

Abstract
Motivation: Next-generation sequencing has become an important tool for genome-wide quantification of DNA and RNA. However, a major technical hurdle lies in the need to map short sequence reads back to their correct locations in a reference genome. Here, we investigate the impact of SNP variation on the reliability of read-mapping in the context of detecting allele-specific expression (ASE).
Results: We generated 16 million 35 bp reads from mRNA of each of two HapMap Yoruba individuals. When we mapped these reads to the human genome we found that, at heterozygous SNPs, there was a significant bias toward higher mapping rates of the allele in the reference sequence, compared with the alternative allele. Masking known SNP positions in the genome sequence eliminated the reference bias but, surprisingly, did not lead to more reliable results overall. We find that even after masking, ∼5–10% of SNPs still have an inherent bias toward more effective mapping of one allele. Filtering out inherently biased SNPs removes 40% of the top signals of ASE. The remaining SNPs showing ASE are enriched in genes previously known to harbor cis-regulatory variation or known to show uniparental imprinting. Our results have implications for a variety of applications involving detection of alternate alleles from short-read sequence data.
Availability: Scripts, written in Perl and R, for simulating short reads, masking SNP variation in a reference genome and analyzing the simulation output are available upon request from JFD. Raw short read data were deposited in GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE18156.
Contact: jdegner@uchicago.edu; marioni@uchicago.edu; gilad@uchicago.edu; pritch@uchicago.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
There has been a great deal of recent interest in identifying genes for which the two alleles in an individual are expressed at different rates (Knight, 2004; Milani et al., 2009; Ronald et al., 2005; Wittkopp et al., 2008; Yan et al., 2002). At least two important biological mechanisms can be uncovered through the identification of allele-specific expression (ASE). For example, studies investigating ASE have uncovered both genes harboring cis-regulatory variation and imprinted genes that are epigenetically silenced in one copy but not the other (Babak et al., 2008; Serre et al., 2008; Wang et al., 2008).
Recently developed sequencing technologies such as the Illumina Genome Analyzer, Roche 454 GS FLX sequencer and Applied Biosystems SOLiD sequencer have the potential to greatly improve our ability to detect ASE and to improve our understanding of cis-regulatory variation and epigenetic imprinting. However, the detection of ASE depends critically on accurate mapping of short reads in the presence of sequence variation. Here, using RNA-Seq data from two HapMap individuals, along with simulation experiments, we characterize the effects of individual SNPs on the quantification of expression levels. Our results are also relevant to other applications of next-generation sequencing, such as SNP discovery, expression QTL mapping and detection of allele-specific differences in transcription factor binding.
2 METHODS
2.1 RNA isolation and sequencing
Total RNA from two HapMap Yoruba lymphoblastoid cell lines (GM19238 and GM19239) was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA) and assessed using an Agilent Bioanalyzer. mRNA was then isolated with Dyna1 oligo-dT beads (Invitrogen, Carlsbad, CA) from 10 μg of total RNA. The mRNA was randomly fragmented using the RNA fragmentation kit from Ambion. First-strand cDNA synthesis was performed using random primers and SuperScriptII reverse-transcriptase (Invitrogen, Carlsbad, CA). This was followed by second-strand cDNA synthesis using DNA Polymerase I and RNaseH (Invitrogen, Carlsbad, CA).
The short cDNA fragments from each sample were prepared into a library for Illumina sequencing. Briefly, the Illumina adaptor was ligated to the ends of the double-stranded cDNA fragments and a 200 bp size selection of the final product was performed by gel-excision, following the Illumina-recommended protocol. To create the final library, 200 bp cDNA template molecules with the adaptor attached were enriched by PCR. Sequencing was performed on the Illumina Genome Analyzer II for 36 cycles (resulting in 35 bp reads after discarding the final base). The images taken during the sequencing reactions were processed using Illumina's standard analysis pipeline (v.1.3.2). Two lanes of a flow-cell were used for each individual yielding 15 579 717 and 16 780 153 total sequence reads for GM19238 and GM19239, respectively.
