.E.
For experiments with 8-NBD-cAMP a fluorescent analog of cAMP (BIOLOG, Bremen, Germany), the sample was excited at 470 nm, and the emission spectra were recorded from 480 to 650 nm. The inner filter correction was carried out similar to the experiments with cAMP and cGMP, except the A at 470 nm was used instead of the A at 295 nm in Equation 1.
To estimate the binding affinity, plots of the change of the peak fluorescence intensities versus total cyclic nucleotide concentration were analyzed as in Cukkemane et al. (60). Briefly, binding of a ligand to a receptor was treated as a simple first order reaction,
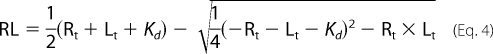 |
where R, L, and RL are concentrations of the free receptor and ligand, and receptor-ligand complex, respectively; Rt and Lt are total receptor and ligand concentrations; ΔF is the peak fluorescence change, and x is a scaling factor. The data analysis and fitting of the plots was performed in Origin (Microcal Software, Inc).
Circular Dichroism Measurements
Circular dichroism (CD) spectra were recorded with an Aviv 62A DS spectrometer (Aviv Associates, Lakewood, NJ) at 22 °C using a 1-mm pathlength cuvette. Three scans were averaged for each sample with data acquired every 1 nm. For the CD experiments, the protein was purified in 150 mm potassium phosphate buffer with 10% glycerol, 1 mm TCEP, pH 7.5. The protein concentration used was 20 μm.
Electrophysiology
The cDNA encoding mHCN2 channels in pGEM vector, and hERG1, hERG1-S631A, and mEAG1 channels in pGH19 vector were kindly provided by S. Siegelbaum (Columbia University, New York, NY) and G. Robertson (University of Wisconsin-Madison, Madison, WI), respectively. The cRNA was transcribed using the T7 mMessage mMachine kit (Ambion). Xenopus laevis oocytes were defolliculated and injected with the cRNA as previously described (44). Following manual removal of the vitelline membrane, currents were recorded in the inside-out patch configuration (45) with an EPC-10 patch-clamp amplifier (HEKA Electronik). Data were acquired with Pulse software (HEKA Elektronik) and analyzed with Igor (WaveMetrics, Inc). Patch pipettes were pulled from borosilicate glass and had resistances of 0.40–1 mΩ after fire polishing. The intracellular (bath) and extracellular (pipette) solutions contained 130 mm KCl, 10 mm HEPES, 0.2 mm EDTA, pH 7.2. cAMP or cGMP were added to the bath solution as indicated. The bath solution was changed with RSC- 100 solution changer (BioLogic). mHCN2 currents were elicited by applying a series of 5-s voltage pulses (ranging from −140 to −70 mV in 10-mV increments) from a holding potential of 0 mV, followed by a 1-s voltage tail pulse to −40 mV. hERG1 currents were elicited by applying a series of 0.25-s voltage pulses (ranging from −100 to +100 mV in 20-mV increments) from a holding potential of −80 mV, followed by a 0.5-s voltage pulse to −100 mV. mEAG1 and hERG1-S631A currents were elicited by applying a series of 0.1-s voltage pulses (ranging from −140 to +50 mV in 10-mV increments) from a holding potential of −100 mV, followed by a 0.5-s voltage pulse to −120 mV. Currents were not leak-subtracted.
RESULTS
The Residues Crucial for Cyclic Nucleotide Binding Are Absent in the CNBD of the EAG Family of K+ Channels
To determine if the CNBD of the EAG channels contain residues implicated in cyclic nucleotide binding we aligned amino acid sequences of the CNBD of several members of the EAG family of K+ channels with the sequences of proteins that are regulated by direct binding of cyclic nucleotides (Fig. 1). A canonical CNBD contains three α-helices and eight β-strands forming an antiparallel β-roll. The general architecture of the CNBD emerged from the crystal structures of CAP (46), PKA (47), and the C terminus of the HCN2 (31) and MlotiK1 (48) channels. Extensive biochemical studies and the crystal structures of the CNBD identified six invariant residues, indicated by black arrows in Fig. 1. The Gly residues are essential for the structural integrity of the β-roll, Glu and Arg residues take part in the binding of cyclic nucleotides, and the role of Ala is not clear (49, 50).
FIGURE 1.

The invariant Glu and Arg residues are not conserved in the EAG family of K+ channels. Amino acid sequence alignment of the CNBD of the EAG family of K+ channels and other cyclic nucleotide-binding proteins. Identical residues are red on yellow background, conserved residues are blue on cyan background, similar residues are black on green background, and weakly similar residues are in green. The α-helices (red rectangles) and β-sheets (blue arrows) represent structural motifs from the crystal structure of the CNBD of mHCN2 channels (31). The six invariant residues are indicated by black arrows. The GenInfo identifier numbers are: mEAG1, 487740; rEAG1, 557265; dEAG, 24642070; hERG1, 103488986; mELK2, 187954689; mHCN2, 148699724; bCNGA1, 231739; MlotiK1, 81779664; bPKAIαB, 145559486; CAP, 168751822.
