

Abstract
Glutaconyl-CoA decarboxylase (Gcd) couples the biotin-dependent decarboxylation of glutaconyl-CoA with the generation of an electrochemical Na+ gradient. Sequencing of the genes encoding all subunits of the Clostridium symbiosum decarboxylase membrane complex revealed that it comprises two distinct biotin carrier subunits, GcdC1 and GcdC2, which differ in the length of a central alanine- and proline-rich linker domain. Co-crystallization of the decarboxylase subunit GcdA with the substrate glutaconyl-CoA, the product crotonyl-CoA, and the substrate analogue glutaryl-CoA, respectively, resulted in a high resolution model for substrate binding and catalysis revealing remarkable structural changes upon substrate binding. Unlike the GcdA structure from Acidaminococcus fermentans, these data suggest that in intact Gcd complexes, GcdA is associated as a tetramer crisscrossed by a network of solvent-filled tunnels.
Glutaconyl-CoA decarboxylase (Gcd,4 EC 4.1.1.70) catalyzes the decarboxylation of glutaconyl-CoA to crotonyl-CoA (Reaction 1 and Equation 1).

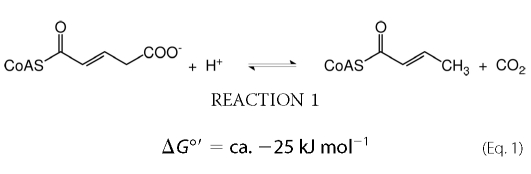 |
In aerobic organisms and respiring anaerobes this decarboxylation represents the irreversible second half-reaction of the FAD-containing homotetrameric enzyme glutaryl-CoA dehydrogenase (1). Fermentative bacteria, however, conserve the small amount of free energy of decarboxylation (Equation 1) as an electrochemical Na+ gradient (sodium-motive force: SMF) (2, 3). These energy-limited organisms contain glutaconyl-CoA decarboxylases, which are Na+-dependent and biotin-containing integral membrane enzymes composed of 4–5 different subunits. Because of generation of SMF, the decarboxylation is reversible as observed in vitro for the related Na+-dependent oxaloacetate (EC 4.1.1.3) and methylmalonyl-CoA decarboxylases (EC 4.1.1.41) (4). In Synthrophus aciditrophicus, this probably happens in vivo, where the synthesis of benzoate from crotonate requires carboxylation of crotonyl-CoA to glutaconyl-CoA (5).
The best-studied Na+-dependent glutaconyl-CoA decarboxylase is involved in the fermentation of glutamate to ammonia, CO2, acetate, butyrate, and hydrogen by the strictly anaerobic gut bacterium Acidaminococcus fermentans (6). The enzyme complex comprises a hydrophilic α-subunit (GcdA, 65 kDa) that catalyzes the Na+-independent transfer of the carboxylate of glutaconyl-CoA to biotin, the latter being covalently bound to the γ-subunit (GcdC, 14 kDa, Equation 2). The hydrophobic β-subunit (GcdB, 35 kDa, 10–12 transmembrane helices) mediates the subsequent decarboxylation of N1-carboxybiotin coupled to the transport of Na+ from the cytoplasm to the outside of the cell (Equation 3).
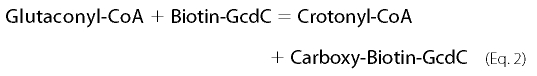 |
The function of the small δ-subunit (GcdD, 12 kDa, one transmembrane helix) remains to be established (7). The gcdA gene coding for the carboxytransferase (GcdA) is part of the 2-hydroxyglutarate operon from A. fermentans (8, 9). The three other genes, gcdDCB, form a separate transcription unit, preceded by mcdA, encoding the putative carboxyltransferase subunit of methylmalonyl-CoA decarboxylase (Fig. 1) (7). Glutaconyl-CoA decarboxylase is readily purified from the solubilized membrane fraction of A. fermentans by affinity chromatography on monomeric avidin Sepharose. On SDS-PAGE, Gcd subunits A, B, and D separate according to their calculated molecular masses, whereas GcdC forms a ladder of Coomassie stainable bands between 120 and 140 kDa, probably due to a stretch of 50 amino acids comprising 34 alanines and 14 prolines (10). The purified enzyme was reconstituted in phospholipid vesicles and shown to pump Na+. In the same manner, the decarboxylase from the glutamate fermenting anaerobic gut bacterium Clostridium symbiosum was characterized as an electrogenic Na+ pump (10). However, on SDS-PAGE gels of purified C. symbiosum Gcd the biotin carrier subunit GcdC migrated as two distinct bands with apparent molecular masses of 22 and 24 kDa.
FIGURE 1.

Arrangement of the genes involved in glutamate fermentation. Gene cluster of the glutaconyl-CoA decarboxylase genes (gcd, dark blue) in A. fermentans, C. symbiosum, and F. nucleatum. Further genes encode the glutaconate-CoA transferase (gct, pale blue) and 2-hydroxyglutaryl-CoA dehydratase (hgd, red) with its activator (hgdC or hgdD, magenta) (43). Additional genes in F. nucleatum code for a regulator R and the glutamate/Na+ symporter S (both in yellow). In A. fermentans the genes of the smaller glutaconyl-CoA decarboxylase subunits D, C, and B are in a separate locus preceded by the gene of subunit A of methylmalonyl-CoA decarboxylase (mmdA, turquoise).
The carboxytransferase subunit GcdA from A. fermentans was obtained as a single protein by overproduction in Escherichia coli (7, 8). The enzyme catalyzes the carboxylation of free biotin with glutaconyl-CoA, albeit with a very high Km of 40 mm for biotin and an about 100-times lower kcat than the native enzyme (11, 12). The recombinant GcdA has been crystallized, and its three-dimensional structure has been determined (13). Both active sites of the homodimeric enzyme lie along the two interfaces between the monomers, where the N-terminal domain provides the glutaconyl-CoA binding site and the C-terminal domain binds biotin attached to GcdC. Recently, the structure of the carboxytransferase domain of oxaloacetate decarboxylase from Vibrio cholerae was reported, which is structurally distinct from that of GcdA (14).
