

Abstract
Agonist-induced ubiquitylation and degradation of heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) play an essential role in surface receptor homeostasis, thereby tuning many physiological processes. Although β-arrestin and affiliated E3 ligases mediate agonist-stimulated lysosomal degradation of the β2-adrenergic receptor (β2AR), a prototypic GPCR, the molecular cues that mark receptors for ubiquitylation and the regulation of receptor degradation by the proteasome remain poorly understood. We show that the von Hippel–Lindau tumor suppressor protein (pVHL)–E3 ligase complex, known for its regulation of hypoxia-inducible factor (HIF) proteins, interacts with and ubiquitylates the β2AR, thereby decreasing receptor abundance. We further show that the interaction of pVHL with β2AR is dependent on proline hydroxylation (proline-382 and -395) and that the dioxygenase EGLN3 interacts directly with the β2AR to serve as an endogenous β2AR prolyl hydroxylase. Under hypoxic conditions, receptor hydroxylation and subsequent ubiquitylation decrease dramatically, thus attenuating receptor degradation and down-regulation. Notably, in both cells and tissue, the abundance of endogenous β2AR is shown to reflect constitutive turnover by EGLN3 and pVHL. Our findings provide insight into GPCR regulation, broaden the functional scope of prolyl hydroxylation, and expand our understanding of the cellular response to hypoxia.
Introduction
β-Adrenergic receptors (βARs) are prototypic GPCRs that play an important role in the regulation of cardiovascular and pulmonary function, and sustained βAR down-regulation (and dysfunction) is associated with diseases such as heart failure and asthma (1, 2). There are three subtypes of βAR (β1, β2, and β3). The β2AR constitutes about 25 to 30% of total βARs in the human heart and is the predominant subtype present in both vascular and airway smooth muscle (1, 2). In contrast, the β1AR predominates in the heart (∼70%). Whereas β1- and β2ARs have overlapping cellular actions (1–4), β2ARs may be especially important in the failing heart where the β1AR is down-regulated. Indeed, overexpression or pharmacological stimulation, or both, of β2ARs increases cardiac contractility and is cardioprotective in animal models (4–8).
Continuous agonist stimulation induces β2AR ubiquitylation, internalization, and degradation (9), thereby decreasing the total number of receptors. Agonist-stimulated removal of receptors from the cell surface involves a pathway in which the adaptor protein β-arrestin binds phosphorylated receptors (after agonist stimulation) and recruits E3 ligases (in particular NEDD4) that subserve ubiquitin-mediated lysosomal degradation (9, 10). Cues other than phosphorylation may also mark receptors for ubiquitylation, internalization, or both, and events that control proteasomal (as opposed to lysosomal) degradation of receptors remain a mystery. In particular, an E3 ligase responsible for proteasomal degradation of the β2AR has not been identified, yet proteasomal inhibitors markedly reduce receptor internalization and degradation (under both resting conditions and after agonist stimulation) (9).
The abundance and balance of βAR subtypes is thought to be regulated primarily by adrenergic state. Thus, for example, the net decreased abundance of cardiac βARs that occurs in response to hypoxia has been attributed to elevated concentrations of catecholamines (11). However, whereas β1AR abundance decreases under severely hypoxic conditions, consistent with heightened adrenergic tone, β2AR abundance actually increases (12), raising the possibility that the β2AR may be regulated directly by O2. Metallo (iron)-sensory enzymes transduce O2-responsive signals through the modification of target proteins with which they interact. Here, we report that the iron-containing dioxygenase EGLN3 serves as an O2-dependent β2AR prolyl hydroxylase. After hydroxylation of the β2AR at Pro382 and Pro395, the von Hippel–Lindau tumor suppressor protein (pVHL)–E3 ligase complex, previously characterized in the context of hypoxia-inducible factor (HIF) regulation, is recruited to and ubiquitylates the β2AR, promoting its down-regulation by proteasomal degradation. Accordingly, hypoxia stabilizes the β2AR.
Results
Hypoxia limits β2AR ubiquitylation
The effect of oxygen tension (partial pressure of oxygen, PO2) on β2AR stability was examined with the use of human embryonic kidney (HEK) 293 cells stably expressing human β2ARs that contain an N-terminal FLAG tag (β2AR-293 cells). Cells were cultured under ambient PO2 (21% O2) or hypoxic conditions (1% O2) in the presence or absence of the βAR agonist, isoproterenol (10 μM, 18 hours). Receptors were immunoprecipitated from whole-cell lysates with agarose beads conjugated to an antibody against FLAG (anti-FLAG) and Western blotted with an antibody against β2AR (anti-β2AR) to assess the total cellular abundance of β2AR. Hypoxia substantially increased the abundance of β2AR in the presence or absence of agonist (Fig. 1, A, B, and D). In addition, the half-life of the β2AR was increased significantly by 1% compared to 21% O2, as measured in pulse-chase experiments (without agonist treatment) after metabolic labeling of cells with 35S[Met] and 35S[Cys] (Fig. 1C).
Fig. 1.
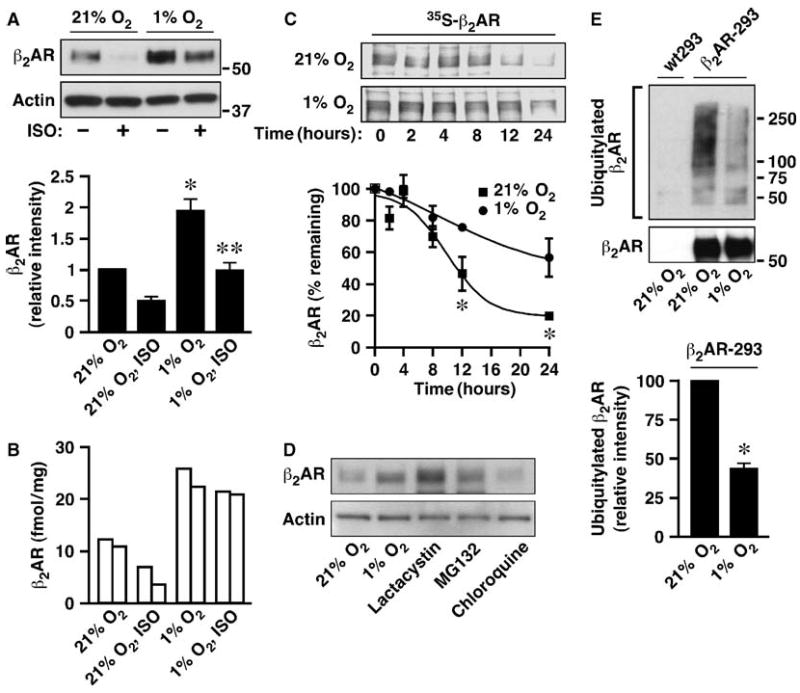
Hypoxia stabilizes the β2AR by inhibiting its ubiquitylation. (A) FLAG-tagged β2AR was isolated with anti-FLAG beads from β2AR-293 cells cultured under 21 or 1% O2 with or without 10 μM isoproterenol (ISO) for 18 hours, and Western blotted with anti-β2AR antibody. Actin was used as the loading control. n = 3 (n = the number of independent experiments, here and elsewhere), *P< 0.01 versus 21%, **P< 0.05 versus 21% + ISO (Student's t test). (B) HUVECs were cultured under 21 or 1% O2 with or without 10 μM ISO for 18 hours. Cell membranes were collected and β2AR density was measured by radioligand binding (n = 2; each bar represents the result of an individual experiment). (C) β2AR-293 cells were metabolically labeled with 35S[Met] and 35S[Cys] for 12 hours followed by a chase period of the indicated durations carried out at 21 or 1% O2. FLAG-β2AR was immunoprecipitated from the cell lysates and signals were detected by SDS-PAGE followed by autoradiography. n = 3, *P < 0.05 versus 1% by analysis of variance (ANOVA). (D) β2AR-293 cells were treated with MG132 (10 μM), lactacystin (20 μM), or chloroquine (20 μM) for 18 hours. Receptor was collected with anti-FLAG beads and Western blotted with anti-β2AR. (E) β2AR was collected with anti-FLAG beads from β2AR-293 cells cultured at 21 or 1% O2 with MG132 (10 μM) for 18 hours and Western blotted with anti-ubiquitin or anti-β2AR. n = 3, *P < 0.01 versus 21% (Student's t test). In (A), (C), and (E), densitometric analysis (mean ± SEM) was performed.