The three invariant Gly residues are conserved in the EAG family of K+ channels, indicating that the overall fold of the CNBD of the EAG channels is similar to the canonical CNBD structure. The invariant Glu residue is conserved in the hERG1 and is replaced by Asp and Cys in the mEAG1 and mELK2 channels, respectively. In a canonical CNBD, the Glu residue directly binds to the ribose of cyclic nucleotides. Mutations of the Glu residue to Ala in HCN2 channels (51), to seven different amino acids in CAP (52, 53) and to Lys in PKA (54) drastically decreased the apparent binding affinity of cyclic nucleotides. Interestingly, substituting Asp for the Glu residue also impaired cAMP binding in CAP (53).
The invariant Arg residue is absent in the EAG family of K+ channels. In other cyclic nucleotide-binding proteins, it forms a salt bridge with the negatively charged phosphate of cyclic nucleotides. Mutations of the Arg residue to Lys or Trp in PKA (54, 55), to several different residues, including Ala, in CAP (52, 53, 56), and to Gln in MlotiK1 channels (57) abolished cAMP binding. Mutation of the Arg residue to Ala in MlotiK1 and HCN2 channels, and to neutral and negatively charged residues in CNGA1 channels drastically decreased apparent cyclic nucleotide binding affinity (51, 58, 59).
The invariant Glu and Arg residues are essential for the binding of cyclic nucleotides; however, there are exceptions. Even though Epac1, a cAMP-dependent small G-protein Rap activator, is known to bind cAMP with high affinity, it has Gln instead of the invariant Glu residue (49). Mutating the invariant Arg to Ala in MlotiK1 and HCN2 channels significantly decreases cyclic nucleotide binding affinity, yet, still allows for the cyclic nucleotide modulation of the channels (51, 58). Thus, the absence of the invariant Glu or Arg residues would be expected to decrease binding affinity of cyclic nucleotides; however, it will not necessarily prevent binding.
Isolated CNBD of mHCN2, mEAG1, and hERG1 Are Monodisperse and Fold Properly
To investigate binding of cyclic nucleotides to the isolated CNBD of mEAG1 and hERG1 channels, mEAG1-(505–702) and hERG1-(666–872) MBP fusion constructs were grown in BL21 (DE3) cells and purified with affinity and size-exclusion chromatography. As a positive control for cyclic nucleotide binding in our assays we used mHCN2J-L586W with a Trp residue substituted for the Leu located on the P-helix near the cyclic nucleotide binding pocket. All of the constructs were monodisperse on size exclusion chromatographs, indicating that the protein was not aggregated and likely properly folded (Fig. 2A). The exact molecular weight of the proteins was confirmed with mass spectroscopy. In addition to the CNBD, mEAG1-(505–702), hERG1-(666–872), and mHCN2J-L586W contain the A′-F′ α-helices, spanning the C-linker region between the end of the last transmembrane domain and the CNBD.
FIGURE 2.

Biochemical studies of the CNBD of the mHCN2, mEAG1, and hERG1 channels. A, size exclusion profiles of the isolated CNBD used in the study. Runs were performed on a Superdex 200 column. The void volume is indicated by the arrow. B–D, CD spectra of the isolated CNBD of the indicated channels in the far-ultraviolet region. 100 μm cAMP was added to the CNBD in C and D as indicated.
To further assess the folding of the CNBD, we measured CD spectra for mHCN2J-L586W and mEAG1-(505–702) proteins. The CD spectra indicated a similar structured fold for mHCN2J-L586W and mEAG1-(505–702) (Fig. 2B). Addition of 100 μm cAMP to mHCN2J-L586W increased α-helical content (Fig. 2C) and had no effect on the CD spectrum of mEAG1-(505–702) (Fig. 2D). This suggests that binding of cAMP causes a conformational rearrangement of the CNBD in mHCN2 channels and has little effect on the structure of the CNBD in mEAG1 channels. Increase in the α-helical content upon binding of cAMP was also reported for the isolated CNBD of MlotiK1 channels (60).
cAMP and cGMP Decrease Trp Fluorescence of mHCN2J-L586W in a Concentration-dependent Manner but Have No Effect on the Fluorescence of mEAG1-(505–702)
Fluorescence of Trp residues has been widely used as an indicator of macromolecular interactions and ligand binding due to its environmental sensitivity (43, 60). To determine if cyclic nucleotides bind to the isolated CNBD of the mEAG1 channels we took advantage of the endogenous Trp residue at position 649 that, based on a homology model of mEAG1 channels, is located in the P helix neighboring the putative cyclic nucleotide binding pocket (Fig. 3A). Binding of cyclic nucleotides in the vicinity of the Trp residue would be expected to change the Trp fluorescence.
