

Abstract
Autophagy has many roles in immunity, including the control of intracellular microbes by a cell-autonomous mechanism. In this issue of Immunity, Shelly et al. (2009) use VSV infection in Drosophila to show the role of autophagy genes in controlling viruses.
Autophagy is important in many health and disease processes, including aging, metabolism, cancer, neurodegeneration, immunity, and inflammatory illnesses. In principle, autophagy is evolution's answer to the demands of keeping the cytoplasm and complex organellar systems of the eukaryotic cells in good repair, with an added bonus of nimble biomass adjustments in response to growth factor withdrawal and starvation. The key emblematic physical manifestation of autophagy (executed by the proteins termed Atg) that can be morphologically observed under the microscope is the formation of autophagosomes inside the cell's cytoplasm. Autophagosomes corral portions of the cytosol for digestion in autolysosomes and reuse it at times of starvation. They also capture defective or obsolete organelles earmarked for removal, such as leaky mitochondria, surplus peroxisomes, excess endoplasmic reticulum, etc. The metabolic aspects of autophagy are under the negative control by growth factors, insulin receptor substrates, type I phosphatidylinositol 3 kinase (PI3K), Akt, Tor, and Atg1 signaling cascades. Autophagic machinery is regulated at yet another key node centered on an ancient stress-signaling enzyme, known as type III PI3K VPS34, via its interacting partner Beclin 1. Many signals affect these two “nerve centers” of autophagic control. In the case of Beclin 1, stress inputs such as activation via JNK kinase, death-associated protein kinase, p14ARF agonist and stabilizer of the tumor suppressor p53, which in turn also regulates autophagy, hypoxia response regulator HIF-1 via the BH3-only protein Bnip3, and perhaps even MyD88, an adaptor downstream of many pattern recognition receptors (PRRs), can lead to autophagy activation.
The multitude of immune functions of autophagy (Deretic, 2009), dubbed once as “immunophagy,” encompass: (1) peripheral and central tolerance including thymic selection along with MHC II presentation of cytoplasm antigens; (2) homeostasis of T cell, B cell, and other specialized immune cells, e.g., Paneth cells of the intestinal crypts; (3) activation or dampening of proinflammatory processes including IL-1b and type I IFN production; (4) effector output of Th1 and Th2 cell polarization in defense against intracellular pathogens; (5) activation of PRR along with carrying out antimicrobial effector functions downstream of PRR stimulation; and (6) cell-autonomous defense against bacterial, protozoan, and viral pathogens. In this issue, Shelly et al. (2009) touch upon the points 5 and 6 from the above list of the immunological functions of autophagy. The processes studied by Shelly et al. probably hark back to the evolutionarily most ancient use of autophagy: to apprehend and destroy by digestion the microbial intruders that manage to erode into the eukaryotic cell's cytosol. A confounding roadblock to uncovering and clearly demonstrating this primordial function of autophagy is that years of evolutions stand in the way when one examines finely tuned present-day host-pathogen pairs. This is because more often than not, the inherent ability of mammalian cells to efficiently deploy their antimicrobial measures is masked by the countermeasures deployed by the highly adapted microbes, either causing disease or evolving all the way to commensalism. The molecular underpinnings of this problem are best illustrated in the examples of Shigella (Ogawa et al., 2005) and herpes simplex virus type 1 (HSV-1) (Orvedahl et al., 2007), both of which possess highly evolved specific factors enabling the microorganisms to efficiently evade autophagy: Shigella virulence protein IcsB blocks a specific bacterial epitope that otherwise induces autophagy, whereas HSV-1 protein ICP34.5 binds the key autophagy factor Beclin 1 and inhibits its function. The work by Shelly et al. (2009) bypasses this problem inherent to evolutionarily finely tuned host-pathogen pairs: the authors employed a mismatch by using Drosophila as a model system to study how autophagy affects vesicular stomatitis virus (VSV). This unlikely pair is not as remote from real-life situations as it may appear at first blush, given that VSV normally alternates between arthropod vectors and mammalian host and can grow in both insect and mammalian cells.