HIFs coordinate transcriptional adaptation to hypoxia by binding to hypoxia-response elements (HREs) within many target genes. However, it is unlikely that the increased abundance of β2AR in the β2AR-293 cells is regulated by HIFs because FLAG-β2AR expression was driven by a cytomegalovirus promoter that is devoid of HREs, and overexpression of a normoxia-stabilized HIF-1α (13) had no effect on β2AR abundance (Fig. 2E and fig. S1). Although agonist-stimulated down-regulation of overexpressed β2AR was largely unaffected by hypoxia in HEK cells (the decrease in β2AR abundance in response to isoproterenol was ∼50% at both 21 and 1% O2; Fig. 1A), hypoxia inhibited receptor down-regulation by agonist under more physiologically relevant conditions. Specifically, in human umbilical venous endothelial cells (HUVECs), hypoxia not only increased the endogenous β2AR from 10 to 22 fmol/mg under basal conditions, but also inhibited isoproterenol-stimulated down-regulation (Fig. 1B). Thus, both constitutive and ligand-induced trafficking of the β2AR may be regulated by PO2 in primary cell lines.
Fig. 2.
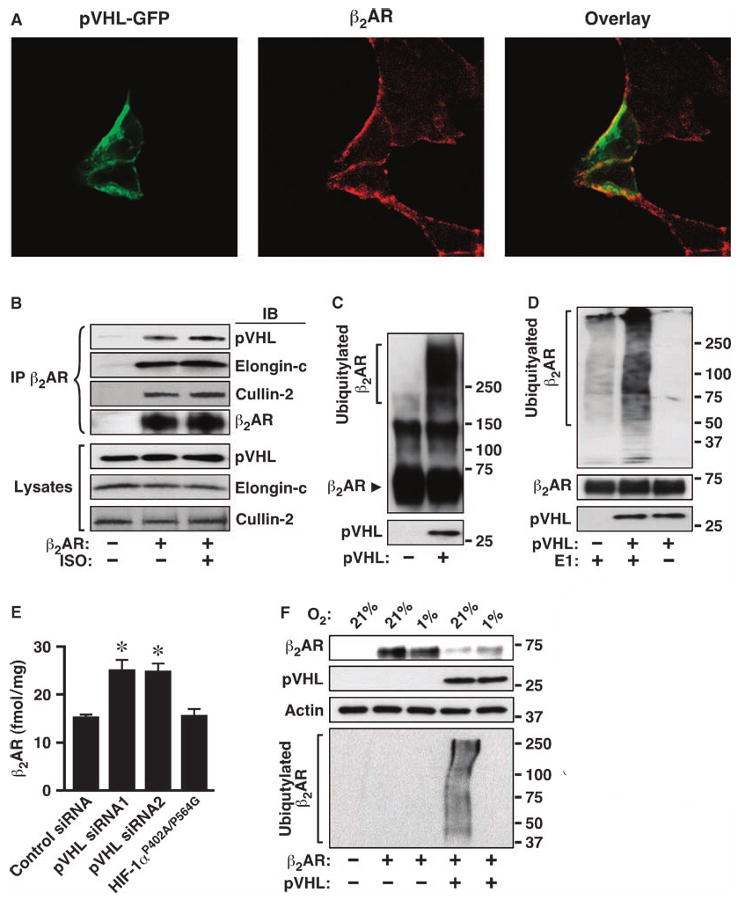
The pVHL complex associates with the β2AR and serves as an E3 ubiquitin ligase. (A) β2AR-293 cells were transiently transfected with pVHL-GFP, fixed, and stained with anti– FLAG M2 (β2AR) and TRITC-conjugated secondary antibody (red). (B) FLAG-pVHL and β2AR or pcDNA3 were cotransfected into HEK293 cells. ISO stimulation was for 15 min. Cell lysates were immunoprecipitated (IP) with anti-β2AR and Western blotted (IB) with anti-β2AR, anti-FLAG (for pVHL), anti–cullin-2, or anti–elongin-C as indicated. (C and D) In vitro ubiquitylation assays were performed for recombinant β2AR (150-kD dimer in C is reflective of high recombinant protein concentration) in the presence or absence of pVHL complex or E1 ubiquitin-conjugating enzyme. Samples were Western blotted with anti-β2AR (C) or anti-ubiquitin (D). Bottom panels show the pVHL input. (E) HUVECs were electrotransfected with scrambled siRNA, siRNAs for human VHL, or plasmid expressing HIF-1αP402A/P564G for 72 hours. Cell membranes were collected and β2AR density was measured by radioligand binding. n = 3, *P < 0.01 versus scrambled siRNA (Student's t test). (F) β2AR was transfected into 786-O or 786-O–pVHL cells; after 24 hours, cells were incubated for an additional 18 hours at 21 or 1% O2. Proteins were Western blotted with specific antibodies as indicated. The illustrated pVHL-dependent effects of PO2 are representative of three experiments, but cotransfection with pVHL had variable effects on the overall abundance of transfected β2AR in 786-O cells.
GPCR endocytosis and degradation may involve both lysosomal and proteasomal mechanisms (9). We found that under steady-state conditions, degradation of the β2AR was blocked by proteasome inhibitors but not by lysosome inhibitors (Fig. 1D). We then examined whether β2AR ubiquitylation was regulated by PO2. β2AR-293 cells were cultured overnight at 21 or 1% O2 in the presence of the proteasome inhibitor MG132, and immunoprecipitated receptors were Western blotted with antibodies against ubiquitin (anti-ubiquitin) or against β2AR. The β2AR was ubiquitylated under normoxic conditions in the absence of agonist stimulation, and the degree of ubiquitylation was dramatically decreased at 1% O2 (Fig. 1E).
pVHL complex serves as the E3 ubiquitin ligase for the β2AR
Agonist stimulation induces rapid ubiquitylation of both the β2AR and the regulatory protein β-arrestin (9, 10). Agonist-coupled ubiquitylation of the β2AR requires receptor phosphorylation and subsequent binding of β-arrestin 2 (9, 10); thus, β-arrestin 2 acts to recruit an E3 ligase to the β2AR. However, the β-arrestins are primarily cytosolic in unstimulated cells, in which β2AR ubiquitylation is prominent (Fig. 1E), and constitutive trafficking of the β2AR also occurs (14). Therefore, we considered the possibility that ubiquitylating enzymes may interact directly with the β2AR to regulate constitutive trafficking of the receptors. The pVHL complex is an O2-responsive E3 ligase for HIF-1α (15), and we hypothesized that it might also serve as an E3 ligase for the β2AR. We first conducted immunofluorescence and coimmunoprecipitation experiments in both β2AR-293 and HEK293 cells to determine whether pVHL and the β2AR interact. Although pVHL, both overexpressed and endogenous, was broadly distributed throughout the cell, pVHL colocalized with β2ARs associated mainly with cellular membranes (Fig. 2A and fig. S2A). Additionally, both overexpressed and endogenous pVHL coimmunoprecipitated with β2AR both in resting cells and after treatment with isoproterenol (Fig. 2B and fig. S2B). Moreover, endogenous elongin C and cullin-2, the components of the native pVHL complex (16), were also present in β2AR immunoprecipitates (Fig. 2B), and the direct interaction between the β2AR and pVHL complex was confirmed by far-Western blot (fig. S2C). Collectively, these findings suggest that the pVHL–E3 ligase complex associates with the β2AR in cultured cells.