2.2 Read-mapping and binomial tests
Reads were initially mapped to the human genome (build 36.3) with (MAQ MAQ v. 0.7.1, Li et al., 2008), using default parameters, excluding random sequence fragments and masking one copy of the pseudo-autosomal regions. In particular, reads were assigned to the location in the genome with the best match, provided that the number of mismatching bases was <3 and that the sum of quality scores at mismatched bases was <70. Reads that mapped to multiple locations equally well according to MAQ's quality-aware alignment algorithm (i.e. had mapping quality scores of 0) were discarded.
At each exonic SNP that was heterozygous according to the HapMap genotype data, we quantified the amount of expression from each allele by counting the number of times each allele was observed [exons defined by RefSeq (Pruitt et al., 2007); HapMap SNPs and genotypes from release r22 (International HapMap Consortium, 2005, 2007)]. Overall, <1.0% of all reads had a base-call at a HapMap SNP position that was inconsistent with the known genotype of the individual and these reads were discarded.
To study the effect of allelic differences between the sequence reads and the reference genome they were mapped against, we classified all read calls in the dataset as matching the reference allele or the non-reference allele. For each individual, in order to include a SNP in our analysis, we required that at least 20 reads mapped to that SNP position in that individual. The two sequenced individuals were analyzed independently such that two separate tests were performed if both individuals had >20 reads overlapping the same SNP. For each individual, we compared the observed distribution of the proportion of mapped reads coming from the reference allele to the expected distribution assuming symmetric binomial sampling. Two one-sided binomial tests were applied to each SNP, to test the complementary alternative hypotheses that expression of the reference allele was greater than or less than 0.5. False discovery rate (FDR) corrections were applied across both individuals to correct for multiple testing such that we allowed an overall FDR of 1%, 5% and 10%. Results in the main text correspond to an FDR of 1%, while results corresponding to an FDR of 5% and 10% are given in Supplementary Tables S1-S3.
Additionally, in an attempt to correct for the bias toward preferentially mapping the reference allele, we created a copy of the human genome in which all SNP positions were masked. SNP locations were obtained from the February 2009 release of the 1000 Genomes project (Kaiser, 2008, www.1000genomes.org). Since currently available mapping algorithms do not allow for ambiguity codes in the reference sequence, masking was accomplished by changing the nucleotide at each SNP position to a third allele that is not known to segregate in humans (e.g. changing A→T in the reference sequence at the position of an A/G SNP).
2.3 Simulations
To better understand the bias toward the reference allele and the amount of this bias that could be attributed to read-mapping, we simulated 1.8 million 35 bp reads. Three simulated sets of reads were created that, at each SNP, consisted of equal numbers of reference and non-reference alleles (Supplementary Fig. S1). Each simulated set started with all 35 bp segments of human chromosome 1 that overlap an exonic HapMap SNP. For each of these 35 bp segments and on each strand, one read matched the reference allele and one read matched the non-reference allele at the SNP position. All base quality scores were assigned as the modal quality score for that position in the real RNA-Seq data. Random ‘sequencing’ errors were added to two of the sets of simulated reads such that each base in the read had a Bernoulli probability of 0.01 or 0.05, respectively, of being changed to a different randomly selected base. These two error rates were chosen to span the range of possible values that might be observed in real data. Additionally, to explore the potential impact of read-mapping biases on studies using longer read lengths, we applied the same procedure to simulate all 50 and 100 bp reads (without additional errors) that overlapped the same SNPs.
To determine if there were differences in the observed bias among three popular mapping algorithms, we mapped the simulated 35 bp reads to the SNP-masked genome using each mapping program [MAQ v. 0.7.1, as used for the analyses of the real data in this article (Li et al., 2008); BOWTIE v. 0.9.9.2 (Langmead et al., 2009); and BWA v. 0.4.6 (Li and Durbin, 2009)]. All programs were downloaded from their respective sources on April 15, 2009. For each mapping algorithm, the settings were left as close to the defaults as possible, while still allowing meaningful comparisons across algorithms. If the program allowed a setting for the size of the sequence used in a heuristic search, the entire read length was chosen (more details about the settings used for each algorithm are given in Supplementary Table S5). For most of our analyses we considered that a read mapped to a particular location in the genome if that location yielded a uniquely best match. Each of the mapping programs allows for some stochastic assignment of ambiguous reads among potential best hits in the genome. However, since allowing this feature would not offer a complete solution to the mapping bias problem and would make the results more difficult to interpret, we did not use this feature in any of our analyses. For MAQ and BWA, which both report a quality score, we tested whether changing the stringency of the quality score cutoff in simulation experiments had any effect on the biases described here. The results of this analysis appear in Supplementary Figure S2.