For crystallization of the whole glutaconyl-CoA decarboxylase complex, we chose the enzyme from C. symbiosum because of its simple availability and the normal behavior of its GcdC subunits. Despite encouraging initial results, we again obtained crystals of GcdA alone, whose structure will be described in this work. The structure revealed a more precise view on substrate binding, catalysis, and the possible quaternary structure of the intact enzyme complex. The genes of all subunits of the C. symbiosum decarboxylase were found to be arranged like those in the genome of the glutamate fermenting Fusobacterium nucleatum (15, 16). However, it was shown that the former enzyme complex indeed contains two distinct GcdC subunits.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Cell Growth C. symbiosum
HB25, kindly provided by Dr. G. C. Mead, Food Research Institute, Norwich, England (17), was grown under anaerobic conditions at 37 °C in the glutamate medium previously described (18). For cloning, sequencing, and gene expression Escherichia coli strains DH5α (Invitrogen, Karlsruhe, Germany) (19) and RosettaTM (DE3)pLysS (Novagen, Darmstadt, Germany) (20) were grown in Luria-Bertani (LB) medium or standard I nutrient broth (Merck, Darmstadt, Germany) supplemented with the required antibiotics at 37 °C.
Purification of Glutaconyl-CoA Decarboxylase from C. symbiosum
The purification of the glutaconyl-CoA decarboxylase complex was carried out as previously described (21). The only exception was the replacement of the detergent Triton X-100 by dodecylmaltoside, to facilitate UV-spectroscopic tracking of purification and subsequent crystallization. After affinity chromatography on a column prepared with Avidin SoftLink® Resins (Promega, Mannheim, Germany), the purified enzyme was concentrated by ultrafiltration and stored at 4 °C. The purity of the eluted protein was monitored by SDS-PAGE with Coomassie Brilliant Blue or silver staining (22).
Preparation of Genomic DNA
Frozen wet cells from C. symbiosum (1 g) were resuspended in 2 ml of 50 mm Tris/HCl pH 8.0, 25% sucrose (23) and incubated while gently shaking with 50 mg of lysozyme for 60 min at 37 °C. Then 2 ml of 50 mm Tris/HCl, pH 8.0/25 mm EDTA were added and incubated for 15 min on ice. After addition of 5 ml of the same buffer supplemented with 1% SDS, 100 mg RNase, and 10 mg proteinase K the solution was shaken for 3 h at 37 °C. Proteins were removed by repeated (3 times) phenolization and extraction with chloroform/isoamylalcohol (24:1). The DNA solution was dialyzed against 5 liters of 10 mm Tris/HCl, 1 mm EDTA, pH 8.0 at 4 °C overnight.
Sequencing of the gcd Genes from C. symbiosum
Construction of recombinant plasmids for sequencing and expression were carried out using standard techniques (23). DNA was restricted and ligated according to the protocols of the enzyme suppliers. PCR fragments were cloned using TOPO TA cloning kit (Invitrogen). Degenerated oligonucleotides for GcdA were used to amplify an internal DNA fragment of 600 bp from the chromosomal DNA of C. symbiosum. Based on this fragment a Southern blot probe (522 bp) was synthesized using a DIG-dNTP labeling kit (Roche Applied Science). Mapping by digestion with different restriction enzymes, Southern blot hybridization, ligation with pBluescript II SK+ (Stratagene, Heidelberg, Germany) and pCR2.1 (Invitrogen, Karlsruhe, Germany), and sequencing increased the known sequence information. The remainders of the sequence of gcdA at the 5′- and 3′-ends were obtained by several direct genomic sequencing walks using the Topo® Walker kit (Invitrogen) (24) and the DNA Walking SpeedUp kit (SeeGene; Seoul, Korea) (25). Confirmation of these data was accomplished by sequencing of PCR products using the proofreading Phusion High-Fidelity DNA polymerase (Finnzymes, Espoo, Finland). In a similar way the sequences of gcdDC2C1B were obtained using the N termini of GcdC1 and C2. DNA sequence data were analyzed using the web-based tool Biology WorkBench 3.2. Data base searches were performed with the basic local alignment search tool (BLAST) (26) at the National Center for Biotechnology Information (NCBI) in the nonredundant protein library.
Cloning of the Genes gcdA, gcdC1, and gcdAC1 from C. symbiosum
The genes gcdA and gcdC1 were amplified by PCR, ligated into the cloning vector pGEM-T (Promega, Mannheim, Germany), and then subcloned into the expression vector pASK-IBA7plus (IBA, Göttingen, Germany). This vector provides an N-terminal Strep-tag II peptide fused protein to enable one-step purification. For the coexpression of gcdA with gcdC1 the vector pCDFDuet-1 (Novagen, Darmstadt, Germany) was used. GcdA was produced including an N-terminal His6 tag peptide.
Gene Expression of gcdA, gcdC1, and gcdAC1 in E. coli
For expression, the transformed E. coli RosettaTM (DE3)pLysS cells were grown in LB medium supplemented with 0.2% glucose, 50 μm biotin, carbenicillin (50 μg ml−1) and chloramphenicol (34 μg ml−1), at 37 °C. Cells transformed with a pCDFDuet-1 construct additionally needed spectinomycin (150 μg ml−1). An overnight preculture was used to inoculate fresh medium. At A578 = 0.5, the cells were induced by adding 0.4 μm anhydrotetracylin for pASK-IBA7plus constructs, and 0.2 mm isopropyl β-d-thiogalactoside for the co-expression construct derived from pCDFDuet-1, respectively. After 3 h, the cells were harvested and stored at −80 °C.
Purification of Recombinant GcdA, GcdC1, and GcdAC1
Purification of recombinant GcdA was carried out according to the manufacturer's specifications using a 1-ml Strep-Tactin Sepharose gravity flow column (IBA). Subsequently, the purified enzyme was dialyzed against 20 mm Tris/HCl, pH 7.4, 100 mm NaCl, and concentrated up to 7 mg ml−1 protein. GcdC1 was purified by avidin-biotin affinity chromatography analogous to the purification of the glutaconyl-CoA decarboxylase complex.
Protein subcomplexes obtained by the co-expression of gcdA and gcdC1 were purified by Ni-NTA affinity chromatography using HIS-Select® HF Nickel Affinity Gel (Sigma) as matrix. The proteins were eluted with a linear gradient up to 500 mm imidazole and subsequently analyzed by Western blotting. For biotinylated proteins the blocking solution contained an avidin-peroxidase conjugate (0.5 mg ml−1, Sigma-Aldrich). Signals were detected with luminol/p-hydroxycoumaric acid chemiluminescence on Fuji 100 NIF medical x-ray films. His-tagged proteins were visualized using the anti-Penta-His-HRP-conjugate (Qiagen, Hilden, Germany) and Amersham Biosciences ECL PlusTM Western blotting Detection Reagents (GE Healthcare).