FIGURE 3.

cAMP and cGMP decrease Trp fluorescence of mHCN2J-L586W and have no effect on the fluorescence of mEAG1-(505–702). A, ribbon representation of the homology model of mEAG1-(505–702) obtained with SWISS-MODEL (70) based on the crystal structure of mHCN2J (31). cAMP, colored in pink, is shown to illustrate a possible binding location as seen in the crystal structure of the CNBD of mHCN2 channels. Enlarged view of the cAMP binding pocket and the Trp residue are shown on the right. B and D, emission spectra of mHCN2J-L586W and mEAG1-(505–702) recorded without and with the indicated concentrations of cAMP. The excitation wavelength was 295 nm. Protein concentration was 4 μm. C and E, plots of change in the peak fluorescence intensity versus total cyclic nucleotide concentration for mHCN2J-L586W and mEAG1-(505–702). The change in the peak fluorescence intensity (ΔF) was calculated by subtracting averaged peak emission intensities for low cyclic nucleotide concentrations (intensities at 0, 0.01, and 0.1 μm cAMP at 342 nm) from the peak emission intensities. Data in C were fitted with Equation 4. The binding affinities were 13 ± 2 μm for cAMP and 62 ± 23 μm for cGMP for mHCN2J-L586W.
We first explored if Trp fluorescence can report on cyclic nucleotide binding to the CNBD of mHCN2 channels that are known to directly bind cyclic nucleotides. There are no endogenous Trp residues in the CNBD of the mHCN2 channels. Therefore, we substituted Trp for Leu residue at the position 586, analogous to 649 in mEAG1 channels, on the P-helix (mHCN2J-L586W). While wild-type mHCN2J showed very little fluorescence upon excitation at 295 nm (supplemental Fig. S1A), mHCN2J-L586W displayed a robust fluorescent signal with a peak at 342 nm (Fig. 3B). The fluorescence intensity decreased with increasing concentration of applied cAMP (Fig. 3, B and C) and cGMP (Fig. 3C). The decrease in the fluorescence intensity is specific to cyclic nucleotide binding to the CNBD as application of cAMP to free tryptophan in solution showed no dose-dependent quenching (supplemental Fig. 1B). To determine cyclic nucleotide binding affinity, we plotted the change in the peak fluorescence intensity versus the total cyclic nucleotide concentration (Fig. 3C). The dose response curves were fitted with Equation 4 as described under “Experimental Procedures.” The analysis revealed the binding affinities of 13 ± 2 μm for cAMP and 62 ± 23 μm for cGMP. These affinities are an order of magnitude lower than the cyclic nucleotide binding affinities of the isolated CNBD of MlotiK1 (60). The difference is due in part to the L586W mutation, as discussed in the next section. Nevertheless, our results indicate that Trp fluorescence can be used as a reporter of cyclic nucleotide binding in mHCN2 channels.
To determine if cyclic nucleotides bind to the CNBD domain of mEAG1 channels we measured the Trp fluorescence spectrum of mEAG1-(505–702). A robust fluorescence signal with a maxima at 342 nm was observed; however, application of up to 10 mm cAMP or cGMP had no effect on the fluorescence spectrum (Fig. 3, D and E). mEAG1-(505–702) has a second Trp residue at position 544 outside of the CNBD. Consistent with two tryptophan residues contributing to the observed fluorescence signal, the fluorescence intensity of mEAG1-(505–702) was about two times larger than the intensity of free Trp in solution at the same concentration (supplemental Fig. 1C).
8-NBD-cAMP Reports Binding to mHCN2J-L586W and hERG1- (666–872)/MBP but Not mEAG1- (505–702)
The absence of concentration-dependent changes in Trp fluorescence suggests that either cyclic nucleotides do not bind to the isolated CNBD of mEAG1 channels or the Trp residue is insensitive to the cyclic nucleotide-induced changes in environment. To test the latter possibility, we employed 8-NBD-cAMP, a fluorescent analog of cAMP. The fluorescence of 8-NBD-cAMP is minimal in solution; however it increases upon binding to protein binding sites in a hydrophobic environment (supplemental Fig. 1D). We observed a concentration-dependent increase in the 8-NBD-cAMP fluorescence upon binding to mHCN2J-L586W (Fig. 4A). To determine the binding affinity of 8-NBD-cAMP, the change in the peak fluorescence with the application of the cyclic nucleotide was plotted versus the total 8-NBD-cAMP concentration (Fig. 4B). Fitting the data with Equation 4 indicated a binding affinity for 8-NBD-cAMP of 3.8 ± 0.6 μm. 8-NBD-cAMP has been show to bind with a few-fold higher affinity than its non-fluorescent analog (58, 60). For example, binding affinities of 8-NBD-cAMP and cAMP to the isolated CNBD of MlotiK1 were 17.6 nm and 73.5 nm, respectively (60). Therefore, the binding affinity of 8-NBD-cAMP of 3.8 μm observed in our study for mHCN2J-L586W correlates well with the binding affinity of 13 μm for cAMP obtained based on the changes in the Trp fluorescence (Fig. 3C).