Finally, for all SNP positions across the genome with >20 reads in the real data, we simulated all potential reads that could overlap these sites (adding no additional errors) and mapped these reads against the SNP-masked genome using MAQ. This set of simulations was used to determine which SNPs have an inherent bias in the mappability of reads between alleles. We then discarded from analysis all SNPs for which a different number of artificial reads mapped to the reference allele compared with the non-reference allele. Further, for all SNPs with >20 reads in the real data, we simulated reads where the coverage at each SNP was 10 × (as compared to the 1 × simulated data described above) incorporating (i) random read-mapping errors and (ii) variable base quality scores. However, we found that the 1 × coverage simulations were so highly correlated with the 10 × coverage simulations that they were sufficient to predict the SNPs that showed an inherent bias (1 × predicted bias had a correlation coefficient r2 > 0.98 with predicted bias in both 10× simulations).
3 RESULTS
Genome-wide RNA-Seq was performed on RNA from lymphoblastoid cell lines from two Yoruba HapMap individuals and reads were mapped to the human reference genome using MAQ (Section 2). In both individuals, 60–65% of total reads mapped uniquely to annotated exons. To identify ASE, we isolated all reads that, after mapping, overlapped heterozygous exonic HapMap SNPs (yielding 104 128 and 97 359 reads for GM19238 and GM19239, respectively). There were 1981 heterozygous SNPs with >20 reads in one individual (averaging 70.5 reads per SNP-individual combination). By applying this minimum read threshold, we enriched for highly expressed genes. Indeed, 62% of the exons which contained the SNPs we tested were in the top 10% of exons when ranked by expression level. We determined the allele for each of these reads based on the observed nucleotide at the SNP position.
These initial data suggested that ASE was widespread (Fig. 1). Out of 1981 tests, 90 tests of the null hypothesis of equal expression yielded binomial test P-values that were less than P = 5.5 × 10−5 corresponding to an FDR of 1%. However, the results indicated a worrying bias. First, averaging across all sites, there was a highly significant bias toward overrepresentation of reference alleles. Overall, 52.2% of reads matched the reference allele (P < 2 × 10−16 for a binomial test against a true frequency of 50%). Secondly, 61 out of 90 significant results showed overrepresentation of the reference allele (Binomial test; P = 0.002) and all eight of the strongest signals were biased toward the reference allele (Fig. 1B). Therefore, we hypothesized that biases introduced at the read-mapping stage might have affected our results.
Fig. 1.

RNA-Seq data show a higher variance in the relative expression of each allele and a skew toward high expression of the reference allele compared with the predicted distribution. (A) Estimated probability densities for the proportion of reads matching the reference allele (i.e. the allele given in the reference human genome sequence) at heterozygous SNPs in exons. Solid lines correspond to the observed distributions for known heterozygous SNPs with more than 20 reads in two Yoruba HapMap individuals. The dashed line shows the predicted distribution without reference bias or ASE. (B) QQ-plots of P-values for one-sided tests that expression of the reference allele is either higher (circles) or lower (triangles) than the non-reference allele. The horizontal dashed line is the P-value threshold corresponding to a FDR of 1.0%. Notice the enrichment of very significant P-values for overexpression of reference alleles.
To explore this hypothesis further, we simulated reads spanning known SNPs, and tested how often each allele was mapped back to the correct location in the genome. For both alleles at each SNP, we generated all 35 bp reads that overlapped the position of the SNP (Section 2; Supplementary Fig. S1). We observed that some positions showed an extremely strong bias toward the reference sequence. For 1% of SNPs, at least 75% of the mapped reads (averaging across all read positions) carried the reference allele, while for 0.7% of SNPs, all mapped reads carried the reference allele.
