

Abstract
Valproate, an anticonvulsant and mood stabilizer, up-regulates Bcl-2, a neurotrophic/neuroprotective protein. In this study, we investigated the molecular mechanism through which Bcl-2 is up-regulated by valproate using cultured human neuron-like cells. Valproate, within therapeutically relevant ranges, induced time- and concentration-dependent up-regulations of both Bcl-2 messenger RNA and protein implicating an underlying gene transcriptional-mediated mechanism. Bcl-2 up-regulations were associated with ERK1/2 and PI3K pathway activations and elevated levels of activated phospho-RSK and phospho-CREB, convergent targets of the ERK1/2 and PI3K pathways. Valproate increased transcriptional activity of a human bcl-2 promoter–reporter gene construct. This effect was attenuated, but not blocked, by mutation of a CREB DNA binding site, a CRE site in the human bcl-2 promoter sequence. ERK and/or PI3K pathway inhibitors and RSK1 small hairpin RNA knockdown reduced, but did not abolish, baseline and valproate-induced promoter activities and lowered Bcl-2 protein levels. These data collectively suggest that valproate induces Bcl-2 regulation partially through activations of the ERK and PI3K cascades and their convergent kinase, RSK, although other unknown mechanism(s) are likely involved. Given the known roles of Bcl-2 in the central nervous system, the current findings offer a partial yet complex molecular mechanistic explanation for the known neurobiological effects of valproate including neurite growth, neuronal survival, and neurogenesis.
Keywords: Valproate, bcl-2, ERK, PI3K, RSK
Introduction
Bcl-2 has been shown to promote cell survival in a diverse array of cell types through the inhibition of programmed cell death (Chuang 2005). More recently, Bcl-2 has been implicated in the regulations of a number of interrelated central nervous system (CNS) functions such as brain development (Shacka and Roth 2005), neuronal process growth and regeneration (Chen et al. 1997), neuronal survival after a variety of insults (Chuang 2005), and adult hippocampal neurogenesis (Kuhn et al. 2005). Promoting Bcl-2 function has been proposed to treat neurodegenerative diseases, CNS trauma, and acute neurological disorders such as stroke (Chuang 2005).
Valproate (VPA) is a commonly used anticonvulsant and mood stabilizer (Belmaker 2004). VPA inhibits N-methyl-d-aspartate-evoked transient depolarizations, thereby increasing gamma-aminobutyric acid (GABA) turnover and potentiating GABAergic function in selective brain regions (Gobbi and Janiri 2006). VPA-induced GABAergic functional potentiation is thought to underlie VPA's antiepileptic actions (Gobbi and Janiri 2006). In contrast to its anticonvulsant action, VPA's antimanic action requires subchronic drug administration, ranging from 2 to 3 days using a high-dose oral and intravenous loading regimen to more than 5 days using a standard regimen (Phrolov et al. 2004). VPA's mechanism of late onset antimanic action is unknown (Belmaker 2004).
Emerging data demonstrate that VPA treatment up-regulates Bcl-2 and promotes Bcl-2-related neuronal protective and neurotrophic effects. VPA treatment increases Bcl-2 messenger RNA (mRNA) or protein levels in cultured cells (Yuan et al. 2001; Laeng et al. 2004; Michaelis et al. 2006; Lai et al. 2006) and in specific regions of animal brains (Chen et al. 1999b; Hao et al. 2004; Sugai et al. 2004). VPA also promotes Bcl-2 functions including neurite growth, neurogenesis, and anti-apoptosis (Chuang 2005; Chen et al. 1999b).
Recent human imaging and postmortem studies reveal discrete regional morphological changes in the brains of bipolar mood disorder (BD) victims (Drevets 2000; Rajkowska 2002). Correspondingly, postmortem studies show decreases in densities, sizes, and/or numbers of neurons and glial cells in brain-region-specific samples from BD patients (Rajkowska 2002) and dysfunctional neurotrophic signaling in the brains of suicide subjects (Dwivedi et al. 2006). These convergent findings suggest that Bcl-2-related neurotrophic and neuroprotective actions play a critical role in the pathogenesis and treatment of BD.