FIGURE 4.

Fluorescence of 8-NBD-cAMP increases upon interaction with mHCN2J-L586W and hERG1-(666–872)/MBP but not mEAG1-(505–702). A, C, and E, emission spectra of 8-NBD-cAMP at the indicated concentrations in the presence of 1 μm mHCN2J-L586W, mEAG1-(666–872), and hERG-(666–872)/MBP, respectively. The excitation wavelength was 470 nm. B, D, and F, plots of the change in the peak fluorescence intensity versus total 8-NBD-cAMP concentration for mHCN2J-L586W, mEAG1-(666–872), hERG-(666–872)/MBP, and MBP, respectively. The change in the peak fluorescence intensity (ΔF) was calculated by subtracting the peak emission intensity at 536 nm in the absence of 8-NBD-cAMP from the peak emission intensities in the presence of 8-NBD-cAMP. The data in B and F were fitted with Equation 4. The binding affinities for 8-NBD-cAMP were 3.8 ± 0.6 μm for mHCN2J-L586W, and ≥ 51 ± 4 μm for mERG1-(666–872)/MBP.
We also observed a concentration-dependent increase in the 8-NBD-cAMP fluorescence upon binding to wild-type mHCN2J (supplemental Fig. 2A). 8-NBD-cAMP binds to mHCN2J tighter than to mHCN2J-L586W as much lower concentrations of 8-NBD-cAMP were needed to elicit a significant increase in the fluorescence (supplemental Fig. 2, A and B). Consistent with the high binding affinity of 8-NBD-cAMP the Kd determined by the fit of the data in supplemental Fig. 2B with Equation 4 was not well defined.
No increase in the fluorescence of 8-NBD-cAMP was observed in the presence of mEAG1-(505–702) (Fig. 4, C and D). This suggests that 8-NBD-cAMP does not bind to the isolated CNBD of mEAG1 channels. Interestingly, the fluorescence of 8-NBD-cAMP increased in a concentration-dependent manner in the presence of hERG1-(666–872)/MBP but not MBP alone (Fig. 4, E and F). This binding was not saturated with up to 100 μm 8-NBD-cAMP. 8-NBD-cAMP has a very high absorbance at concentrations higher than 100 μm making it impossible to properly correct for the inner filter effect and analyze data at concentrations higher than 100 μm. Fitting the plots of the change in the peak fluorescence versus total 8-NBD-cAMP concentration gave Kd of ≥ 51 ± 4 μm (Fig. 4F). However, the estimate of 51 μm for the Kd of 8-NBD-cAMP binding represents a lower limit. Nevertheless, our data indicate that the isolated CNBD of hERG1 channels binds cAMP. This observation is in agreement with the study by Cui et al. (40) where authors observed binding of a 3H-labeled cAMP to intact hERG channels.
Cyclic Nucleotides Do Not Modulate Currents from mEAG1 and hERG1 Channels in Excised Inside-out Macro Patches
Experiments in the previous sections explored binding of cyclic nucleotides to the isolated CNBD. Here we tested for direct cyclic nucleotide modulation in the context of the intact channels. mHCN2, mEAG1, and hERG1 channels were expressed in X. laevis oocytes and currents were recorded in the inside-out patch configuration. Wild-type hERG1 channels are characterized by a fast inactivation that makes excised patch-clamp studies of the channels difficult (11–13) and might obscure the effect of cyclic nucleotides. Therefore, experiments were carried out on both wild-type hERG1 and hERG1-S631A channels with a S631A mutation in the pore that significantly slowed the rate of inactivation (61).
As it has been reported previously (25, 28, 32, 62), currents from mHCN2 channels were activated by cyclic nucleotides and hyperpolarization (Fig. 5A and Table 1). Application of 1 mm cAMP and cGMP increased the current and shifted the conductance-voltage relations of the mHCN2 channels to the right (Fig. 5A). Currents from mEAG1 and hERG1-S631A channels exhibited a characteristic increase in amplitude with membrane depolarization (33, 61); however application of cAMP or cGMP had no effect on the current amplitude or kinetics even at concentrations as high as 10 mm (Fig. 5, B and C and Table 1). Furthermore, application of cAMP and cGMP had no effect on currents from the wild-type hERG1 channels (supplemental Fig. S3 and Table 1), indicating that the absence of cyclic nucleotide effect on hERG1-S631A channels is not due to the altered inactivation properties of the channels. Thus, cAMP and cGMP modulate mHCN2 channels but have no effect on the currents from mEAG1 and hERG1 channels.