Shelly et al. (2009) first showed that VSV replicated in Drosophila S2 cells. The virus released from S2 cells was found to replicate in mammalian cells, thus showing that the virus produced in this system is functional without an apparent penalty to its host range. Having established this, Shelly et al. (2009) first knocked down three of the key autophagy factors, Atg1, Atg5, and Atg8a, and later on, a panel of other Atg factors (Atg1, Atg2, Atg4, Atg6, Atg7, Atg8b, and Atg9) and showed that this increased VSV production in infected cells. The changes detected by two measures, including viral titers, were small but statistically significant. Furthermore, the authors found that VSV induced autophagy in Drosophila cells. Interestingly, this induction occurred whether the cells were infected with replication-competent or UV-inactivated VSV. This gave authors a hint that replication intermediates were not needed to induce autophagy; such a finding is of significance because in experiments by others, using different virus-dendritic cell pairs, viral replication was a key to the engagement of the autophagic pathway (Lee et al., 2007). As an upshot from these observations, Shelly et al. (2009) considered the possibility that a preformed viral molecule (rather than a replicating virus) acted as a pathogen-associated molecular pattern (PAMP) possibly stimulating an unidentified PRR. This is a reasonable assumption because PAMPs and PRRs are known to engage the autophagosomal pathway in a variety of ways: by inducing autophagy upon PRR stimulation with PAMPs (Deretic, 2009), by employing autophagy as a topological inversion device to deliver PAMPs to endosomal PRRs (Lee et al., 2007), or even by using autophagy to dampen potentially excessive response of RIG-I-like receptors (RLRs) in the context of infection with VSV (Tal et al., 2009). Shelly et al. (2009) went on to challenge GFP-LC3 (used as a probe for autophagy) reporter cells with VSV glycoprotein G (VSVG)-induced membrane blebs or vesicular particles and observed that this material can induce formation of GFP-LC3 puncta, which are conventionally taken as a sign of autophagosome formation. On the basis of these observations, the authors proposed that VSVG is a PAMP that can induce autophagy, although the precise composition of PAMP and the identity of its cognate PRR remained to be established. A hint for a likely participation of TLRs comes from the mammalian hosts pointing to TLR4 as a PRR for VSVG. This dovetails with the reports that TLR4 can induce autophagy (Xu et al., 2007).
Next, Shelly et al. (2009) used the VSV-Drosophila model to demonstrate a requirement for autophagy to render VSV infection nonpathogenic in adult flies. When flies, which normally survive infection with VSV, were depleted of Atg8, 90% of them died by day 12 after infection. The virus in the Atg18-depleted flies replicated to higher titers prior to lethality. The authors went an extra mile and showed that Atg7 or Atg12 depletion in flies lead to similar phenotypes in terms of lethality and viral replication in vivo, albeit some variances vis-à-vis Atg18 were observed. Potentially relevant for any therapeutic ideas that may emanate form this work, Shelly et al. (2009) showed that there was a role for the Akt pathway (and probably Tor) in controlling the virus in infected flies. In an elegant conclusion to the paper, the authors resorted to a clever experimental trick bypassing some difficulties inherent to the system. The authors gave flies insulin (which activates Akt and inhibits autophagy in many cells) and showed that infection with the virus abrogated insulin-dependent Akt activation. This observation suggests that signaling leading to induction of autophagy in response to VSV infection also includes inhibitory effects on Akt. Thus, there seems to be a link between atophagy induction and inhibition of pathways associated with nutrition and growth-factor-dependent increase in cellular biomass (Figure 1). It brings a new meaning to the myth “feed a cold and starve a flu.”
Figure 1. Autophagy in Antiviral Immunity.