VPA affects several intracellular signaling and gene expression mechanisms that modulate bcl-2 expression. Klein and his associates discovered that VPA is a potent histone deacetylase (HDAC) inhibitor (Phiel et al. 2001). Greenberg and her associates have demonstrated that valproate inhibits de novo biosynthesis of myoinositol, thereby reducing inositol content, an effect that is known to be induced by lithium through inhibition of inositol phosphatases (Shi et al. 2005). We and other investigators have shown that valproate directly or indirectly inhibits glycogen synthase kinase-3 (GSK-3; Chen et al. 1999a; De Sarno et al. 2002). Jope and several other groups have found that valproate activates the phosphatidyl inositol 3 kinase/protein kinase B (PI3K/PKB) pathway (De Sarno et al. 2002). Additionally, we and several other groups have found that valproate activates the extracellular signal-regulated kinase (ERK) pathway in cultured cells and in the brain (Hao et al. 2004; Michaelis et al. 2006; Yuan et al. 2001; Di Daniel et al. 2006; Einat et al. 2003). p90 ribosomal S6 kinase (RSK) is a serine–threonine dual kinase that requires phosphorylation input by both the ERK and PI3K pathways for its full activation and is known to directly phosphorylate ser133CREB (Frodin and Gammeltoft 1999; Xing et al. 1998). In this study, we show that VPA activates both ERK and PI3K pathways and enhances RSK1 expression in our cell culture system and that perturbations of VPA-induced activations of either or both pathways as well as knock down of RSK1 attenuate bcl-2 expression.
Given the role of Bcl-2 in the CNS, the brain morphological findings in BD, and the known effects of valproate on Bcl-2 and signaling pathways, we undertook the current project to further elucidate the molecular mechanisms by which valproate regulates bcl-2 expression.
Materials and Methods
Cell Culture
The human neuroblastoma SH-SY5Y cell line has been used in a variety of studies because of its human origin and because of its usefulness as an experimental model of neuronal differentiation, growth, and apoptosis. Additionally, previous studies from investigators including us have shown that valproate increases Bcl-2 levels and activates the ERK and PI3K pathways in this cell line (Yuan et al. 2001; De Sarno et al. 2002). These findings are similar to those conducted with cultured cortical cells and with brain region samples from rodents chronically treated with VPA (Hao et al. 2004; Einat et al. 2003), indicating that the SH-SY5Y cell line is a suitable system to study the specific mechanisms of action of VPA. SH-SY5Y cell culture was conducted as previously described (Yuan et al. 2001). Briefly, SH-SY5Y cells were cultured in a humidified incubator with 95% air/5% CO2 at 37°C in high glucose Dulbecco's modified Eagle medium (DMEM) plus 10% fetal bovine serum, 100 IU/ml penicillin, 50 μg/ml streptomycin, and 1 mM sodium pyruvate (Gibco, Carlsbad, CA, USA). When cells reached 80–90% confluence, serum-free medium was replaced overnight and treated as indicated in the following experiments.