FIGURE 5.

Cyclic nucleotides do not modulate currents from mEAG1 and hERG1 channels. Currents and conductance-voltage relations for mHCN2 (A), mEAG1 (B), and hERG1-S631A (C) channels recorded in the inside-out patch configuration with cAMP (red), cGMP (green), and without the cyclic nucleotides (black). cAMP and cGMP were applied at 1 mm to the mHCN2 channels and 10 mm to mEAG1 and hERG1-S631A channels. The conductance-voltage relations were obtained from tail currents.
TABLE 1.
Parameters of conductance-voltage relations for the mHCN2, mEAG1, and hERG1 channels
V1/2 and slopes ± S.E. are from the Boltzmann fits of the conductance-voltage relations with and without 1 mm cAMP and cGMP for the mHCN2 channels, 4 mm cAMP and 16 mm cGMP for the hERG1 channels, and 10 mm cAMP and cGMP for the mEAG1 and hERG1 channels. The number of averaged experiments is indicated in parentheses.
| Channel | −cAMP |
+cAMP |
−cGMP |
+cGMP |
||||
|---|---|---|---|---|---|---|---|---|
| V1/2 | Slope | V1/2 | Slope | V1/2 | Slope | V1/2 | Slope | |
| mV | mV | mV | mV | |||||
| mHCN2 (3) | −130 ± 3.3 | 3.6 ± 0.3 | −109 ± 2.4 | 4.5 ± 0.2 | −130 ± 3.3 | 3.6 ± 0.3 | −112 ± 2.2 | 4.5 ± 0.43 |
| mEAG1 (5) | −37 ± 4.7 | 16.7 ± 1.2 | −38 ± 4.5 | 17 ± 1.0 | −38 ± 5.5 | 17.3 ± 1.4 | −37 ± 5.4 | 17.8 ± 1.0 |
| hERG1 (4) | −33 ± 13.8 | 26.4 ± 5.1 | −42 ± 8.1 | 22.1 ± 2.5 | −35 ± 4.8 | 27.4 ± 3.6 | −34 ± 7.2 | 20.7 ± 8.7 |
| hERG1-S631A (3) | −29 ± 5.4 | 13.1 ± 0.3 | −29 ± 5.3 | 13.1 ± 0.4 | −29 ± 5.8 | 12.8 ± 0.4 | −30 ± 6.0 | 12.8 ± 0.3 |
DISCUSSION
In this study we investigated whether cAMP and cGMP directly bind and modulate mEAG1 and hERG1 channels. Direct binding of cyclic nucleotides to the isolated CNBD of mEAG1 and hERG1 channels was tested with fluorescence-based methods. Physiological modulation by cyclic nucleotides of the full-length mEAG1 and hERG1 channels was explored with electrophysiological recordings of currents from mEAG1 and hERG1 channels expressed in oocytes. For the isolated CNBD of mEAG1, we observed no cyclic nucleotide-dependent changes in the fluorescence of the Trp residue located near the hypothetical cyclic nucleotide binding site and no increase in the fluorescence of 8-NBD-cAMP upon addition of the isolated CNBD. Furthermore, application of cyclic nucleotides to inside-out patches had no effect on the currents from mEAG1 channels. Taken together, these results indicate that cAMP and cGMP do not bind or modulate mEAG1 channels.
Concentration-dependent increase in the fluorescence of 8-NBD-cAMP upon addition of the isolated CNBD of hERG1 channels indicated low affinity binding (Kd of ≥ 51 μm). Interestingly, cAMP had no effect on the currents from hERG1 channels. Although we cannot exclude that the cyclic nucleotide binding site is not exposed in the intact channels the more likely explanation could be that the binding of cAMP does not induce conformational changes necessary to facilitate hERG1 channel opening. Also, without a crystal structure we do not know if the binding site of cAMP in hERG1-(666–872) is the same as the cyclic nucleotide binding pocket resolved in the structure of mHCN2J (31).