We performed an in vitro ubiquitylation assay to determine whether the pVHL–E3 ligase complex directly ubiquitylated β2AR. FLAG-pVHL was transiently overexpressed in HEK293 cells, immunoprecipitated, and mixed with purified recombinant β2AR in a cell-free ubiquitylation system. As revealed by Western blotting with anti-β2AR antibody, native receptor migrated at ∼55 kD (monomer) or 110 kD (dimer) (Fig. 2C). Multiple smeared bands of higher molecular weight, representing polyubiquitylated β2ARs, appeared only when immunoprecipitate from cells expressing pVHL was added to the in vitro system (Fig. 2C). In vitro ubiquitylation of the β2AR was verified by Western blotting with an antibody against ubiquitin (anti-ubiquitin) (Fig. 2D) and by control experiments in which omitting the E1 ubiquitin-activating enzyme from the in vitro assay completely eliminated the ubiquitin signal (Fig. 2D). Silencing small interfering RNA (siRNA) studies provided further evidence that the pVHL complex serves as the endogenous E3 ligase, because knockdown of endogenous pVHL in HUVECs resulted in a significant increase in the abundance of endogenous β2AR (Fig. 2E and fig. S2E).
The role of pVHL in O2-dependent ubiquitylation of the β2AR was further corroborated by studies in pVHL-deficient 786-O cells in which the stabilizing effect of hypoxia on the β2AR was absent (Fig. 2F). Introduction of pVHL restored O2 responsivity and thereby decreased β2AR abundance in normoxia relative to 1% O2 (Fig. 2F). Additionally, β2AR ubiquitylation was greatly increased at 21% O2 and inhibited at 1% O2, and this effect was dependent on pVHL (Fig. 2F). These findings suggest that, as is the case for HIF-1α, the pVHL complex is an E3 ligase for the β2AR.
Hydroxylation of the β2AR at proline residues P382 and P395
HIF is a dimer of α and β subunits, with the α subunit regulated by oxygen and the β subunit constitutively present (17). Oxygen dependence is mediated by a family of enzymes (EGLN 1 to 3) that catalyze hydroxylation of specific prolyl residues within the α subunits of HIF (18, 19). The hydroxylated α subunit is then targeted by pVHL for ubiquitylation and proteasomal degradation under normoxic conditions. Hypoxia decreases HIF-1α prolyl hydroxylation, which is required for the interaction between pVHL and HIF-1α (20, 21), and thus stabilizes HIF. We noted that hypoxia similarly decreases the interaction between pVHL and the β2AR (Fig. 3, A and B, and fig. S2D), raising the possibility that proline hydroxylation might also mediate the interaction of β2AR with pVHL. Proline hydroxylation of HIF-1α occurs at a conserved motif L[XX]LAP (18, 22). Although a substitution of flanking Leu or Ala residues has little effect on HIF-1α hydroxylation (23), an acidic amino acid at the +5 position (relative to Pro) seems to be important (24). There are three Pro residues within intracellular domains of the β2AR and acidic amino acids are found downstream of two of them (Pro382 and Pro395). Mutation of these two proline residues (β2ARP382A/P395A) decreased the interaction between receptor and pVHL relative to that of wild-type β2AR (Fig. 3B). In addition, exposing cells expressing the proline mutant receptor to hypoxia or treatment with dimethyloxalyglycine (DMOG), a broad-spectrum prolyl hydroxylase inhibitor, failed to increase the abundance of β2ARP382A/P395A (Fig. 3C).
Fig. 3.
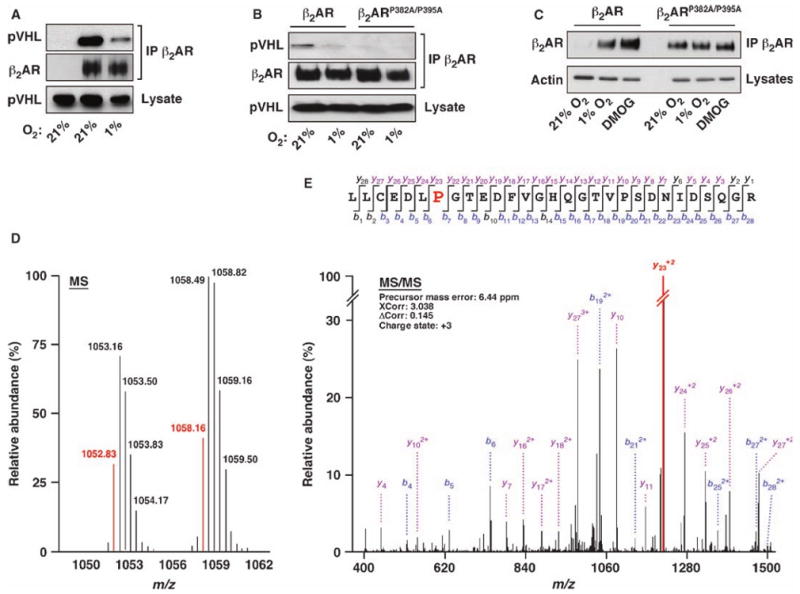
The β2AR is hydroxylated at proline residues P382 and P395. (A and B) FLAG-pVHL and β2AR [(A) and (B)] or β2ARP382A/P395A (B) were cotransfected into HEK293 cells; in (A), the leftmost lane shows the result of control transfection with pcDNA3 rather than pcDNA3-β2AR. After 24 hours, cells were incubated for an additional 18 hours at either 21 or 1% O2 in the presence of 10 μM MG132. Cell lysates were immunoprecipitated with anti-β2AR and Western blotted with anti-FLAG (for pVHL) or anti-β2ARs. (C) HEK293 cells stably expressing FLAG-β2AR or FLAG-β2ARP382A/P395A were incubated at 21 or 1% O2 or treated with 1 mM DMOG for 18 hours at 21% O2. β2AR or β2ARP382A/P395A was isolated with anti-FLAG beads and Western blotted with anti-β2AR antibody. Actin was used as loading control. (D) FLAG-β2AR purified from β2AR-293 cells cultured at 21% O2 was digested with trypsin and analyzed by LC-MS. Monoisotopic precursor ions (shown in red) at m/z = 1052.83 and 1058.16 correspond to [M+3H]+3 of the unhydroxylated and hydroxylated LLCEDLPGTEDFVGHQGTVPSDNIDSQGR peptides, respectively. (E) Tandem mass spectrum of the precursor ion at m/z = 1058.16. Large red “P” indicates a hydroxylated proline residue. The peak heights are the relative abundances of the corresponding fragment ions, with the annotation of the identified matched N terminus–containing ions (b ions) in blue and C terminus–containing ions (y ions) in magenta. For clarity, only the major identified peaks are labeled (a complete table of fragment ions for this spectrum is presented in fig. S3B).