Imunoblotting Experiments to Monitor the Effects of VPA on Levels of Bcl-2 and Signaling Molecules
To detect the effects of VPA on levels of Bcl-2 and relevant signaling molecules, confluent SH-SY5Y cells were treated with sodium valproate (Sigma-Aldrich, St. Louis, MO, USA) for the durations or at the concentrations indicated in the figures. To investigate the involvement of signaling pathways in the effects of VPA on levels of Bcl-2 and other signaling molecules, confluent SH-SY5Y cells were treated with PD98059 (50 μM), LY294002 (10 μM), or dimethyl sulfoxide (DMSO; 0.1% final) vehicle control followed 2 h later with sodium valproate (0.8 mM) or vehicle (DMEM) control for 48 h. Treated cells were then washed three times with phosphate-buffered saline and homogenized by brief pulsed sonication in an extraction buffer. The buffer contained 20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 1% Triton-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, protease inhibitor cocktail (Sigma-Aldrich), and phosphatase inhibitor cocktail I and II (Sigma-Aldrich). The homogenates were centrifuged at 14,000×g for 15 s to remove undissolved debris. Subsequent immunoblotting was performed using amounts of protein demonstrated by Bradford analyses to be within the linear range for immunoblotting. Equal amounts of total protein from each sample were loaded on each gel for comparative analyses. The antibodies for immunoblots were diluted according to the manufacturer's recommendations. Antibodies against Bcl-2 and total RSK1 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Phospho-ERK1/2 (Thr202/Tyr204), total ERK1/2, phospho-RSK1 (Thr359/Ser363), phospho-Akt (Ser473), total Akt, phospho-CREB (Ser133), total CREB, acetyl-histone H3 (lys9), and total histone H3 antibodies were from Cell Signaling Technology (Danvers, MA, USA). GAPDH antibody was from Abcam (Cambridge, MA, USA). The immunocomplex was detected using chemiluminescence (ECL kit, Amersham Pharmacia Biotech). Quantitations of the immunoblots were performed by densitometric scanning of the film using a Kodak Image Station (4000R Digital Imaging System).
mRNA Isolation and Quantitative PCR
Total cellular RNA was isolated from VPA-treated cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and purified using RNeasy Mini Kit (Qiagen, Germantown, MD, USA). The RNA concentrations were spectrophotometrically determined in duplicate, and the quality of RNA was evaluated after electrophoresis by visual inspection of an aliquot of the RNA samples. Complementary DNA samples were prepared from cellular total RNA samples as previously described using the oligo dT method of a SuperScript™ First-Strand Synthesis System (Invitrogen). For quantitative polymerase chain reaction (qPCR), a GAPDH primer set (Rn99999916_s1) was purchased from ABI (Foster City, CA, USA) and LUX Primers for Bcl-2 (GATAACGGAGGCTGGGATGC; CGGCGAG AAATCAAACAGAGGC(FAM)G) were designed on the web-based D-LUX Designer (orf.invitrogen.com/lux/) and purchased from Invitrogen. These primers were designed to amplify the human sequences spanning two or more exons. An aliquot of samples used in qPCR reactions was within the range of the standards. qPCR reactions were conducted according to the manufacturer's specifications using the 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA).
Human bcl-2 Promoter–Reporter Gene Construct and Site-Directed Mutagenesis of its CRE Site
Bcl-2 gene has two promoters. The major bcl-2 promoter, P1, is located 1,386–1,423 bp upstream of the translation start site. It contains a cyclic AMP (cAMP) response element (CRE; Wilson et al. 1996) which is essential for bcl-2 expression in neuronal cells (Riccio et al. 1999). A minor promoter, P2, utilized in some cell types, is located 1.3 kb downstream from the first one (Wilson et al. 1996). To investigate the role of CRE in the effects of VPA on bcl-2 expression, a human bcl-2 gene fragment (1,640 bp upstream from translation start site) was produced from a template (AC021803-human chromosome 18 clone RP11-495C15 from CHORI) by PCR using the Advantage GC Genomic PCR kit (Clontech, Mountain View, CA, USA) with the following primers: 5′-TTT AACCCGGGCCAGGGAG-3′ and 5′-CCTTCCCAGAGG AAAAGCA-3′. The PCR fragment was cloned into pGEM-T Easy vector (Promega, Madison, WI, USA) and then subcloned into pTATA-Luc (ATCC, Manassas, VA, USA) between the BamH1 and HindIII restriction enzyme sites. A point mutation within the CRE site of the promoter was made using the QuikChange® Site-directed mutagenesis kit (Stratagene, LaJolla, CA, USA) with the following oligonucleotides (CRE site in italics and mutation site in boldface): TTGAATGAACCGTGTGCCGTTACGCACAG GAAAC and GTTTCCTGTGCGTAACGGCACACGGTT CATTCAA. The sequences of the bcl-2 promoters were confirmed by sequencing conducted at NINDS DNA Sequencing Facility (NIH, Bethesda, MD, USA) using T7/SP6 primers for pGEM-T Easy constructs and using the above bcl-2 specific primers for pTATA-Luc constructs with optimized reaction conditions for GC-rich sequencing.