The conclusions of our study are in agreement with the results of Robertson et al. (33) and Sanguinetti and co-workers (11) that demonstrated no modulation by cyclic nucleotides of currents from excised macropatches expressing mEAG1 channels and currents recorded with a two-electrode voltage clamp from oocytes expressing hERG1 channels, respectively. However, Cui et al. (39, 40) reported direct binding of cAMP to the hERG1 channels and inhibition of currents from hERG1 channels recorded in a whole-cell configuration by cAMP. We also observed direct binding of cAMP to the isolated CNBD of hERG1, however this binding was insufficient to affect currents from hERG1 channels. The discrepancy between the studies could be due to the limitations of the whole-cell configuration. To test the effects of cyclic nucleotides on the channels Sanguinetti and co-workers (11) and Cui et al. (39, 40) applied membrane-permeable analogs of cAMP to the extracellular solution. The membrane-permeable analogs could act on the channels directly or they can activate various signaling pathways that could affect the channels indirectly. One of the most important roles of cyclic nucleotides in eukaryotes is to regulate activity of protein kinases (63). In turn, protein kinases have been shown to regulate activity of the ERG1 channels (40, 64–66). Therefore, in a whole-cell setting it is difficult to distinguish between a direct effect of cyclic nucleotides on hERG1 channels and an indirect effect through protein kinases. This holds true even in the presence of protein kinase inhibitors since it is impossible to control the exact concentration of the protein kinase inhibitors and the membrane-permeable analogs of cyclic nucleotides in a whole-cell configuration.
What is the role of the CNBD in the EAG family of K+ channels? It has been shown that mutations in the CNBD affect trafficking of the hERG channels (19, 67, 68) and can lead to long-QT syndrome (69). However, the most anticipated function of the CNBD in the EAG family of K+ channels would be to modulate activity of the channels via direct binding of a ligand. Evidence is mounting that cAMP and cGMP do not modulate currents from mEAG1 and hERG1 channels by direct binding to the CNBD. Future studies will reveal whether the CNBD can function as a ligand binding module or only represents an evolutionary link between the EAG family of K+ channels and HCN and CNG channels.
Supplementary Material
Acknowledgments
We thank Galen Flynn for providing the current traces and conductance-voltage relations for the mHCN2 channels and members of the Zagotta laboratory for helpful discussions. We thank K. D. Black, S. Simmons, S. Cunnington, and G. Sheridan for excellent technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants 2R01EY010329-16 (to W. N. Z.) and F32 HL095241 (to A. E. C.). This work was also supported by the Howard Hughes Medical Institute.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- EAG
- ether-à-go-go
- ERG
- EAG-related gene
- CNBD
- cyclic nucleotide binding domain
- ELK
- EAG-like K+ channel
- HCN
- hyperpolarization-activated cyclic nucleotide-modulated
- CNG
- cyclic nucleotide-gated
- MBP
- maltose-binding protein.
REFERENCES
- 1.Warmke J. W., Ganetzky B. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauer C. K., Schwarz J. R. (2001) J. Membr. Biol. 182, 1–15 [DOI] [PubMed] [Google Scholar]
- 3.Farias L. M., Ocaña D. B., Diaz L., Larrea F., Avila-Chávez E., Cadena A., Hinojosa L. M., Lara G., Villanueva L. A., Vargas C., Hernández-Gallegos E., Camacho-Arroyo I., Dueñas-González A., Pérez-Cárdenas E., Pardo L. A., Morales A., Taja-Chayeb L., Escamilla J., Sánchez-Peña C., Camacho J. (2004) Cancer Res. 64, 6996–7001 [DOI] [PubMed] [Google Scholar]
- 4.Hemmerlein B., Weseloh R. M., Mello , de , Queiroz F., Knötgen H., Sánchez A., Rubio M. E., Martin S., Schliephacke T., Jenke M., Heinz-Joachim-Radzun , Stühmer W., Pardo L. A. (2006) Mol. Cancer 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouadid-Ahidouch H., Le Bourhis X., Roudbaraki M., Toillon R. A., Delcourt P., Prevarskaya N. (2001) Receptors Channels 7, 345–356 [PubMed] [Google Scholar]