Overall, 50.7% of the mapped reads in the simulated data carried the reference allele. This is actually a significantly smaller bias than the 52.2% observed in the real data (P < 2 × 10−16 for a binomial test of the null hypothesis that the proportion in the real data is 50.7%). However, by incorporating random sequencing errors into our simulations, we were able to generate the degree of bias observed in the real data. We found that the magnitude of the bias toward the reference allele rose with increasing sequencing error rates; error rates of 0.01 and 0.05 mutations per base increased the average proportion of mapped reads that matched the reference allele to 51.4% and 59.0%, respectively (Fig. 2A; Section 2).
Fig. 2.

Magnitude of read-mapping biases in simulated data. (A) The distribution (across SNPs) of the proportion of correctly mapped reads that carry the reference allele, compared with the non-reference allele. The y-axis is broken into two segments to show more clearly the rates of highly biased SNPs. Three different rates of sequencing errors are shown. (B) Read-mapping was performed as in (A), except that the reads were aligned against a version of the genome sequence in which all SNP locations were masked. Notice that for both analysis methods, some SNPs are strongly biased, and that SNP masking does not clearly improve the results. Sequencing errors can substantially increase the extent of bias.
One plausible method for removing this bias might be to mask all known SNP positions in the reference genome prior to read-mapping. We found that this did eliminate the overall reference bias in both simulated and real data (Figs 2B and 5A). However, perhaps unexpectedly, this correction failed to reduce the number of individual SNPs with very strong biases (Fig. 2). After masking, 2% of SNPs had at least 75% of reads derived from one allele, and for 1.4% of SNPs all mapped reads came from one allele. As before, sequencing errors increased the fraction of SNPs that had unequal mapping rates for the two alleles, but there was not a substantial average bias toward the reference allele. In summary, we do not find a clear advantage to masking over not masking; however, our subsequent analyses do use masking due to the slight improvements for higher rates of sequencing errors.
Fig. 5.
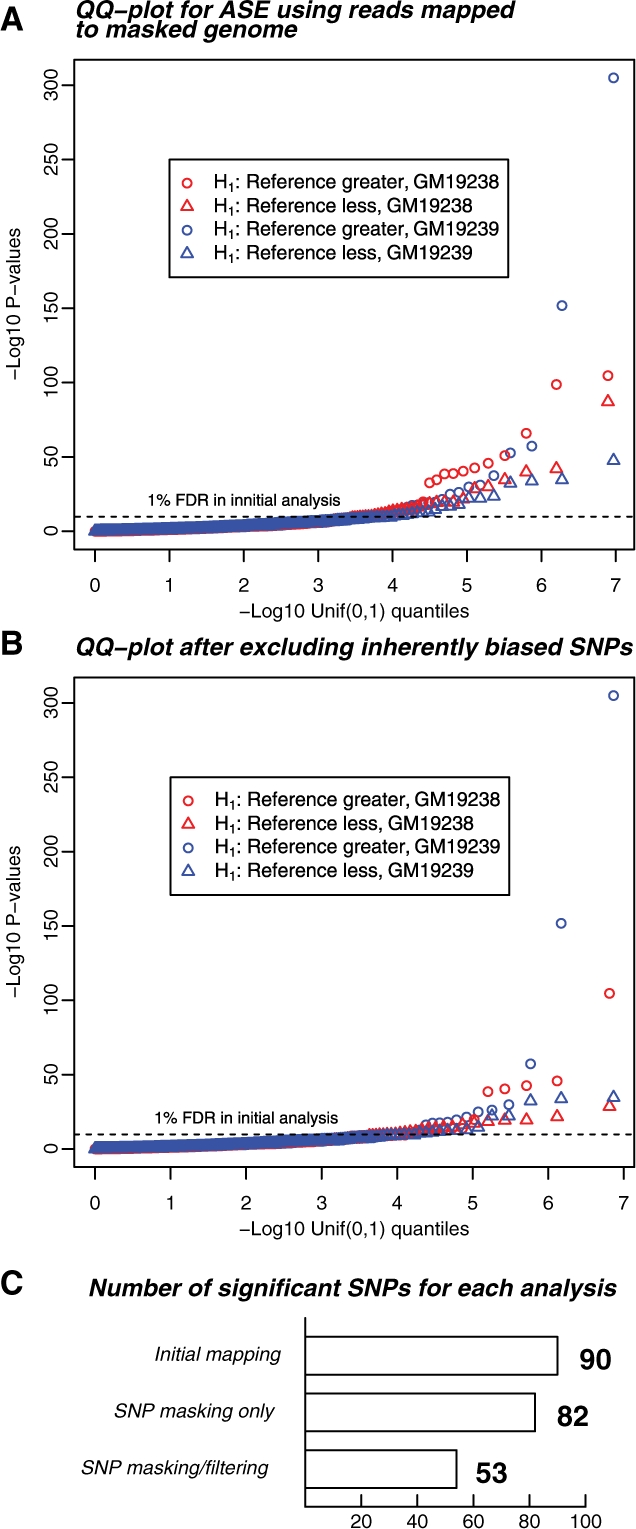
Summary of the ASE results after SNP masking, and after excluding inherently biased SNPs. (A) Distribution of ASE P-values after masking known SNP variation. Masking has largely eliminated bias toward the reference allele (circles: overrepresentation of reference allele; triangles, overrepresentation of non-reference allele), however, the number of significant results is not reduced. Display is as in Figure 1B. The horizontal dashed line represents the P-value threshold of 5.5 × 10−5 that allowed an FDR of 1% in the analysis presented in Figure 1. The FDR for this analysis using the initial P-value threshold was also 1%. (B) Distribution of P-values after excluding SNPs with an inherent bias toward one allele, as determined by simulations of perfect reads. This set of significant results is likely much more reflective of genes that show genuine ASE. The FDR for this analysis using the initial P-value threshold here was 1.4%. (C) Barplot showing the number of significant results for the three read-mapping strategies used in this article, corresponding to Figures 1B, 5A and 5B, using a P-value cutoff of 5.5 × 10−5, corresponding to FDRs of 1.0%, 1.0% and 1.4%, respectively.