Determination of Enzyme Activity of GcdA
The carboxytransferase activity of glutaconyl-CoA decarboxylase subunit A was determined using the assay for native glutaconyl-CoA decarboxylase (7, 18) in the presence of d-biotin. After adding recombinant GcdA, the increasing absorbance of NADH was measured at 340 nm, ϵ = 6.3 mm−1 cm−1 (27).
Synthesis of CoA-esters and Analysis by MALDI-TOF Mass Spectrometry
Glutaconyl-CoA was enzymatically synthesized from acetyl-CoA and glutaconate as catalyzed by glutaconate CoA-transferase (28). Crotonyl-CoA and glutaryl-CoA were chemically synthesized by acylation of CoASH with the corresponding anhydrides (29). The calculated amount of anhydride (20% excess) was dissolved in 0.5 ml of acetonitrile and added to 25 mg of CoASH in 4.5 ml of 0.2 m NaHCO3 at ambient temperature.5 Completion of the reaction was checked by the 5,5′-dithiobis(2-nitrobenzoate) (DTNB) assay (30), the reaction mix was acidified to pH 2. The CoA-thioester was isolated by passing the mixture through a SepPak C18 column, which was conditioned with methanol and equilibrated with 0.1% trifluoroacetic acid. The column was washed with 5–10 column volumes of 0.1% trifluoroacetic acid, and the CoA-thioester was eluted with 0.1% trifluoroacetic acid in 50% acetonitrile. After evaporation of the acetonitrile under vacuum, the concentration of the CoA-thioester was determined from its absorbance at 260 nm and by the enzymatic DTNB assay (28). The samples were lyophilized and stored at −80 °C. The molecular mass of the CoA-thioester was confirmed by MALDI-TOF MS. A drop of the CoA-ester solution was mixed on a gold-plated target with the matrix 4-hydroxy-α-cyanocinnamic acid in equal amounts, dried under air and analyzed by using a Voyager-DE/RP-MALDI-TOF MS in positive and reflector mode (31).
Other Biochemical Techniques
The subunits of the isolated glutaconyl-CoA decarboxylase complex were separated by SDS-PAGE (32), blotted on polyvinylidene difluoride membranes (33), and N-terminally sequenced by Edman degradation (34). Protein concentrations were determined with the Bio-Rad Protein Assay. Bovine serum albumin was used as standard.
Crystallization, Data Collection, and Processing
Prior to crystallization the purified C. symbiosum GcdA was either dialyzed or subjected to size exclusion chromatography on a Superdex 200 HR 10/30 gel filtration column (GE Healthcare) using a buffer containing 10 mm MES, pH 6.5, and 100 mm NaCl. The protein was concentrated to 20–30 mg/ml on a 50-kDa Vivascience Centricon unit. All crystallization experiments were carried out applying the vapor diffusion method at room temperature. In the case of co-crystallization trials, 100–200 mm solutions of biocytin, crotonyl-CoA, glutaryl-CoA, and glutaconyl-CoA in 10 mm MES, pH 6.5, 100 mm NaCl were prepared. These solutions were individually added to the protein samples prior to crystallization. For soaking experiments the substrate solutions were added directly to the respective crystallization condition. Co-crystallization and soaking experiments were carried out at final ligand concentrations of 5–10 mm (CoA-esters) and 5–50 mm (biocytin). Initial crystallization screening was performed in 500 + 500 nl sitting drops using a Microsys 4004 Cartesian crystallization robotics system and a set of commercial crystallization screens, followed by manual optimization in hanging drops.
Crystals were flash-frozen in their respective crystallization buffers supplemented with 20–30% glycerol as cryoprotectant. X-Ray data were collected at 100 K to a maximum resolution of 2.70 to 1.75 Å using beamline X13, Deutsches Elektronen Synchrotron, Hamburg, beamlines ID14-1 and ID23-2, European Synchrotron Radiation Facility, Grenoble, France, and a MAR345dtb imaging plate coupled to a Bruker AXS FR591 x-ray generator with a copper rotating anode (wavelength, 1.5418 Å). The diffraction data were indexed and integrated using MOSFLM (35). The integrated data were scaled, merged, and truncated using the SCALA and TRUNCATE programs of the CCP4 suite (36). Data collection statistics are summarized in supplemental Table S1.
Structure Determination and Refinement
The structure was solved by molecular replacement using the program MOLREP (37) with the atomic coordinates of the previously determined structure of A. fermentans GcdA (Protein Data Bank code 1PIX) as a search model. Subsequent model building and refinement were completed using the programs COOT (38) and REFMAC5 (39). Progress in the structural refinement was evaluated at each stage by inspection of the free R factor and the stereochemical parameters calculated by the program PROCHECK (40). Statistics of refinement and structure validation are presented in supplemental Table S2. Secondary structure elements were assigned using the program STRIDE (41). Protein interfaces and intermolecular interactions were calculated using the Protein Interfaces, Surfaces, and Assemblies Service at the European Bioinformatics Institute (42). The coordinates and structure factors have been deposited at the Protein Data Bank (PDB), Research Collaboratory for Structural Bioinformatics (RCSB), with the accession codes 3GF7 for apo-GcdA, 3GF3, 3GLM, and 3GMA for GcdA co-crystallized with glutaconyl-CoA, crotonyl-CoA, and glutaryl-CoA.
RESULTS AND DISCUSSION
Silver-stained SDS-PAGE of the intact Gcd complex revealed five different subunits with apparent molecular masses of GcdA, 65 kDa; GcdB, 35 kDa; GcdC1, 24 kDa; GcdC2, 23 kDa; and GcdD, 15 kDa (Fig. 2A). Comparison of the N-terminal sequences of GcdA, C1 and C2 with those of the corresponding subunits from A. fermentans (7) and F. nucleatum (15) confirmed their assignment. As with the decarboxylase from A. fermentans, the N termini of GcdB and D could not be sequenced by Edman degradation. For sequencing of the corresponding genes PCR primers were deduced from the N termini and from highly conserved regions of the proteins (see “Experimental Procedures”). After sequencing, it was observed that the 3′-end of gcdA had been previously sequenced during cloning of the genes coding for 2-hydroxyglutaryl-CoA dehydratase (HgdCAB) from C. symbiosum (43). Hence gcdA is located upstream of hgdCAB as already observed in A. fermentans and F. nucleatum. Similarly, upstream of gcdA the two gctAB genes coding for glutaconate CoA-transferase were detected (9). The sequence of the DNA fragment comprising gcdA was deposited in GenBankTM under accession number FJ390113. Unlike in A. fermentans but similar to F. nucleatum (15), all four genes coding for the smaller subunits are located upstream of gctAB (Fig. 1, accession number FJ390114).