- 6.Pardo L. A., del , Camino D., Sánchez A., Alves F., Brüggemann A., Beckh S., Stühmer W. (1999) EMBO J. 18, 5540–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber C., Mello , de , Queiroz F., Downie B. R., Suckow A., Stühmer W., Pardo L. A. (2006) J. Biol. Chem. 281, 13030–13037 [DOI] [PubMed] [Google Scholar]
- 8.Mello , de , Queiroz F., Suarez-Kurtz G., Stühmer W., Pardo L. A. (2006) Mol. Cancer 5, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavrilova-Ruch O., Schönherr K., Gessner G., Schönherr R., Klapperstück T., Wohlrab W., Heinemann S. H. (2002) J. Membr. Biol. 188, 137–149 [DOI] [PubMed] [Google Scholar]
- 10.Gómez-Varela D., Zwick-Wallasch E., Knötgen H., Sánchez A., Hettmann T., Ossipov D., Weseloh R., Contreras-Jurado C., Rothe M., Stühmer W., Pardo L. A. (2007) Cancer Res. 67, 7343–7349 [DOI] [PubMed] [Google Scholar]
- 11.Spector P. S., Curran M. E., Zou A., Keating M. T., Sanguinetti M. C. (1996) J. Gen. Physiol. 107, 611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith P. L., Baukrowitz T., Yellen G. (1996) Nature 379, 833–836 [DOI] [PubMed] [Google Scholar]
- 13.Trudeau M. C., Warmke J. W., Ganetzky B., Robertson G. A. (1995) Science 269, 92–95 [DOI] [PubMed] [Google Scholar]
- 14.Schönherr R., Heinemann S. H. (1996) J. Physiol. 493, 635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curran M. E., Splawski I., Timothy K. W., Vincent G. M., Green E. D., Keating M. T. (1995) Cell 80, 795–803 [DOI] [PubMed] [Google Scholar]
- 16.Kiehn J., Lacerda A. E., Wible B., Brown A. M. (1996) Circulation 94, 2572–2579 [DOI] [PubMed] [Google Scholar]
- 17.Sanguinetti M. C., Jurkiewicz N. K., Scott A., Siegl P. K. (1991) Circ. Res. 68, 77–84 [DOI] [PubMed] [Google Scholar]
- 18.Li X., Xu J., Li M. (1997) J. Biol. Chem. 272, 705–708 [DOI] [PubMed] [Google Scholar]
- 19.Zhou Z., Gong Q., Epstein M. L., January C. T. (1998) J. Biol. Chem. 273, 21061–21066 [DOI] [PubMed] [Google Scholar]
- 20.Smith G. A., Tsui H. W., Newell E. W., Jiang X., Zhu X. P., Tsui F. W., Schlichter L. C. (2002) J. Biol. Chem. 277, 18528–18534 [DOI] [PubMed] [Google Scholar]
- 21.Meyer R., Heinemann S. H. (1998) J. Physiol. 508, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crociani O., Guasti L., Balzi M., Becchetti A., Wanke E., Olivotto M., Wymore R. S., Arcangeli A. (2003) J. Biol. Chem. 278, 2947–2955 [DOI] [PubMed] [Google Scholar]
- 23.Becchetti A., De Fusco M., Crociani O., Cherubini A., Restano-Cassulini R., Lecchi M., Masi A., Arcangeli A., Casari G., Wanke E. (2002) Eur. J. Neurosci. 16, 415–428 [DOI] [PubMed] [Google Scholar]
- 24.Guy H. R., Durell S. R., Warmke J., Drysdale R., Ganetzky B. (1991) Science 254, 730. [DOI] [PubMed] [Google Scholar]
- 25.DiFrancesco D., Tortora P. (1991) Nature 351, 145–147 [DOI] [PubMed] [Google Scholar]
- 26.Fesenko E. E., Kolesnikov S. S., Lyubarsky A. L. (1985) Nature 313, 310–313 [DOI] [PubMed] [Google Scholar]
- 27.Haynes L., Yau K. W. (1985) Nature 317, 61–64 [DOI] [PubMed] [Google Scholar]
- 28.Ludwig A., Zong X., Jeglitsch M., Hofmann F., Biel M. (1998) Nature 393, 587–591 [DOI] [PubMed] [Google Scholar]
- 29.Nakamura T., Gold G. H. (1987) Nature 325, 442–444 [DOI] [PubMed] [Google Scholar]
- 30.Finn J. T., Grunwald M. E., Yau K. W. (1996) Annu. Rev. Physiol. 58, 395–426 [DOI] [PubMed] [Google Scholar]
- 31.Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., Gouaux E. (2003) Nature 425, 200–205 [DOI] [PubMed] [Google Scholar]
- 32.Santoro B., Liu D. T., Yao H., Bartsch D., Kandel E. R., Siegelbaum S. A., Tibbs G. R. (1998) Cell 93, 717–729 [DOI] [PubMed] [Google Scholar]
- 33.Robertson G. A., Warmke J. M., Ganetzky B. (1996) Neuropharmacology 35, 841–850 [DOI] [PubMed] [Google Scholar]
- 34.Ludwig J., Terlau H., Wunder F., Brüggemann A., Pardo L. A., Marquardt A., Stühmer W., Pongs O. (1994) EMBO J. 13, 4451–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frings S., Brüll N., Dzeja C., Angele A., Hagen V., Kaupp U. B., Baumann A. (1998) J. Gen. Physiol. 111, 583–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanguinetti M. C., Jiang C., Curran M. E., Keating M. T. (1995) Cell 81, 299–307 [DOI] [PubMed] [Google Scholar]