To better understand the sources of read-mapping bias, we examined more closely a number of the most strongly biased SNPs. We find that the strong biases occur at SNPs for which the flanking sequence shares sequence identity with another region of the genome (Fig. 3). When we do not mask the SNP location, problems arise when the non-reference allele matches the alternative location as well as, or better, than the correct location (Fig. 3A). With masking, both alleles have an 1 bp mismatch against the correct location, but either allele might match the corresponding position in the alternative location, thereby biasing against correct mapping of the allele that matches elsewhere (Fig. 3B).
Fig. 3.

Two examples in which homology with other genomic locations leads to read-mapping biases. (A) Example of a SNP where there is a bias toward the reference allele before and after SNP masking (rs506008) and (B) example of a SNP where there is a bias toward the non-reference allele after SNP masking (rs11585481). Each example shows the variable sites in: (top row) the reference version of the genome sequence in the true location; (next six rows) three sample reads carrying the reference and three sample reads carrying the non-reference alleles at the SNP and (bottom row) the sequence in a region of homology elsewhere in the genome. The right-hand columns show how each read is mapped with, and without SNP masking. In these examples a read is mapped to a particular location if it has a unique best match at that location, and is unmapped if there is a tie between possible locations. The SNP masking generates an 1 nt mismatch between both alleles and the reference sequence at the masked site.
Next, we investigated whether any of three popular read-mapping programs showed less bias than the others. For the simulated set with a per-bp error rate of 0.01, MAQ seemed to slightly outperform BWA and BOWTIE, in that it produced the highest proportion of SNPs (94%) for which an approximately equal number of reads were mapped from each allele (within 5%; Fig. 4A). However, it remains unclear from this analysis whether this subtle difference between algorithms was mostly due to our parameter choices or if it represents inherent differences between the algorithms themselves. Additionally, we investigated whether changing the quality score thresholds required for mapping by MAQ and BWA reduced the amount of bias. We found that for any particular choice of quality score threshold, there were SNPs that showed an inherent bias. In fact, there was no noticeable improvement in the extent of mapping bias for increasing quality score cutoffs (Supplementary Fig. S2).
Fig. 4.