FIGURE 2.

A, silver-stained 15% SDS-PAGE analysis of the purified glutaconyl-CoA decarboxylase from C. symbiosum. Lane Gcd contains 30 μg of purified enzyme (lane M: molecular mass marker). B, comparison of the amino acid sequences of GcdC1 and GcdC2 from C. symbiosum. The N-terminal domain was sequenced by Edman degradation (underlined). Conserved residues are marked in yellow. Bold amino acids indicate the AP-rich linker region and the biotin-binding site. The highly conserved motif is highlighted by a blue box while the lysine to which biotin is attached is shown in red. C, Western blot analysis of a Ni-NTA purification of the co-expression of GcdA and biotin-containing GcdC1. The proteins were detected with avidin-peroxidase (lane B) or with anti-Penta-His-HRP conjugate (lane H). Each lane contains 5 μg of protein.
The amino acid sequence of the deduced carboxytransferase subunit GcdA matches well with those from A. fermentans (7), F. nucleatum ssp. nucleatum (15), and F. nucleatum ssp. vincentii (16) (69% identity for each). The sequence of the hydrophobic subunit GcdB is also very similar to those listed above (67, 73, and 73%, respectively) and includes the conserved amino acids supposedly involved in Na+ transport. Interestingly, the two GcdC subunits (GcdC1 and GcdC2) are encoded by slightly different genes (79% sequence identity) (Fig. 2B). Similar to A. fermentans (7) their calculated molecular masses (15 and 14 kDa, respectively) as well as that of the GcdD subunit (10 kDa) were substantially lower than those observed on SDS-PAGE. The C-terminal region of GcdC1 and GcdC2 (A78-N149 and A68-N139, respectively) corresponds to the biotin carrier domain, whereas the N-terminal region (M1-A31) is presumably involved in immobilizing the GcdA subunits on the GcdAB subcomplex (13). In the GcdC1 sequence the linker between these two domains comprises a stretch of 47 amino acid residues that is composed of 25 alanines and 14 prolines. In GcdC from A. fermentans (58% sequence identity), this apparently highly flexible stretch is similar in length (50 amino acids) and likewise composed of 34 alanines and 14 prolines. The main difference between the two GcdCs from C. symbiosum is the shortening of the GcdC2 linker by ten amino acids (Fig. 2B).
In contrast to the highly conserved β-subunits, the δ-subunits are variable; the closest identity (33%) was found between the GcdDs of C. symbiosum and F. nucleatum. Thus, the gene cluster for the specific part of the glutamate fermentation pathway in C. symbiosum from (R)-2-hydroxyglutarate, via (R)-2-hydroxyglutaryl-CoA, and glutaconyl-CoA to crotonyl-CoA (6) has been established. This cluster matches well with that in the genome of the neotype strain of C. symbiosum ATCC 14940 (Genome Sequencing Center, Washington University, St. Louis), which is slightly different from the strain HB25 used in this work (44).
SDS-PAGE analysis of the purified GcdA heterologously expressed in E. coli revealed pure protein with an apparent molecular mass of 66 kDa (calculated mass, 65 kDa). With the multienzyme assay system used for the glutaconyl-CoA decarboxylase, activity was observed only in the presence of d-biotin. This assay generates glutaconyl-CoA in situ and determines formed crotonyl-CoA via the NAD+-dependent oxidation to acetyl-CoA and acetylphosphate (18). Under saturating glutaconyl-CoA concentrations (0.1 mm) an apparent Km of 2.8 mm for biotin and a specific activity of Vmax of 0.11 units/mg were determined. The Km was 14-times lower than that observed with GcdA from A. fermentans and Vmax was about 1000 times lower than that of the complete decarboxylase (21).
Partial co-purification of subcomplexes of His6-GcdA and biotinylated GcdC1 was demonstrated by Western blotting using an anti-Penta-His-HRP-conjugate and an avidin-peroxidase conjugate, respectively (Fig. 2C) Hence, E. coli is able to biotinylate recombinant GcdC1 at the conserved lysine between two methionines (MKM), the recognition sequence for biotin ligases (EC 6.3.4.-). Unfortunately, the GcdAC1 subcomplex was not pure enough and lacked stability due to an intrinsic aggregation tendency of GcdC1, which impeded its crystallization.
Crystallization
First crystals of apo-GcdA were grown in 0.1 m MES, pH 6.5, 1.3–1.6 M MgSO4, 5–25% glycerol (Condition A). Co-crystals with crotonyl-CoA were readily obtained under the same conditions. Although complete data sets could be collected for both an apo-GcdA and a crotonyl-CoA·GcdA co-crystal, reproducibility of these crystals was rather poor and the crystal quality made the use of synchrotron radiation indispensable. Thus, for co-crystallization with glutaryl-CoA and glutaconyl-CoA, robot supported screening was repeated, resulting in the identification of two additional crystallization conditions. Glutaryl-CoA co-crystals were obtained in 0.1 m MES, pH 6.5, 10–11% polyethylene glycol 20000, 15–20% glycerol (Condition B). The highest quality crystals of GcdA were grown in the presence of glutaconyl-CoA in 0.1 m sodium citrate, pH 5.6–6.5, 1 m ammonium phosphate 27.5% glycerol (Condition C). Interestingly, the latter condition yielded only co-crystals with glutaconyl-CoA. The addition of crotonyl- or glutaryl-CoA either prevented crystal growth or resulted in crystals of significantly lesser quality with none of the ligands bound.