- 37.Brüggemann A., Pardo L. A., Stühmer W., Pongs O. (1993) Nature 365, 445–448 [DOI] [PubMed] [Google Scholar]
- 38.Zhong Y., Wu C. F. (1993) J. Neurosci. 13, 4669–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui J., Kagan A., Qin D., Mathew J., Melman Y. F., McDonald T. V. (2001) J. Biol. Chem. 276, 17244–17251 [DOI] [PubMed] [Google Scholar]
- 40.Cui J., Melman Y., Palma E., Fishman G. I., McDonald T. V. (2000) Curr. Biol. 10, 671–674 [DOI] [PubMed] [Google Scholar]
- 41.Chen G. Q., Gouaux E. (1997) Protein Eng. 10, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 42.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 43.Lakowicz J. R. (2006) Principles Of Fluorescence Spectroscopy, 3rd Ed., pp. 56 and 551, Springer, New York, NY [Google Scholar]
- 44.Zagotta W. N., Hoshi T., Aldrich R. W. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 7243–7247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 46.Passner J. M., Steitz T. A. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 2843–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., Varughese K. I. (1995) Science 269, 807–813 [DOI] [PubMed] [Google Scholar]
- 48.Clayton G. M., Silverman W. R., Heginbotham L., Morais-Cabral J. H. (2004) Cell 119, 615–627 [DOI] [PubMed] [Google Scholar]
- 49.Rehmann H., Wittinghofer A., Bos J. L. (2007) Nat. Rev. Mol. Cell Biol. 8, 63–73 [DOI] [PubMed] [Google Scholar]
- 50.Shabb J. B., Corbin J. D. (1992) J. Biol. Chem. 267, 5723–5726 [PubMed] [Google Scholar]
- 51.Zhou L., Siegelbaum S. A. (2007) Structure 15, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belduz A. O., Lee E. J., Harman J. G. (1993) Nucleic Acids Res. 21, 1827–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore J., Kantorow M., Vanderzwaag D., McKenney K. (1992) J. Bacteriol. 174, 8030–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogreid D., Døskeland S. O., Gorman K. B., Steinberg R. A. (1988) J. Biol. Chem. 263, 17397–17404 [PubMed] [Google Scholar]
- 55.Bubis J., Neitzel J. J., Saraswat L. D., Taylor S. S. (1988) J. Biol. Chem. 263, 9668–9673 [PubMed] [Google Scholar]
- 56.Gronenborn A. M., Sandulache R., Gärtner S., Clore G. M. (1988) Biochem. J. 253, 801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nimigean C. M., Pagel M. D. (2007) J. Mol. Biol. 371, 1325–1337 [DOI] [PubMed] [Google Scholar]
- 58.Altieri S. L., Clayton G. M., Silverman W. R., Olivares A. O., De la Cruz E. M., Thomas L. R., Morais-Cabral J. H. (2008) J. Mol. Biol. 381, 655–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tibbs G. R., Liu D. T., Leypold B. G., Siegelbaum S. A. (1998) J. Biol. Chem. 273, 4497–4505 [DOI] [PubMed] [Google Scholar]
- 60.Cukkemane A., Grüter B., Novak K., Gensch T., Bönigk W., Gerharz T., Kaupp U. B., Seifert R. (2007) EMBO Rep. 8, 749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herzberg I. M., Trudeau M. C., Robertson G. A. (1998) J. Physiol. 511, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flynn G. E., Black K. D., Islas L. D., Sankaran B., Zagotta W. N. (2007) Structure 15, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson D. A., Akamine P., Radzio-Andzelm E., Madhusudan M., Taylor S. S. (2001) Chem. Rev. 101, 2243–2270 [DOI] [PubMed] [Google Scholar]
- 64.Kiehn J., Karle C., Thomas D., Yao X., Brachmann J., Kübler W. (1998) J. Biol. Chem. 273, 25285–25291 [DOI] [PubMed] [Google Scholar]
- 65.Thomas D., Zhang W., Karle C. A., Kathöfer S., Schöls W., Kübler W., Kiehn J. (1999) J. Biol. Chem. 274, 27457–27462 [DOI] [PubMed] [Google Scholar]
- 66.Cayabyab F. S., Schlichter L. C. (2002) J. Biol. Chem. 277, 13673–13681 [DOI] [PubMed] [Google Scholar]
- 67.Akhavan A., Atanasiu R., Noguchi T., Han W., Holder N., Shrier A. (2005) J. Cell Sci. 118, 2803–2812 [DOI] [PubMed] [Google Scholar]
- 68.Aydar E., Palmer C. (2001) J. Physiol. 534, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berthet M., Denjoy I., Donger C., Demay L., Hammoude H., Klug D., Schulze-Bahr E., Richard P., Funke H., Schwartz K., Coumel P., Hainque B., Guicheney P. (1999) Circulation 99, 1464–1470 [DOI] [PubMed] [Google Scholar]
- 70.Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.