To verify directly hydroxylation of the β2AR, the receptor was purified from β2AR-293 cells (cultured at 21 or 1% O2) with alprenolol-Sepharose affinity resin, digested with trypsin, and then analyzed by liquid chromatography–mass spectrometry (LC-MS). A +16-dalton mass shift, indicative of hydroxylation, was identified for a peptide corresponding to β2AR(376–404), which contains P382 and P395, but does not contain any free Cys thiol or Met, thus excluding S-oxidation (Fig. 3D). MS/MS analysis showed that both proline residues could be hydroxylated in normoxia (Fig. 3E and fig. S3); hydroxylation was not detected under hypoxic conditions. Collectively, these data suggest that either or both prolines are hydroxylated.
EGLN3 interacts with the β2AR and promotes receptor hydroxylation
Considering the similarities between the O2-dependent regulation of HIF-1α and the β2AR, we examined whether the HIF prolyl hydroxylase family called EGLN might also serve as a hydroxylase for the β2AR. EGLN2 is a nuclear protein and thus not likely to hydroxylate β2AR, whereas both EGLN1 and EGLN3 are cytosolic (25). In HEK293 cells, EGLN3, both overexpressed (Fig. 4A) and endogenous (Fig. 4F), but not EGLN1 or 2, specifically coimmunoprecipitated with the β2AR; agonist stimulation did not affect this interaction (fig. S4A). The C-terminal double-stranded β-helix (DSBH) fold is the defining structure of the 2OG dioxygenase superfamily (26) and this structure is highly conserved among the EGLN proteins, which are 86% identical and share about 98% similarity. Therefore, it would not be expected that the β2AR would interact with this DSBH domain of EGLN3 without similarly binding EGLN1 and 2. To further delineate the region of EGLN3 responsible for β2AR binding, we made a set of truncated EGLN3 glutathione S-transferase (GST) fusion proteins and found that only GST-EGLN3(73-116) could pull down recombinant β2AR (fig. S4, B and D). Additional deletion experiments confirmed that residues 73 to 116 of EGLN3 are required for interaction with the β2AR (fig. S4C). Further deletion experiments showed that EGLN3 residues from 88 to 104 were sufficient and required for its interaction with the β2AR (Fig. 4, B and C). The two hydroxylated prolines are located in the C-terminal tail of the β2AR, and the GST-β2AR C-terminal tail [GST-β2AR(330–413)] pulled down EGLN3 from cell lysates (Fig. 4D). We then developed a peptide based on human EGLN3(89–104) and a mutant peptide EGLN(4R) with four residues changed to Arg, which served as a control (see Materials and Methods for details). Only peptide EGLN3(89–104), but not EGLN(4R), competed with recombinant EGLN3 for binding to GST-β2AR(330–413) (Fig. 4E). Both peptides were efficiently taken up by β2AR-293 cells (fig. S4E). However, only peptide EGLN3(89–104), but not EGLN3(4R), significantly increased the cellular abundance of the β2AR (Fig. 4G). Moreover, EGLN3(89–104) did not affect HIF-1α abundance (fig. S4F). Coimmunoprecipitation experiments further confirmed that endogenous EGLN3 associated with β2AR and their interaction was eliminated by peptide EGLN3(89–104) (Fig. 4F).
Fig. 4.

EGLN3 interacts specifically with the β2AR. (A) HEK293 cells were cotransfected with FLAG-EGLN1, 2, or 3 and with β2AR or pcDNA3 control. Cell lysates were immunoprecipitated with anti-β2AR and Western blotted with anti-β2AR or anti-FLAG (for EGLNs). (B) β2AR-293 cells were transfected with truncated FLAG-EGLN3 constructs. Cell lysates were immunoprecipitated with anti-β2AR and Western blotted with anti-FLAG or anti-β2AR. (C) GST or truncated GST-EGLN3(89–104) fusion protein (5 μg) were incubated with an equal amount of β2AR before GST pulldown. Pulldowns and inputs were separated by SDS-PAGE and Western blotted with anti-β2AR or stained with Coomassie blue (for GST or the fusion protein). (D) GST or GST-β2AR(330–413) (GST-β2AR-c-tail) (5 μg) were used to pull down EGLN3. Pulldowns were Western blotted with anti-EGLN3 antibody. (E) 100 nM recombinant EGLN3 and 100 nM GST or GST-β2AR-c-tail were incubated in the presence of different concentrations of FITC-labeled peptide derived from human EGLN3(89–104) or mutant peptide EGLN3(4R). GST or GST fusion were then pulled down and Western blotted with anti-EGLN3. (F) HEK293 cells were transfected with pcDNA3 or FLAG-β2AR and treated with 30 μM EGLN3(89–104) peptide as indicated for 4 hours. Cell lysates were immunoprecipitated with anti-FLAG beads and Western blotted with anti-EGLN3 or anti-β2AR as indicated. (G) β2AR-293 cells were treated with different concentrations of EGLN3(89–104) and EGLN3(4R) mutant peptides or 1 mM DMOG as indicated for 18 hours. β2ARs were collected by anti-FLAG beads and Western blotted with anti-β2AR. Actin was the loading control. Densitometric analysis (mean ± SEM) was performed; n = 3, *P < 0.01 versus control, +P < 0.05 versus control (Student's t test).
Finally, we assessed the functional role of EGLN3 as a β2AR hydroxylase in cells and animals. Depletion of endogenous EGLN3 (but not EGLN1) with siRNA markedly increased the abundance of β2AR in β2AR-293 cells under normoxic conditions (Fig. 5A). Thus, the O2-dependent turnover of the β2AR can be associated specifically with EGLN3 activity. Capture of pVHL has been used as a surrogate assay for HIF-1α hydroxylation (18); we developed a similar strategy to assay for hydroxylation of the β2AR. Immobilized GST-β2AR(330–413) was incubated with [35S]pVHL produced by in vitro transcription and translation either directly or after incubation with lysates from HEK293 cells. The amount of pVHL trapped by the C terminus of the β2AR was increased by exposure to lysates (Fig. 5B), consistent with the interpretation that cellular extracts exhibit β2AR hydroxylase activity. To identify this activity directly with EGLN3, we examined with matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectrometry a GST-β2AR(330–413) that had been exposed either to native HEK293 cell lysates or to lysates depleted of EGLN3 with siRNA. Notably, the β2AR(376–404) peptide exposed to native lysate exhibited a +16-dalton shift in mass indicative of hydroxylation, and this modification was largely eliminated by knockdown of EGLN3 with siRNA (Fig. 5C). Thus, β2AR is a substrate for EGLN3.
Fig. 5.

EGLN3 serves as an endogenous hydroxylase for the β2AR. (A) β2AR-293 cells (expressing FLAG-β2AR) were transfected with EGLN1 or EGLN3 siRNA for 48 hours, and cell lysates were Western blotted with anti-β2AR, anti-EGLN1, anti-EGLN3, and anti-actin. Densitometric analysis (mean ± SEM) was performed. n = 3, *P < 0.01 versus siRNA-scramble (control). (B) GST-β2AR(330–413) immobilized on GSH-Sepharose was incubated with [35S]pVHL (60 min, 4°C) either directly or after three 60-min incubations with fresh HEK293 cell lysate. The beads were eluted and the eluants were analyzed by SDS-PAGE and autoradiography. The bottom panel shows the inputs of GST-β2AR(330–413). (C) Immobilized GST-β2AR(330–413) was incubated with lysates from HEK293 cells transfected with siRNA to EGLN3 or control (scrambled) siRNA and digested with trypsin (37°C, 16 hours). Digests were analyzed by MALDI-TOF. Peaks at m/z = 3099.414 or 3099.375 and 3115.402 correspond to unhydroxylated and hydroxylated LLCEDLPGTEDFVGHQGTVPSDNIDSQGR peptides, respectively. (D) βARs densities on membranes derived from hearts of wild-type (wt) and EGLN3 knockout mice were measured by radioligand binding. n = 7, *P < 0.02 versus wild type and **P < 0.01 versus wild type (Student's t test).