Quality of the Structural Models
GcdA crystals suitable for diffraction data collection were obtained in three different space groups. Apo-GcdA crystals from condition A belonged to space group F222. The final model contained one molecule per asymmetric unit and was refined to an Rfactor of 18.9% and an Rfree of 22.7% at a resolution of 2.4 Å. Co-crystals with crotonyl-CoA were obtained under the same condition. Interestingly, in the presence of crotonyl-CoA the space group changed to C2 with four molecules in the asymmetric unit despite only minor changes in overall crystal packing. The final Rfactor/Rfree values for this structure were 20.0%/23.8% at a resolution of 2.5 Å. In condition B co-crystals of glutaryl-CoA were grown in space group P3121 with two molecules in the asymmetric unit. Refinement resulted in an Rfactor/Rfree of 19.2%/21.8% for data up to 2.7 Å. The best quality crystals, however, were obtained in condition C with the addition of glutaconyl-CoA. These crystals also belonged to space group F222. This structural model comprises one molecule per asymmetric unit and was refined to an Rfactor/Rfree of 16.4%/17.8% at a maximum resolution of 1.75 Å.
Evaluation using PROCHECK (40) revealed good stereochemistry for all four structures, with more than 98% of the residues in the most favored and allowed regions of the Ramachandran plot and with RMSD values for bond distances and angles below 0.010 Å and 1.3°, respectively. A comprehensive list of the data collection and refinement statistics is presented in supplemental Tables S1 and S2.
The substrates of the complex structures are clearly defined by electron density (Fig. 3). However, the structure obtained from crystals grown in the presence of glutaconyl-CoA contained only the product of the decarboxylation reaction, crotonyl-CoA, although all crystals were frozen in cryo-buffers containing the freshly prepared CoA-esters prior to data collection. Unless stated otherwise, the highest resolution structure obtained from condition C was used in the following description and interpretation of the GcdA structure.
FIGURE 3.

A, final 2Fo-Fc electron density corresponding to GcdA-bound crotonyl-CoA contoured at 3 ς (1.75 Å resolution). Additional electron density found in the vicinity of the supposed biotin binding site was assigned to a chloride ion (yellow sphere). The positions of the oxyanion holes OAH1 and OAH2 are indicated in orange. B, Fo-Fc map of glutaryl-CoA contoured at 1 ς (2.7 Å resolution). This figure and Figs. 4–7 were prepared by PyMOL (58).
Monomer Structure
Fig. 4A shows a ribbon representation of the structure of the GcdA monomer with a bound crotonyl-CoA molecule. The secondary structure elements comprise 20 α-helices (α1-α20) and 6 310-helices (η1-η6) and 22 β-strands (β1-β22) altogether. A structural alignment carried out using the secondary structure matching (SSM) server (45) showed the similarity between the presented model and the A. fermentans structure used as a search model in molecular replacement. The 552 aligned residues sharing a sequence identity of 71% superposed with an RMSD of 0.71 Å with all secondary structure motifs conserved. A superposition of the monomer structures of A. fermentans and C. symbiosum GcdA is shown in supplemental Fig. S1. Both enzymes belong to the SCOP family of biotin-dependent carboxylases/carboxyltransferases that build up their active sites by two different homologous subunits or domains of the ClpP/crotonase-like fold. Similar to the A. fermentans structure (13), GcdA from C. symbiosum can be subdivided into two structurally and sequentially equivalent domains of virtually the same size, the N-terminal part comprising Met3-Ala291 and the remaining C-terminal portion of the polypeptide chain. The striking structural characteristic of the GcdA monomer is a bundle of two α-helices (α16/α17) that sticks out from the C-terminal domain and a single α-helix (α8) that similarly protrudes from the N-terminal domain. These helix fingers are involved in the dimerization of GcdA and are a key feature of the active site architecture.
FIGURE 4.

A, overall structure of the C. symbiosum GcdA monomer. α- and 310-helices of the N-terminal domain are colored blue, the respective β-strands are shown in lighter blue. The secondary structure motifs of the C-terminal domain are represented in dark green (helices) and light green (β-strands). The bound crotonyl-CoA is displayed as a magenta stick model and the chloride ion bound to OAH2 is shown as a yellow sphere. Residues missing in the electron density are indicated by broken lines. B, GcdA dimer. N and C terminus of the symmetry-equivalent chain B are colored dark and light gray, respectively. The crotonyl-CoA molecules at the dimer interfaces are represented as CPK models. The second chloride ion is displayed as a green sphere. The positions of the active sites are highlighted by dashed lines. C, two orthogonal orientations of the 222-symmetric GcdA tetramer deduced from the crystal packing. Chains A to D are colored blue, green, red, and yellow, respectively.
Generally, the residues of all three structures are well defined by electron density. However, there are significant exceptions regarding the helix fingers. Here, a number of consecutive residues are missing in the electron density (supplemental Table S2). These discontinuities vary among the structural models and are most pronounced in the apo-structure of GcdA, where the complete helix α8 (Gly218-Lys251) and residues Leu502-Gln513 are missing. The gaps are least distinct in the glutaconyl-CoA co-crystal structure comprising only residues Gly221-Ala237 and Ala505-Gln513. Apparently, the two helix fingers are structurally very flexible until the substrate is bound. Interestingly, the equivalent residues in A. fermentans GcdA were structurally well resolved by electron density. In the latter structure the participation of α8, α16, and α17 in crystal contact formation leads to their relatively high rigidity, while in our C. symbiosium structures they protrude freely into the bulk solvent.
Quaternary Structure
According to the A. fermentans structure, GcdA forms C2-symmetric dimers (Fig. 4B) (13). However, in size-exclusion chromatography GcdA from C. symbiosum elutes at an apparent molecular mass of 232 kDa, indicating a tetrameric assembly (calculated molecular mass, 260 kDa). Moreover, an analysis of the crystal packing of all three C. symbiosum structures including that of A. fermentans revealed that each is built up of identical 222-symmetric tetramers. The same arrangement was observed in a low resolution structure of GcdA that was obtained from a crystal grown from a preparation of intact, membrane-resident Gcd that was isolated as a detergent-solubilized complex from C. symbiosum cells (data not shown). These findings were further confirmed by results of the following web-based programs for the prediction of quaternary protein structures: the Protein Interfaces and Assemblies server Pita (46), the Protein Quaternary Structure server (PQS), and the Protein Interfaces, Surfaces and Assemblies Service (PISA) at European Bioinformatics Institute (42). Enzymes of the crotonase superfamily adopt a variety of different quaternary structures (47) such as monomers (1fc7: photosystem II D1 CTPase (48)), dimers (1od4: acetyl-CoA carboxylase carboxyltransferase subunit from yeast (49)), hexamers (1on3: transcarboxylase 12S (50)) or even tetradecamers (1tyf: proteolytic subunit of caseinolytic protease (51)). Interestingly, the tetrameric carboxyltransferases of acetyl-CoA carboxylase from Staphylococcus aureus and E. coli (2f9i and 2f9y (52)) which also belong to the structural subfamily of biotin-dependent carboxylases are not related in their quaternary arrangement with GcdA.