Bias for three short-read alignment programs and for three read lengths. (A) The plot shows the distribution of the fraction of mapped reads that carry the reference allele. Simulated reads with an error rate of 0.01 were mapped to the masked genome using MAQ (black), BOWTIE (dark blue) and BWA (light blue). Other details as in Figure 2B. (B) Mapped with MAQ as in (A) except that reads contained no additional errors and read lengths were as indicated.
Because next-generation sequencing technologies are improving and longer read lengths are becoming possible, we explored the extent of read-mapping bias for read lengths of 50 and 100 bp. We find that for sequences without read-mapping errors, while the read-mapping bias decreases for increasing read lengths, even reads with 100 bp show SNPs with some bias (Fig. 4B). We also find that there is decreasing bias for increasing read lengths when random errors are added and the default thresholds of MAQ are relaxed (Supplementary Fig. S5).
Armed with an understanding of the effect of biases introduced by SNP variation, we used this knowledge to reanalyze our RNA-Seq data to find loci displaying evidence of ASE. We observed that 1920 SNPs had at least 20 × coverage across one individual after mapping to the masked reference genome. Of these, 82 showed significant deviation from equal expression after masking SNP locations, using a P-value cutoff of 5.5 × 10−5 corresponding to an FDR of 1% in the initial analysis and an FDR of 1% here.
However, as we have noted above, mapping to the SNP-masked genome does not eliminate mapping bias on a SNP-by-SNP basis (Figs 2B, 3 and 5). Simulations show that 185 of the 1920 SNPs have an inherent bias in the mappability of reads coming from one of the alleles (see Section 2 and Fig. 3). Of these inherently biased SNPs, 29 were among the 82 most significant SNPs. This represents a strong enrichment for the inherently biased SNPs among the SNP set that appears to show ASE (Fisher's exact test; P = 2.1 ×10−7). Furthermore, the biases observed in the simulated dataset correlated well with the biases observed at these SNPs in the RNA-Seq data, suggesting that the read-mapping biases described above were contributing to the original signal of ASE (Supplementary Fig. S3).
After excluding these biased SNPs, we were left with 53 SNPs in 47 genes that were significant at the P-value threshold corresponding to an FDR of 1% in the initial analysis. We consider these remaining SNPs as candidates for representing true cases of ASE (see Supplementary Table S1 for a list of these loci).
To verify that the significant results in our final analysis were biologically relevant, we analyzed the overlap of genes in this set with genes previously identified as having cis-regulatory variation or genetic imprinting. Using an eQTL (expression quantitative trait locus) browser that we have developed (http://eQTL.uchicago.-edu; J.F.D. and J.T. Bell), we analyzed the extent of overlap of our significant results with the genes known to have a cis-eQTL in lymphoblasts. Veyrieras et al. (2008) tested for cis-eQTL in 11 466 genes in the HapMap lymphoblastoid cell lines and found that 419 of these genes contained strong evidence for an eQTL (using a posterior probability cutoff of >0.9). Our list of significant genes at an FDR of 1% included 19 of the genes tested by Veyrieras et al. (2008), of which three showed evidence for an eQTL using the same cutoff. We find that this fraction (3/19) supports an enrichment of genes in our set that were previously found to have an eQTL in lymphoblasts (P=0.04; Fisher's exact test). Further, our set of significant genes contained two examples of genes known to be imprinted in humans (annotated imprinted genes obtained from www.geneimprint.com). One gene (SNURF/SNRPN) is located within human chromosome 15q11-15q12, the same region that is involved in Prader–Willi and Angelman Syndromes, and is known to be paternally imprinted (reviewed in Horsthemke and Wagstaff, 2008). A second gene (GNAS) at 20q 13.3 is known to be maternally imprinted and, when disrupted, can cause Albright hereditary osteodystrophy and other complications (reviewed in Weinstein et al., 2004). Thus, we find that there is a significant enrichment among genes showing ASE for genes known to be imprinted in humans (P=0.01; Fisher's exact test). Further supporting the biological relevance of the final results is the fact that in four genes (HLA-DPB1, PIP4K2A, GYPC and PTK2B), we find ASE in both individuals and in four other genes (CRYZ, ATF5, HLA-DRA and SEPT9), two SNPs in the same individual give significant results for ASE in the same direction (i.e. the higher expressed alleles are in phase in the HapMap data; Supplementary Table S1). Finally, we find that heterozygous SNPs within the same genes as our top results, although not all significant by the same threshold, generally support the same direction of ASE as the top results (i.e. the higher expressed alleles are in phase in the HapMap dataset; Supplementary Fig. S4). Taken together, these results suggest that after filtering our data to exclude inherently biased SNPs, we are able to identify real signals of both cis-regulatory DNA variation and genetic imprinting.