To support a role for hydroxylation in regulation of β2AR abundance in vivo, we compared βAR abundance in wild-type and EGLN3-knockout mice. The abundance of β2AR in hearts from EGLN3-knockout mice was significantly increased compared to that from wild-type littermates. In contrast, we observed a minor decrease in β1AR abundance (Fig. 5D). Thus, the classic predominance of β1AR over β2AR in the heart reflects the constitutive turnover of the β2AR by endogenous EGLN3. Together with the demonstration that endogenous β2AR is regulated by endogenous pVHL (Figs. 1B and 2E), these studies make a compelling case for physiological relevance of the regulation of βAR abundance by oxygen-responsive EGLN3 and pVHL.
Discussion
The cellular response to physiological concentrations of O2 is characterized by sequential actions of EGLN and pVHL–E3 ligase complex that destabilize HIF protein, a master regulator of hypoxic genes. Hydroxylation of proline residues in HIF-1α promotes binding of the pVHL ubiquitylation complex, which then targets HIF-1α for proteasomal degradation. Thus, under resting O2 concentrations, HIF is negatively regulated by O2-dependent hydroxylation (mediated by EGLN) and ubiquitylation (mediated by pVHL). Interestingly, RNA polymerase II and collagen IV were recently shown to interact with pVHL in a prolyl hydroxylase–dependent manner (27, 28), raising the possibility that pVHL may have additional substrates (29). However, physiological roles for these interactions have not been well demonstrated. Thus, whether the EGLN and pVHL system might serve more broadly in hypoxic signaling has remained an open question. The present findings suggest that both constitutive and agonist-stimulated GPCR abundance can be controlled by the O2-regulated EGLN and pVHL system and, in particular, that the β2AR is a physiological substrate of EGLN3 and the pVHL–E3 ligase complex. Our data highlight a transcription-independent role for the EGLN and pVHL system, which mediates the influence of O2 on receptor-mediated cellular responsiveness.
The type 2 form of von Hippel–Lindau disease (a hereditary cancer syndrome caused by inactivating mutations of von Hippel–Lindau tumor suppressor gene) is characterized by the presence of pheochromocytomas, a neuroendocrine tumor of the adrenal glands, and that accordingly, type 2 patients often show excessive secretion of catecholamines (endogenous β2AR ligands) and increased sympathetic nervous system activity (16). It is of interest that mice deficient in EGLN3 also display an adrenergic phenotype characterized by alterations in blood pressure and cardiac contractility (30). β2AR dysregulation resulting from type 2 mutations might contribute to the symptomology and etiology of von Hippel–Lindau disease, as well as to the EGLN3−/− phenotype.
All mammalian EGLN isoforms can hydroxylate HIF in vitro, but only EGLN1 regulates HIF-1α abundance in vivo (31, 32). Whether EGLN proteins exhibit functional redundancy or play distinct roles is largely unknown (25, 33). Our finding that only EGLN3 binds and regulates the β2AR and that the interaction is mediated through the N-terminal region, which is highly variable among EGLN isoforms, suggests that each isoform may have unique substrates.
EGLN3 is most abundant in cardiac and smooth muscle (33, 34), where the β2AR is highly abundant in vivo. βAR activation increases cardiac contractility and relaxes vascular and airway smooth muscle, and βAR dysregulation may be broadly involved in the pathogenesis of cardiovascular and airway diseases, including heart failure, hypertension, and asthma (2, 35, 36). Notably, the β2AR, in particular, enhances bronchodilation and alveolar fluid clearance (which increase O2 uptake), enhances cardiac output and peripheral vasodilation (which increase O2 delivery), and enhances cardioprotection and angiogenesis under ischemic conditions (35–39), thereby effectively recapitulating the integrated physiological response to hypoxia. Thus, our results showing up-regulation of the β2AR in response to hypoxia puts the function of the receptor in new light. The ability of the EGLN-pVHL hydroxylation and ubiquitylation pathway to regulate the β2AR and the implications of that regulation for the response to ischemia and hypoxia suggest previously unidentified targets in the treatment of cardiovascular and respiratory diseases. In addition, our findings may implicate age-dependent increases in EGLN3 expression, observed in both humans and mice (40), in multiple degenerative diseases.
Materials and Methods
Cell culture and mouse tissue
HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). 786-O cells were maintained in RPMI 1640 with 10% FBS. HUVECs were cultured in EBM-2 medium with 10% FBS supplemented with EGM-2 SingleQuots Kit. PO2 was controlled by incubating cells at 37°C in humidified, O2-CO2– regulated incubators (Sanyo) adjusted to 5% CO2 and the indicated PO2 (balanced by N2), or in a standard CO2-controlled incubator maintained at 5% CO2–95% air. EGLN3 knockout mice were described previously (41). The hearts of knockout mice and their wild-type littermates at 6 weeks old were removed and perfused with cold 0.9% saline. Hearts were then flash-frozen in liquid nitrogen and stored at −80°C.
siRNA and EGLN peptides
ON-TARGET plus siRNAs for suppressing human VHL (J-003936-09-0020, J-003936-10-0020) were purchased from Thermo Scientific. siRNA duplexes designed to suppress the expression of human EGLN1 (sense, 5′-CAAGGUAAGUGGAGGUAUAUU-3′; antisense, 5′-UAUACCUCCACUUACCUUGUU-3′; GenBank accession number, EGLN1 NM_022051) and EGLN3 (sense, 5′-GCAAAUACUACGU-CAAGGAUU-3′; antisense, 5′-UCCUUGACGUAGUAUUUGCUU-3′ and sense, 5′-UUCAGGAAUUUAACUAGGAUU-3′; antisense, 5′-UCCUAGUUAAAUUCCUGAAUU-3′; GenBank accession number, EGLN3 NM_022073) and scrambled siRNA control were purchased from Dharmacon. HEK293 cells at 40 to 50% confluency were transfected with siRNAs (50 nM) by exposure to lipofectamine 2000 (Invitrogen) for 48 hours. HUVECs were electrotransfected by a Bio-Rad GenePulser Xcell2 for 72 hours. The effects of suppression were validated by Western blotting with antibodies that recognized pVHL (BD Biosciences), EGLN1, or EGLN3 (Novus-Biologicals). Fluorescein isothiocyanate (FITC)– labeled peptide derived from human EGLN3(89–104) (EAISFLLSLID-RLVLY) or mutant peptide EGLN3(4R) (EARSFLRSLIRRLRLY) with four amino acids in EGLN3(89–104) mutated to Arg were purchased from EZBiolab.
Pulse-chase analysis
β2AR-293 cells were radiolabeled in Met-Cys–free medium supplemented with [35S]Met] and [35S]Cys (100 μCi/ml) for 12 hours at 21 or 1% O2. Cells were washed with DMEM containing excess cold Met and Cys (3 mM), then cells were chased in complete DMEM medium plus excess cold Met and Cys (3 mM) at 21 or 1% O2. FLAG-β2AR was immunoprecipitated from the cell lysates and resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) followed by autoradiography.
In vitro ubiquitylation assay
Recombinant β2AR (2 μg) reconstituted in phospholipid vesicles (9) was used as the substrate. pVHL–E3 ubiquitin ligase complex was immunoprecipitated with anti-FLAG beads (Sigma) from HEK293 cells transfected with FLAG-pVHL (cells transfected with empty vector as the control) and eluted with FLAG peptide (Sigma). The reactions were carried out at 30°C for 60 min in buffer containing 50 mM tris (pH 7.4), 2 mM adenosine triphosphate–Mg2+, 2 mM dithiothreitol (DTT), and 2.5 mM MgCl2 supplemented with 250 ng of human recombinant E1 (BostonBiochem), 500 ng of human recombinant UbcH5b (Boston-Biochem), and 10 μg of ubiquitin (BostonBiochem).