The 222-symmetric tetramer is obtained by simple rotation around the crystallographic symmetry axes and has a surface area of 68,280 Å2, thus occluding an area of 36,310 Å2 (9,078 Å2 per monomer) from solvent access. The GcdA tetramer corresponds to a dimer of dimers with a surface area of 25,030 Å2 for each monomer and associates pairwise to the dimers AB and CD (Fig. 4C) with contact areas of 4,232 Å2, respectively. Further contact areas of 1,743 Å2 and 1,307 Å2 are generated along the A-C/B-D and A-D/B-C interfaces. The tetramer comprises 106 hydrogen bonds altogether. More than 95% of these interactions are conserved with respect to the A. fermentans structure.
Active Site Architecture
The active site of GcdA is formed by a C2-symmetric “head-to-tail” dimerization as shown in Fig. 4B. The N-terminal domain of one monomer tightly interacts with the C-terminal domain of the second molecule, while the crossing helix fingers cover the active site cavity like a lid (Fig. 5A). This bi-domain active site arrangement is common among members of the crotonase superfamily (47) and is characteristic for all structurally characterized biotin-dependent decarboxylases/carboxyltransferases (53).
FIGURE 5.

Active site architecture. A, surface representation of the A-B dimer of GcdA facing the entangled helix fingers. The color code is in accordance to Fig. 4B. The orange circles indicate the position of the two active site clefts leading to OAH1 and OAH2. B, superposition of the crotonyl-CoAG·cdA and the apo-GcdA structure, displayed as a close-up view of the glutaconyl-CoA binding site at the A-B dimer interface. The corresponding chains of the apo-structure are displayed in orange. The broken lines illustrate the “movement” of helix α17 upon substrate binding.
On each side of the entangled “fingers” there is a cavity leading to one of two different oxyanion holes (OAH) at the bottom of the reactive center. The groove-like entry (cleft A) building the N-terminal part of the active site represents the glutaconyl-CoA binding-site and contains OAH1 (Val153, Gly196). In the complex structures this part of the active site contained electron density that was clearly assigned to the substrate analogue glutaryl-CoA and the product crotonyl-CoA, respectively (Fig. 3). The second, more voluminous cavity (cleft B) is funnel-shaped and leads to the biotin binding pocket containing OAH2 (Ile420, Ala460, Ala461); it is supposed to act as the interface for interaction with the biotin carrier GcdC (13). In apo-GcdA and the glutaryl-CoA co-crystal structure OAH2 is occupied by a water molecule, whereas in the case of the crotonyl-CoA complex extra electron density at this position was allocated to a chloride ion (Fig. 3A).
A superposition of the crotonyl-CoA complex and the apo-structure reveals some remarkable structural changes upon substrate binding (Fig. 5B). The most obvious example for these modifications is the immersion of helix finger A (α8) that was only hinted at by featureless electron density in apo-GcdA. Moreover, in comparison to the apo-structure, α17 moves about 13° toward the bound crotonyl-CoA. This movement might be part of a dynamic process coupling the progress of the transcarboxylation reaction to conformational changes on the surface of the GcdA subcomplex.
The Glutaconyl-CoA Binding Site
The bottom of the glutaconyl-CoA binding site (cleft A) is formed by the 310-helicesη1 and η2, the connecting loop regions and β-strand β6. The cavity is covered up by the crossed helix fingers and a series of positively charged residues of α-helices α3 and α16 “guard” the entrance of the otherwise mostly uncharged binding pocket. The substrate-protein interactions are predominantly hydrophobic and comprise mainly residues of the N-terminal domain of one monomer. The observed binding mode of the CoA-esters in C. symbiosum GcdA is exemplified by crotonyl-CoA in Fig. 6. The U-shaped conformation of the CoA moiety is very similar to biotin-dependent carboxylases, such as the carboxyltransferase domain of yeast acetyl-CoA carboxylase (49) and transcarboxylase 12S (50). The similarities of the binding motif with the A. fermentans structure are illustrated in Fig. 6 showing most of the specific interactions to be conserved. The adenine moiety of CoA is sandwiched between Met118, Ala121, and Phe155 and Met497 from the C-terminal domain. The pantothenate unit is deeply buried in a pocket formed by residues of β6, η2, and α16. Both the ribose 3′-phosphate and the diphosphate moieties are more solvent exposed and thus structurally more flexible as indicated by considerably higher B-factors than the rest of the molecule. There are a number of hydrogen bonds in this region specifically interacting with the phosphate groups of the product. The acyl part of the CoA-thioester sticks deeply into the binding pocket with the carbonyl oxygen hydrogen bonding to OAH1. In the glutaryl-CoA co-crystal structure the carboxyl group points toward OAH2, which is supposed to bind the ureido oxygen of the co-substrate biotin (Fig. 3B).
FIGURE 6.

Stereo view of the glutaconyl binding site displayed with amino acid residues within 4 Å of the crotonyl-CoA product. Residues of the N- and C-terminal domains of GcdA are colored blue and gray, respectively. Structurally equivalent residues of A. fermentans GcdA are displayed in orange. Non-conserved residues are marked with orange labels. The dashed lines indicate hydrogen bonds.