4 DISCUSSION
We have shown here that differential mapping of SNP alleles can greatly affect inferences that rely on quantifying DNA or RNA with next-generation sequencing data. This may be especially problematic in studies that aim to detect allele-specific differences in gene expression, transcription factor binding or other related applications. It may also cause problems in other contexts, for example, in QTL mapping of exon expression levels, or for discovery of new sequence variants. Our results also highlight the complexities that may arise when using short read sequences to study organisms with poor quality genome sequences or whose actual genome sequence differs from the reference individual. Although not considered here, it is likely that small insertions and deletions will cause problems at least as severe as we have described here for SNPs.
Perhaps surprisingly, we found that masking known SNPs does little to eliminate inherent biases in read-mapping. However, using simulated sequence reads, we were able to identify individual SNPs that are inherently biased due to problems in read-mapping. In so doing, we were able to identify and remove a large number of false positive results that were present in a naive analysis (Fig. 5C). Although our final analysis makes use of knowledge of SNP variation in the human genome, the simulations that determined the ‘mappability’ of each allele were the key to identifying and removing false positive results. Thus, a similar approach could be taken in organisms with a less complete annotation of SNP variation. This article highlights a clear need for the development of more detailed statistical models that can incorporate knowledge of SNP variation into read-mapping and explicitly model uncertainty in the mapping locations for reads when testing for allele-specific effects.
ACKNOWLEDGEMENTS
We thank three anonymous reviewers for thoughtful comments. We are grateful to the other members of the Jonathan Pritchard, Molly Przeworski and Matthew Stephens labs for helpful advice on this project. Solexa GAII sequencing was performed at sequencing centers at Yale and Argonne, and we thank Paul Zumbo and Mark Domanus for their support.
Funding: National Institute of Health (grant RO1 MH084703-01 to Jon.K.P. and grant GM077959 to Y.G.); the Howard Hughes Medical Institute; the National Institutes of Health Genetics and Regulation Training (grant T 532 GM007197-34 to J.F.D., A.A.P. and Jos.K.P.).
Conflict of Interest: none declared.
REFERENCES
- Babak T, et al. Global survey of genomic imprinting by transcriptome sequencing. Curr. Biol. 2008;18:1735–1741. doi: 10.1016/j.cub.2008.09.044. [DOI] [PubMed] [Google Scholar]
- Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A. 2008;146A:2041–2052. doi: 10.1002/ajmg.a.32364. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. DNA sequencing. A plan to capture human diversity in 1000 genomes. Science. 2008;319:395. doi: 10.1126/science.319.5862.395. [DOI] [PubMed] [Google Scholar]
- Knight JC. Allele-specific gene expression uncovered. Trends Genet. 2004;20:113–116. doi: 10.1016/j.tig.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani L, et al. Allele-specific gene expression patterns in primary leukemic cells reveal regulation of gene expression by CpG site methylation. Genome Res. 2009;19:1–11. doi: 10.1101/gr.083931.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, et al. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald J, et al. Simultaneous genotyping, gene-expression measurement, and detection of allele-specific expression with oligonucleotide arrays. Genome Res. 2005;15:284–291. doi: 10.1101/gr.2850605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, et al. Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet. 2008;4:e1000006. doi: 10.1371/journal.pgen.1000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras JB, et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS ONE. 2008;3:e3839. doi: 10.1371/journal.pone.0003839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS, et al. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–5464. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- Wittkopp P, et al. Independent effects of cis- and trans-regulatory variation on gene expression in Drosophila melanogaster. Genetics. 2008;178:1831–1835. doi: 10.1534/genetics.107.082032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, et al. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]