In vitro hydroxylation and pVHL capture assay
To assay hydroxylation in vitro, 5 μg of GST-β2AR(330–413) captured on glutathione (GSH)-Sepharose beads (Sigma) were incubated with cell lysates from HEK293 cells (transfected with siRNA for EGLN3 or scrambled siRNA) in hypotonic extraction buffer [HEB: 20 mM tris (pH 7.5), 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT] supplemented with 2 mM ascorbate, 0.2 mM 2-oxoglutarate, and 0.1 mM Fe2+ at 30°C for 60 min; this was repeated twice more with fresh cell lysate. The beads were then washed three times with 20 mM ammonium bicarbonate (pH 8.0) and digested with trypsin (Trypsin Gold; Promega) at 37°C for 16 hours. Proline hydroxylation was confirmed by MALDI-TOF (Applied Biosystems, Model 4700).
To assay pVHL capture, GST-β2AR(330–413) was mixed with [35S]pVHL produced by in vitro transcription and translation, either directly or after incubation with HEK293 cell extract as above. The mixture was incubated with rotation in binding buffer [50 mM tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, and 10% glycerol] for 60 min at 4°C. Bound [35S]pVHL was eluted and analyzed by SDS-PAGE and autoradiography.
EGLN3 binding to β2AR: GST pull-down assay and coimmunoprecipitation
Complementary DNA (cDNA) corresponding to EGLN3 residues 1 to 36, 37 to 72, 73 to 116, 89 to 104, or 222 to 239 or β2AR(330–413) residues were amplified by polymerase chain reaction and cloned into pGEX-2T vector. GST or GST fusion proteins were purified from Escherichia coli with GSH-Sepharose (Sigma). GST or GST fusion proteins (5 μg) were incubated overnight with equal amounts of β2AR (from β2AR-293) or EGLN3 (derived from transiently transfected HEK293 cells or recombinant protein from SF9 cells) at 4°C in 0.5 ml of binding buffer [50 mM tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, and 10% glycerol]. Thirty microliters of 50% GSH-Sepharose slurry was added to the mixture followed by rotation for another hour at 4°C. Beads were then washed five times with 1 ml of binding buffer. Proteins were separated by SDS-PAGE and analyzed by Western blot. For coimmunoprecipitations of β2AR and/or EGLNs, cells were lysed [50 mM tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 10% glycerol, and protease inhibitor cocktail] and lysates were clarified by centrifugation at 14,000g. After addition of 1 to 2 μg of antibody, lysates were incubated with rotation overnight at 4°C and for an additional hour after the addition of 25 μl of 50% protein A or G agarose. Beads were washed five times with lysis buffer and proteins were analyzed by Western blotting.
Far-Western analysis
One microgram of recombinant β2AR reconstituted in lipid vesicles or empty vesicles was separated by nonreducing SDS-PAGE and either immunoblotted with anti-β2AR or incubated with 100 nM recombinant pVHL–elongin C–elongin B complex (VCB) (42) in phosphate-buffered saline (PBS), 0.1% Tween-20, and 5% milk. Bound VCB was detected by anti-pVHL.
Immunofluorescence and confocal microscopy
Immediately after removal from the incubator, cells were washed with PBS and fixed and permeabilized in phosphate-buffered 2% paraformaldehyde and 0.2% Triton X-100 for 30 min at 4°C. Immunofluorescence labeling was performed with mouse anti-FLAG M2 (1:500 dilution; Sigma) or anti-pVHL (BD Biosciences) followed by tetramethyl rhodamine isothiocyanate (TRITC)–conjugated goat secondary antibody against mouse (Jackson ImmunoResearch Laboratories). Images were acquired by confocal laser-scanning microscopy (LSM5 Pascal; Carl Zeiss, Inc.).
Radioligand binding
Membranes of HUVECs cultured under 21 or 1% O2 and membrane preparations from hearts of wild-type or EGLN3-knockout mice were prepared, and 125I-labeled cyanopindolol binding was performed in triplicate in the presence or absence of the antagonist propranolol or cGP-20712A as described previously (9, 43).
Liquid chromatography–tandem mass spectrometry analysis
Human β2AR was purified from stably overexpressing HEK293 cells with alprenolol-Sepharose affinity resin, as described previously (9). After reduction with DTT (10 mM, 37°C, 30 min) and alkylation with iodoacetamide (20 mM for 30 min at room temperature in the dark), purified receptors were digested with trypsin in solution at a working concentration of 5 ng/μl. Digested peptides were purified by Stage tip chromatography, lyophilized, and reconstituted in 5% acetonitrile–5% formic acid. Liquid chromatography–tandem mass spectrometry (LC– MS/MS) experiments were performed on a hybrid linear quadrapole ion trap/FT-ICR (LTQ FT) mass spectrometer (Thermo Electron, San Jose, CA) equipped with a Finnigan Nanospray II electrospray ionization source (Thermo Electron), an Agilent 1100 Series binary high-performance liquid chromatography (HPLC) pump (Agilent Technologies, Palo Alto, CA), and a Famos autosampler (LC Packings, San Francisco, CA). Peptide mixtures were loaded onto a 125-μm inside diameter fused-silica microcapillary column packed in-house with C18 reversed-phase resin (Magic C18AQ; particle size, 5 μm; pore size, 200 Å; Microsom Bioresources, Auburn, CA), and separation was achieved with a 75-min gradient at a flow rate of 300 nl/min provided across a flow splitter by HPLC pumps. The LTQ-FT mass spectrometer was operated in the data-dependent mode with the FT10 strategy (44). In brief, a scan cycle was initiated with a full survey scan of high mass accuracy [mass/charge ratio (m/z), 350 to 1700) in the FT-ICR mass spectrometer. This was followed by MS/MS scans in the linear ion trap of the 10 most abundant ions in the survey scan, with dynamic exclusion of previously selected ions. Singly charged ions were excluded from MS/MS analysis.
Database search for MS/MS
Instrument control and primary data processing were carried out with the Xcalibur software package, Version 1.4 SR1 (Thermo Electron). Raw data were converted to the mzXML format and in-house Perl scripts were used to identify the charge state and the monoisotopic m/z for peptide ions chosen for MS/MS analysis. This information was used to generate .dta files that were subjected to SEQUEST search analysis against a small database containing the human β2AR. Searches were performed with the following parameters: full-trypsin specificity, a mass tolerance of 1.1 Da, static modifications of oxidized Met (+15.9949), carboxyamidomethylated Cys (+57.0215), and dynamic modifications of hydroxylated Pro (+15.9949). All peptide matches were filtered by XCorr, mass accuracy, and dCn′ (defined as the normalized difference between XCorr values of the top-ranked candidate peptide and the next candidate with a different amino acid sequence).
Supplementary Material
www.sciencesignaling.org/cgi/content/full/2/78/ra33/DC1
Fig. S1. Normoxia-stablilized HIF-1α (HIF-1αP402A/P564G) has no effect on β2AR abundance in β2AR-293 cells. β2AR-293 cells (expressing FLAG-β2AR) were transfected with plasmid expressing HIF-1αP402A/P564G or pcDNA3 control plasmid for 24 hours. β2AR was isolated with anti-FLAG beads and Western blotted with anti-β2AR antibody. Actin was used as the loading control.