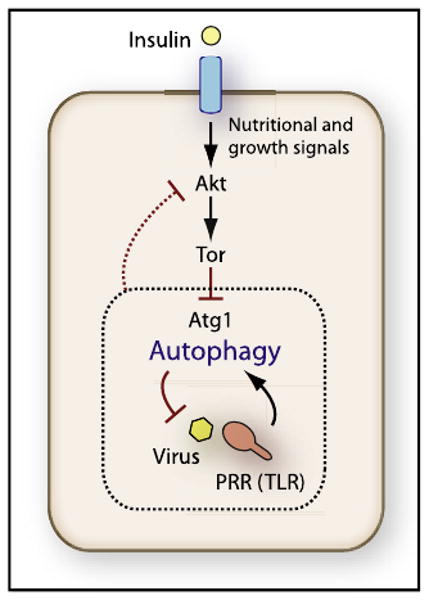
Induction of autophagy as a cell-autonomous antiviral defense in metazoans may require simultaneous stimulation with PRR agonists and inhibition of the Akt pathway. Autophagy, as demonstrated by Shelly et al. (2009), controls VSV in vivo. This process occurs via viral PAMP-dependent induction of autophagy through a PRR (possibly TLR). Nutritional signals, such as insulin, can counteract autophagy induction by stimulating Akt and Tor (Tor inhibits autophagy), but Shelly et al. (2009) observed that during viral PAMP stimulation of autophagy, Akt pathway was inhibited by an unknown mechanism (dashed line); this could be related to the feedback loops known to exist between Tor targets and Akt function (not depicted).
An interesting question arises as to whether these systems are coordinated in all organisms and whether this is perhaps a key primordial reaction of infected eukaryotic or metazoan cell to limit growth of microbes by simultaneously blocking protein synthesis and inducing cell-autonomous systems that can capture and degrade intracellular pathogens or their biosynthetic intermediates. If so, this would resonate well with the studies by Levine and colleagues (Orvedahl et al., 2007), where HSV-1 was found to have evolved ways to inhibit both of these defense mechanisms (represented by PKR and Beclin 1) in the mammalian host. Perhaps this dual action will become a common theme in the context of autophagy and host-pathogen interactions. Furthermore, this could be an Achilles' heel of this defense process, as seen with the stimulation of the Akt pathway downstream of IL-4 and IL-13, when Th2 cell response inhibits autophagic control of Mycobacterium tuberculosis (Harris et al., 2007).
In conclusion, the study by Shelly et al. (2009) used a clever approach to unmasking the role of autophagy in vivo. A question arises: can such processes be observed in finely tuned host-pathogen pairs without disarming the microbe of its antiautophagic adaptations? The answer is yes: the first paper that showed that autophagy control viruses comes from a study in plants—in which Beclin 1, Atg3, and Atg7 were necessary to limit tobacco mosaic virus in the infected leaves. However, it is easier to appreciate this role of autophagy if the pathogen and the host are not perfectly matched. In vivo studies akin to those by Shelly et al. (2009) in Drosophila have shown that autophagy controls bacterial infections (e.g., Listeria) (Yano et al., 2008). M. tuberculosis falls prey to autophagy in human cells (Gutierrez et al., 2004). Interestingly, M. tuberculosis, albeit a big-time scourge of human population, has had evolutionarily speaking a relatively short time to fully adapt to its host given the slow doubling time. This seems to bring to the fore one of the potential take-home messages (if not a rule) that can be extracted from the study by Shelly et al. (2009): the ease of detecting autophagic control of intracellular microbes increases if the host-pathogen pair under study has not yet reached an evolutionary equilibrium, i.e., before a pathogen has had a chance to perfect defenses against autophagy.
References
- Deretic V. Curr Opin Immunol. 2009 doi: 10.1016/j.coi.2009.02.002. in press. Published online March 5, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. Immunity. 2007;27:505–517. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Immunity. 2009;30(this issue):588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Proc Natl Acad Sci USA. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, et al. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]