Implications on the Quaternary Structure of Gcd
In the previous symmetric model of the Gcd complex (13), the AB dimer of GcdA was postulated to be docked onto the membrane-resident GcdB subunit(s) which mediate(s) decarboxylation-dependent sodium translocation (Fig. 7B). Given the intrinsic stability of the GcdA tetramer and the presence of two different biotin-carrier subunits, GcdC1 and GcdC2, we propose an alternative, asymmetric model (Fig. 7A). Using as restraint that the distances of the GcdC subunits relative to the membrane subunit(s) GcdB have to be similar, only one orientation is feasible. Here the AC dimer packs with its flat, almost rectangular and ∼50 × 70 Å2 wide surface against GcdB and the membrane surface. Sodium ion flow through the GcdA tetramer might still be possible or even depend on allosterically induced changes of its quaternary structure. An initial analysis using the MolAxis server (54) revealed a pronounced spherical cavity in the center of the GcdA tetramer that is connected to the protein surface by a network of tunnels. A subsequent examination with the program CAVER (55) identified four basic types of solvent filled tunnels protruding from the inner sphere (Fig. 7C). In accordance to the hypothetical arrangement of the tetramer relative to the membrane surface the horizontal and vertical tunnels were labeled H1/2 and V1/2, respectively. A close-up view of each channel is displayed in supplemental Fig. S2. The most distinctive of the tunnels, H1, was already mentioned in the discussion of the A. fermentans structure (13). It is built up of residues of chains A(B) and C(D), respectively, and contains a positively charged bottleneck comprising Arg166A(C), Arg166B(D), Arg174A(C), and Arg174B(D) thus blocking the passage of Na+ ions. Similarly, access to H2 is also guarded by the positively charged side chains of residues Arg408A(B), Arg408D(C), Lys343A(B), Lys343D(C), Lys446A(B), and Lys446D(C). By contrast, the vertical tunnels seem to be suitable for cation translocation despite their smaller diameters. The inner surfaces of tunnels V1 and V2 are lined with carbonyl oxygen atoms of the corresponding residues providing opportunity for transient coordination of monovalent cations. Additionally, all helical segments surrounding the V1/V2 channels (α4, α5, α6, α7, α9, α10, α12, α14, and α19 of each chain) point with their C termini toward the channel thus providing potential stabilization for translocating cations by dipole-ion interaction as observed for KcsA and related channels (Fig. 7D).
FIGURE 7.

Hypothetical model of the Gcd complex. A, asymmetric and B, symmetric models of Gcd. The GcdA subunits are shown in surface representation. Dimeric and tetrameric GcdA have the same orientation as in Fig. 4, B and C, respectively. The subunits B, C, and D are colored dark gray, orange, and light gray. The hypothetical movement of the GcdC subunits is indicated by bended arrows. The coloring of the GcdA chains is in accordance to Fig. 4C. C, GcdA tetramer and its network of interior channels. The tunnels were delineated by CAVER (55) and are labeled with respect to their relative positions to the hypothetical membrane plane: the horizontal tunnels H1 and H2 (blue), the transversal tunnels V1 and V2 (green). The membrane-spanning GcdB is indicated as a gray disc. The orange meshwork indicates the binding site of the biotin carriers GcdC1 and GcdC2 and the cavities leading to OAH2. D, A-C surface and the penetrating tunnels V1 and V2 viewed from the membrane plane. Of the α-helices surrounding the channels only the ones close to the membrane interface are labeled for clarity.
In the asymmetric model the locations for the interaction sites with biotin carrier domains differ by about 12 Å relative to the GcdA-GcdB interface, where decarboxylation of the biotin co-substrate is postulated to take place (Fig. 7A). Accordingly, this asymmetry might also explain why two GcdC subunits with different linker lengths between their N-terminal anchor region and their C-terminal biotin carrier domain are incorporated into the intact Gcd complex.
The requirement for broken symmetry in the assembly of an intact Gcd complex and the coupling of a catalyzed chemical reaction with ion-translocation is not without precedent. In F-type ATPases the extramembranous α3β3 subcomplex shows broken symmetry upon interaction with the γ subunit and the subunits (b, b′) of the second stalk (56). Likewise, the trimeric AcrB efflux pump, a member of the RND family, adopts asymmetric transport-dependent conformational states (57). Certainly, solution of the structure of the intact Gcd complex is required to clarify whether this sodium ion pump indeed forms an asymmetric complex analogous to F-type ATPases.
Supplementary Material
Acknowledgments
We thank the beamline staff at the synchrotron beamlines X13, EMBL-Hamburg, and ID14-1 and ID23-2, ESRF, EMBL-Grenoble.
This work was supported by the Deutsche Forschungsgemeinschaft (Priority Program 1070) (to L.-O. E. and W. B.).
The atomic coordinates and structure factors (codes 3GF7, 3GF3, 3GLM, and 3GMA) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The nucleotide sequence(s) reported in this paper has been submitted to the Gen-BankTM/EBI Data Bank with accession number(s) FJ390113 and FJ390114.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Tables S1 and S2.
T. Selmer, personal communication.
- Gcd
- glutaconyl-CoA decarboxylase
- PDB
- Protein Data Bank
- MES
- 4-morpholineethanesulfonic acid
- SMF
- sodium-motive force
- RMSD
- root mean-squared deviation.
REFERENCES
- 1.Härtel U., Eckel E., Koch J., Fuchs G., Linder D., Buckel W. (1993) Arch. Microbiol. 159, 174–181 [DOI] [PubMed] [Google Scholar]
- 2.Buckel W. (2001) Biochim. Biophys. Acta 1505, 15–27 [DOI] [PubMed] [Google Scholar]
- 3.Dimroth P., Schink B. (1998) Arch. Microbiol. 170, 69–77 [DOI] [PubMed] [Google Scholar]
- 4.Hilpert W., Dimroth P. (1984) Biochemistry 23, 5360–5366 [Google Scholar]