Fig. S2. β2AR colocalizes and interacts with endogenous pVHL. (A) HEK293 cells transfected with β2AR-GFP were fixed and stained with anti-pVHL antibody and TRITC-conjugated secondary antibody (red). (B) Flag-β2AR or pcDNA3 were transfected into HEK293 cells. Cell lysates were immunoprecipitated with anti-FLAG-M2-beads and Western blotted with anti-pVHL or anti-β2AR antibodies as indicated. (C) Lipid vesicles reconstituted with recombinant β2AR or empty (control) vesicles were solubilized and separated on non-reducing SDS-PAGE, and either immunoblotted with anti-β2AR antibodies or incubated with recombinant VBC complex or buffer alone. Bound VCB was detected by anti-pVHL antibody (lower band is a β2AR degradation product). (D) Flag-pVHL was transfected into β2AR-293 cells and, after 24 hours, cells were incubated for another 18 hours at either 21% or 1% O2 in the presence of MG132 (10 μM). Cell lysates were immunoprecipitated with anti-pVHL antibody and Western blotted with anti-β2AR or anti-FLAG (for pVHL). (E) HUVEC cells were electro-transfected with control (scrambled) siRNA, pVHL siRNA1 or pVHL siRNA2 for 72 hours. Cell lysates were resolved by SDS-PAGE and Western blotted with anti-pVHL or anti-actin antibodies as indicated.
Fig. S3. Tandem MS/MS spectra of peptide LLCEDLPGTEDFVGHQGTVPSDNIDSQGR and the corresponding proline-hydroxylated peptides OH-Pro382 and OH-Pro395. Analyses of peptide LLCEDLPGTEDFVGHQGTVPSDNIDSQGR, OH-Pro382 and OH-Pro395 are shown in (A), (B) and (C) respectively. The peak heights are the relative abundances of the corresponding fragment ions, with the annotation of the matched N-terminus containing ions (b-ions) in blue and C-terminus containing ions (y-ions) in red. Non-matched product ions are indicated in grey. “#” indicates hydroxylated proline residues. The tables list the theoretical masses of all product ions from precursor peptides with matched masses indicated by the color code described above. “#” indicates hydroxylated proline residues.
Fig. S4. Mapping the interaction between β2AR and EGLN3. (A) β2AR-293 cells were transfected with FLAG-EGLN3 and treated with isoproterenol (ISO; 10μM) for the indicated time. Cell lysates were immunoprecipitated with anti-β2AR and Western blotted with anti-FLAG (for EGLN3) or anti-β2AR antibodies. (B) Schematic illustration of full-length and truncated EGLN3 fusion proteins. Fusion proteins carried an N-terminal GST or FLAG tag. (C) β2AR-293 cells were transfected with truncated FLAG-EGLN3 constructs. Cell lysates were immunoprecipitated with anti-β2AR antibody and Western blotted with anti-FLAG or anti-β2AR antibody. (D) GST or truncated GST-EGLN3 fusion proteins (5 μg) were incubated with an equal amount of β2AR prior to GST pulldown. Pull-downs and inputs were separated by SDS-PAGE and Western blotted with anti-β2AR antibody or stained with coomassie blue (for GST or fusion proteins). (E) 30 μM FITC-labeled peptide derived from human EGLN3(89-104) or mutant peptide EGLN(4R) was incubated with HEK293 cells for 4 hours. Images of internalized peptides were acquired with a Nikon TE300 fluorescence microscope. (F) HEK293 cells were treated with 30 μM peptide EGLN3 (89-104) under 21% O2, or cultured under 2% O2 for 18 hours. Total cell lysates were resolved by SDS-PAGE and Western blotted with anti-HIF-1α or anti-actin antibodies as indicated.
Acknowledgments
This work was supported by P01-HL075443, U19-ES012496, RO1-NS034400 and an RJL Award for Scientific Innovation. We thank D. T. Hess for discussion, advice, and assistance with the manuscript and L. A. Desouza for technical help. The authors have no conflicting financial interests.
References and Notes
- 1.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 2.Johnson M. The β-adrenoceptor. Am J Respir Crit Care Med. 1998;158:S146–S153. doi: 10.1164/ajrccm.158.supplement_2.13tac110. [DOI] [PubMed] [Google Scholar]
- 3.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW., II Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: Critical role for expression level. Circulation. 2000;101:1707–1714. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 5.Penna C, Abbadessa G, Mancardi D, Tullio F, Piccione F, Spaccamiglio A, Racca S, Pagliaro P. Synergistic effects against post-ischemic cardiac dysfunction by sub-chronic nandrolone pretreatment and postconditioning: Role of β2-adrenoceptor. J Physiol Pharmacol. 2008;59:645–659. [PubMed] [Google Scholar]
- 6.Tong H, Bernstein D, Murphy E, Steenbergen C. The role of β-adrenergic receptor signaling in cardioprotection. FASEB J. 2005;19:983–985. doi: 10.1096/fj.04-3067fje. [DOI] [PubMed] [Google Scholar]
- 7.Foerster K, Groner F, Matthes J, Koch WJ, Birnbaumer L, Herzig S. Cardioprotection specific for the G protein Gi2 in chronic adrenergic signaling through β2-adrenoceptors. Proc Natl Acad Sci U S A. 2003;100:14475–14480. doi: 10.1073/pnas.1936026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patterson AJ, Zhu W, Chow A, Agrawal R, Kosek J, Xiao RP, Kobilka B. Protecting the myocardium: A role for the β2 adrenergic receptor in the heart. Crit Care Med. 2004;32:1041–1048. doi: 10.1097/01.ccm.0000120049.43113.90. [DOI] [PubMed] [Google Scholar]
- 9.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 10.Shenoy SK, Xiao K, Venkataramanan V, Snyder PM, Freedman NJ, Weissman AM. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J Biol Chem. 2008;283:22166–22176. doi: 10.1074/jbc.M709668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hainsworth R, Drinkhill MJ. Cardiovascular adjustments for life at high altitude. Respir Physiol Neurobiol. 2007;158:204–211. doi: 10.1016/j.resp.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 12.de Jong W, Lukovic L, Jones CR, Petty MA. Receptor density in the rat heart following ischaemia and prolonged reperfusion. Clin Exp Pharmacol Physiol Suppl. 1995;22:S279–S280. doi: 10.1111/j.1440-1681.1995.tb02916.x. [DOI] [PubMed] [Google Scholar]
- 13.Xie L, Johnson RS, Freeman RS. Inhibition of NGF deprivation-induced death by low oxygen involves suppression of BIMEL and activation of HIF-1. J Cell Biol. 2005;168:911–920. doi: 10.1083/jcb.200407079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 16.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 17.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007:cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 18.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 19.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 20.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 21.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 22.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1α for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277:39792–39800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Hirsilä M, Koivunen P, Brenner MC, Xu L, Yang C, Kivirikko KI, Myllyharju J. Many amino acid substitutions in a hypoxia-inducible transcription factor (HIF)-1α-like peptide cause only minor changes in its hydroxylation by the HIF prolyl 4-hydroxylases: Substitution of 3,4-dehydroproline or azetidine-2-carboxylic acid for the proline leads to a high rate of uncoupled 2-oxoglutarate decarboxylation. J Biol Chem. 2004;279:55051–55059. doi: 10.1074/jbc.M410287200. [DOI] [PubMed] [Google Scholar]
- 25.Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Bürgel T, Jelkmann W, Acker H, Fandrey J. Intra-cellular localisation of human HIF-1α hydroxylases: Implications for oxygen sensing. J Cell Sci. 2003;116:1319–1326. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- 26.Aravind L, Koonin EV. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2001;2:RESEARCH0007. doi: 10.1186/gb-2001-2-3-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuznetsova AV, Meller J, Schnell PO, Nash JA, Ignacak ML, Sanchez Y, Conaway JW, Conaway RC, Czyzyk-Krzeska MF. von Hippel–Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci U S A. 2003;100:2706–2711. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grosfeld A, Stolze IP, Cockman ME, Pugh CW, Edelmann M, Kessler B, Bullock AN, Ratcliffe PJ, Masson N. Interaction of hydroxylated collagen IV with the von Hippel–Lindau tumor suppressor. J Biol Chem. 2007;282:13264–13269. doi: 10.1074/jbc.M611648200. [DOI] [PubMed] [Google Scholar]
- 29.Frew IJ, Krek W. pVHL: A multipurpose adaptor protein. Sci Signal. 2008;1:pe30. doi: 10.1126/scisignal.124pe30. [DOI] [PubMed] [Google Scholar]
- 30.Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, Grosfeld A, Aragones J, Schneider M, van Geyte K, Teixeira D, Diez-Juan A, Lopez-Barneo J, Channon KM, Maxwell PH, Pugh CW, Davies AM, Carmeliet P, Ratcliffe PJ. Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol. 2008;28:3386–3400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG., Jr Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–3244. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freeman RS, Hasbani DM, Lipscomb EA, Straub JA, Xie L. SM-20, EGL-9, and the EGLN family of hypoxia-inducible factor prolyl hydroxylases. Mol Cells. 2003;16:1–12. [PubMed] [Google Scholar]
- 34.Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80:421–426. doi: 10.1139/o02-115. [DOI] [PubMed] [Google Scholar]
- 35.Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac β-adrenergic receptors, and heart failure. Circulation. 2000;101:1634–1637. doi: 10.1161/01.cir.101.14.1634. [DOI] [PubMed] [Google Scholar]
- 36.Iaccarino G, Cipolletta E, Fiorillo A, Annecchiarico M, Ciccarelli M, Cimini V, Koch WJ, Trimarco B. β2-adrenergic receptor gene delivery to the endothelium corrects impaired adrenergic vasorelaxation in hypertension. Circulation. 2002;106:349–355. doi: 10.1161/01.cir.0000022690.55143.56. [DOI] [PubMed] [Google Scholar]
- 37.Mieno S, Watanabe F, Sawa Y, Horimoto H. Gene transfer of β2 adrenergic receptor enhances cardioprotective effects of ischemic preconditioning in rat hearts after myocardial infarction. Interact Cardiovasc Thorac Surg. 2005;4:163–167. doi: 10.1510/icvts.2004.096792. [DOI] [PubMed] [Google Scholar]
- 38.Mutlu GM, Factor P. Alveolar epithelial β2-adrenergic receptors. Am J Respir Cell Mol Biol. 2008;38:127–134. doi: 10.1165/rcmb.2007-0198TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, Piscione F, Priante O, Pastore L, Chiariello M, Salvatore F, Koch WJ, Trimarco B. Ischemic neoangiogenesis enhanced by β2-adrenergic receptor overexpression: A novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182–1189. doi: 10.1161/01.RES.0000191541.06788.bb. [DOI] [PubMed] [Google Scholar]
- 40.Rohrbach S, Simm A, Pregla R, Franke C, Katschinski DM. Age-dependent increase of prolyl-4-hydroxylase domain (PHD) 3 expression in human and mouse heart. Biogerontology. 2005;6:165–171. doi: 10.1007/s10522-005-7950-9. [DOI] [PubMed] [Google Scholar]
- 41.Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor a levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 43.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 44.Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villén J, Gygi SP. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencesignaling.org/cgi/content/full/2/78/ra33/DC1
Fig. S1. Normoxia-stablilized HIF-1α (HIF-1αP402A/P564G) has no effect on β2AR abundance in β2AR-293 cells. β2AR-293 cells (expressing FLAG-β2AR) were transfected with plasmid expressing HIF-1αP402A/P564G or pcDNA3 control plasmid for 24 hours. β2AR was isolated with anti-FLAG beads and Western blotted with anti-β2AR antibody. Actin was used as the loading control.
Fig. S2. β2AR colocalizes and interacts with endogenous pVHL. (A) HEK293 cells transfected with β2AR-GFP were fixed and stained with anti-pVHL antibody and TRITC-conjugated secondary antibody (red). (B) Flag-β2AR or pcDNA3 were transfected into HEK293 cells. Cell lysates were immunoprecipitated with anti-FLAG-M2-beads and Western blotted with anti-pVHL or anti-β2AR antibodies as indicated. (C) Lipid vesicles reconstituted with recombinant β2AR or empty (control) vesicles were solubilized and separated on non-reducing SDS-PAGE, and either immunoblotted with anti-β2AR antibodies or incubated with recombinant VBC complex or buffer alone. Bound VCB was detected by anti-pVHL antibody (lower band is a β2AR degradation product). (D) Flag-pVHL was transfected into β2AR-293 cells and, after 24 hours, cells were incubated for another 18 hours at either 21% or 1% O2 in the presence of MG132 (10 μM). Cell lysates were immunoprecipitated with anti-pVHL antibody and Western blotted with anti-β2AR or anti-FLAG (for pVHL). (E) HUVEC cells were electro-transfected with control (scrambled) siRNA, pVHL siRNA1 or pVHL siRNA2 for 72 hours. Cell lysates were resolved by SDS-PAGE and Western blotted with anti-pVHL or anti-actin antibodies as indicated.
Fig. S3. Tandem MS/MS spectra of peptide LLCEDLPGTEDFVGHQGTVPSDNIDSQGR and the corresponding proline-hydroxylated peptides OH-Pro382 and OH-Pro395. Analyses of peptide LLCEDLPGTEDFVGHQGTVPSDNIDSQGR, OH-Pro382 and OH-Pro395 are shown in (A), (B) and (C) respectively. The peak heights are the relative abundances of the corresponding fragment ions, with the annotation of the matched N-terminus containing ions (b-ions) in blue and C-terminus containing ions (y-ions) in red. Non-matched product ions are indicated in grey. “#” indicates hydroxylated proline residues. The tables list the theoretical masses of all product ions from precursor peptides with matched masses indicated by the color code described above. “#” indicates hydroxylated proline residues.
Fig. S4. Mapping the interaction between β2AR and EGLN3. (A) β2AR-293 cells were transfected with FLAG-EGLN3 and treated with isoproterenol (ISO; 10μM) for the indicated time. Cell lysates were immunoprecipitated with anti-β2AR and Western blotted with anti-FLAG (for EGLN3) or anti-β2AR antibodies. (B) Schematic illustration of full-length and truncated EGLN3 fusion proteins. Fusion proteins carried an N-terminal GST or FLAG tag. (C) β2AR-293 cells were transfected with truncated FLAG-EGLN3 constructs. Cell lysates were immunoprecipitated with anti-β2AR antibody and Western blotted with anti-FLAG or anti-β2AR antibody. (D) GST or truncated GST-EGLN3 fusion proteins (5 μg) were incubated with an equal amount of β2AR prior to GST pulldown. Pull-downs and inputs were separated by SDS-PAGE and Western blotted with anti-β2AR antibody or stained with coomassie blue (for GST or fusion proteins). (E) 30 μM FITC-labeled peptide derived from human EGLN3(89-104) or mutant peptide EGLN(4R) was incubated with HEK293 cells for 4 hours. Images of internalized peptides were acquired with a Nikon TE300 fluorescence microscope. (F) HEK293 cells were treated with 30 μM peptide EGLN3 (89-104) under 21% O2, or cultured under 2% O2 for 18 hours. Total cell lysates were resolved by SDS-PAGE and Western blotted with anti-HIF-1α or anti-actin antibodies as indicated.