- 5.Mouttaki H., Nanny M. A., McInerney M. J. (2007) Appl. Environ. Microbiol. 73, 930–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckel W. (2001) Appl. Microbiol. Biotechnol. 57, 263–273 [DOI] [PubMed] [Google Scholar]
- 7.Braune A., Bendrat K., Rospert S., Buckel W. (1999) Mol. Microbiol. 31, 473–487 [DOI] [PubMed] [Google Scholar]
- 8.Bendrat K., Buckel W. (1993) Eur. J. Biochem. 211, 697–702 [DOI] [PubMed] [Google Scholar]
- 9.Mack M., Bendrat K., Zelder O., Eckel E., Linder D., Buckel W. (1994) Eur. J. Biochem. 226, 41–51 [DOI] [PubMed] [Google Scholar]
- 10.Buckel W., Semmler R. (1983) Eur. J. Biochem. 136, 427–434 [DOI] [PubMed] [Google Scholar]
- 11.Buckel W., Liedtke H. (1986) Eur. J. Biochem. 156, 251–257 [DOI] [PubMed] [Google Scholar]
- 12.Berger S., Braune A., Buckel W., Härtel U., Lee M. L. (1996) Angew. Chem. Int. Ed. Engl. 35, 2132–2133 [Google Scholar]
- 13.Wendt K. S., Schall I., Huber R., Buckel W., Jacob U. (2003) EMBO J. 22, 3493–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Studer R., Dahinden P., Wang W. W., Auchli Y., Li X. D., Dimroth P. (2007) J. Mol. Biol. 367, 547–557 [DOI] [PubMed] [Google Scholar]
- 15.Kapatral V., Anderson I., Ivanova N., Reznik G., Los T., Lykidis A., Bhattacharyya A., Bartman A., Gardner W., Grechkin G., Zhu L., Vasieva O., Chu L., Kogan Y., Chaga O., Goltsman E., Bernal A., Larsen N., D'Souza M., Walunas T., Pusch G., Haselkorn R., Fonstein M., Kyrpides N., Overbeek R. (2002) J. Bacteriol. 184, 2005–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapatral V., Ivanova N., Anderson I., Reznik G., Bhattacharyya A., Gardner W. L., Mikhailova N., Lapidus A., Larsen N., D'Souza M., Walunas T., Haselkorn R., Overbeek R., Kyrpides N. (2003) Genome Res. 13, 1180–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mead G. C. (1971) J. Gen. Microbiol. 67, 47–56 [DOI] [PubMed] [Google Scholar]
- 18.Buckel W. (1986) Methods Enzymol. 125, 547–558 [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D. (1983) J. Mol. Biol. 166, 557–580 [DOI] [PubMed] [Google Scholar]
- 20.Novy R., Drott D., Yaeger K., Mierendorf R. (2001) Innovations 12, 1–3 [Google Scholar]
- 21.Beatrix B., Bendrat K., Rospert S., Buckel W. (1990) Arch. Microbiol. 154, 362–369 [DOI] [PubMed] [Google Scholar]
- 22.Blum H., Beier H., Gross H. J. (1987) Electrophoresis 8, 93–99 [Google Scholar]
- 23.Sambrook J., Russell D. W. (2001) Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 24.Heiner C. R., Hunkapiller K. L., Chen S. M., Glass J. I., Chen E. Y. (1998) Genome Res. 8, 557–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang I. T., Kim Y. J., Kim S. H., Kwak C. I., Gu Y. Y., Chun J. Y. (2003) BioTechniques 35, 1180–1184 [DOI] [PubMed] [Google Scholar]
- 26.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegenhorn J., Senn M., Bücher T. (1976) Clin. Chem. 22, 151–160 [PubMed] [Google Scholar]
- 28.Buckel W., Dorn U., Semmler R. (1981) Eur. J. Biochem. 118, 315–321 [DOI] [PubMed] [Google Scholar]
- 29.Simon E. J., Shemin D. (1953) J. Am. Chem. Soc. 75, 2520–2520 [Google Scholar]
- 30.Riddles P. W., Blakeley R. L., Zerner B. (1979) Anal. Biochem. 94, 75–81 [DOI] [PubMed] [Google Scholar]
- 31.Selmer T., Buckel W. (1999) J. Biol. Chem. 274, 20772–20778 [DOI] [PubMed] [Google Scholar]
- 32.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 33.Textor S., Wendisch V. F., De Graaf A. A., Müller U., Linder M. I., Linder D., Buckel W. (1997) Arch. Microbiol. 168, 428–436 [DOI] [PubMed] [Google Scholar]
- 34.Michel C., Buckel W., Linder D. (1991) J. Chromatogr. 587, 93–99 [DOI] [PubMed] [Google Scholar]
- 35.Powell H. R. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 1690–1695 [DOI] [PubMed] [Google Scholar]
- 36.CCP4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 37.Vagin A., Teplyakov A. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 1622–1624 [DOI] [PubMed] [Google Scholar]
- 38.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 40.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 41.Frishman D., Argos P. (1995) Proteins 23, 566–579 [DOI] [PubMed] [Google Scholar]
- 42.Krissinel E., Henrick K. (2007) J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 43.Hans M., Sievers J., Müller U., Bill E., Vorholt J. A., Linder D., Buckel W. (1999) Eur. J. Biochem. 265, 404–414 [DOI] [PubMed] [Google Scholar]
- 44.Kaneuchi C., Watanabe K., Terada A., Benno Y., Mitsuoka T. (1976) Int. J. Syst. Bacteriol. 26, 195–204 [Google Scholar]
- 45.Krissinel E., Henrick K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 46.Ponstingl H., Kabir T., Thornton J. M. (2003) J. Appl. Crystallogr. 36, 1116–1122 [Google Scholar]
- 47.Hamed R. B., Batchelar E. T., Clifton I. J., Schofield C. J. (2008) Cell Mol. Life Sci. 65, 2507–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao D. I., Qian J., Chisholm D. A., Jordan D. B., Diner B. A. (2000) Nat. Struct. Biol. 7, 749–753 [DOI] [PubMed] [Google Scholar]
- 49.Zhang H., Yang Z., Shen Y., Tong L. (2003) Science 299, 2064–2067 [DOI] [PubMed] [Google Scholar]
- 50.Hall P. R., Wang Y. F., Rivera-Hainaj R. E., Zheng X., Pustai-Carey M., Carey P. R., Yee V. C. (2003) EMBO J. 22, 2334–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J., Hartling J. A., Flanagan J. M. (1997) Cell 91, 447–456 [DOI] [PubMed] [Google Scholar]
- 52.Bilder P., Lightle S., Bainbridge G., Ohren J., Finzel B., Sun F., Holley S., Al-Kassim L., Spessard C., Melnick M., Newcomer M., Waldrop G. L. (2006) Biochemistry 45, 1712–1722 [DOI] [PubMed] [Google Scholar]
- 53.Diacovich L., Mitchell D. L., Pham H., Gago G., Melgar M. M., Khosla C., Gramajo H., Tsai S. C. (2004) Biochemistry 43, 14027–14036 [DOI] [PubMed] [Google Scholar]
- 54.Yaffe E., Fishelovitch D., Wolfson H. J., Halperin D., Nussinov R. (2008) Nucleic Acids Res. 36, W210–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petrek M., Otyepka M., Banás P., Kosinová P., Koca J., Damborský J. (2006) BMC Bioinformatics 7, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedersen P. L. (2007) J. Bioenerg. Biomembr. 39, 349–355 [DOI] [PubMed] [Google Scholar]
- 57.Pos K. M. (2009) Biochim. Biophys. Acta 1794, 782–793 [DOI] [PubMed] [Google Scholar]
- 58.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.