Reporter Gene Experiments
Transfections of SH-SY5Y cells with promoter–reporter constructs were conducted as previously described (Yuan et al. 2001). The cells were then treated with VPA (0 to 2 mM) for 2 days. The transfected cells were also treated alone or in combination with PD98059 (50 μM; Encinas et al. 1999), LY294002 (10 μM; Encinas et al. 1999), scriptaid (3 μM; Keen et al. 2003) or Ro31-8220 (1 μM; Dajas-Bailador et al. 2002; Calbiochem, San Diego, CA, USA), L-690,488 (10 μM; Atack et al. 1994) or SB216763 (5 μM; King et al. 2006; TOCRIS, Ellisville, MO, USA), or DMSO (0.1%) vehicle control followed 2 h later with VPA (0.8 mM) or DMEM vehicle control (Sigma-Aldrich) for 2 days. After the treatments, the cells were lysed and luciferase expression levels detected with a luciferase assay system (Promega) using a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA).
RSK1 Plasmid-Mediated shRNA Experiments
Human RSK1-specific (NM_002953) small interfering RNAs (siRNAs) were initially designed on an siRNA database (http://www.ambion.com/catalog/sirna_search.php, Ambion, Austin, TX, USA) and converted to small hairpin RNAs (shRNAs) with the BLOCK-iT™ RNAi Designer tool (https://rnaidesigner.invitrogen.com/rnaiexpress, Invitrogen). The shRNA sequences were as follows: Sequence 12—5′CACCGGATCCTTTGGCAAAGTCTTCCGAA GAAGACTTTGCCAAAGGATCC and 5′-AAAAGG ATCCTTTGGCAAAGTCTTCTTCGGAAGACTTTGCCAAAGGATCC
Sequence 23—5′CACCGCTCTATCTCATTCTGGACTT AACGAAGTCCA GAATGAGATAGAGC and 5′ AAAAGCTCTATCTCATTCTGGACTTCGTTAAGTCCAGAATGAGATAGAGC
Sequence 23mut—5′CACCGCTCTATCTTATTCTGG ACTTAACGAAGTCCAGAATAAGATAGAGC 5′ AAAAGCTCTATCTTATTCTGGACTTCGTTAAGTCCAG AATAAGATAGAGC.
Following the manual's instructions, shRNA oligonucleotides were annealed and cloned into BLOCK-iT™ U6 RNAi Entry Vectors (Invitrogen) for transient transfection into SH-SY5Y cells. Cells were grown as described previously and transfected with the RSK1-specific vectors using Lipofectamine 2000 (Invitrogen) for 48 h. Cells were processed for RSK1 or Bcl-2 immunoblot analysis or bcl-2 reporter gene expression as outlined in previous sections.
Statistical Analyses
Statistical analyses were performed using analysis of variance with Tukey or Bonferroni post hoc tests or unpaired t test. A value of p<0.05 was considered significant. Data are expressed as means± SEM.
Results
VPA Treatment Increased Bcl-2 Protein Levels in a Time- and Concentration-Dependent Manner
Our previous study showed that incubation of SY5Y cells with VPA for 5 days, but not 1 day, resulted in significant increases in Bcl-2 levels (Yuan et al. 2001). Considering that the clinical antimanic actions of VPA can be observed within 2 or 3 days of treatment with a high-dose loading regimen, we further examined the time–response issue of VPA-induced increases in Bcl-2 protein levels. Again, treatment with VPA (0.8 mM) for 1 day did not significantly alter protein levels of Bcl-2 in SH-SY5Y cells. However, the same treatment for 2 or 3 days significantly enhanced bcl-2 protein levels to 200% and more than 400% of vehicle control for 2 and 3 days of treatment, respectively (Fig. 1A and B). Additionally, treatment with VPA for 2 days increased Bcl-2 levels in a concentration-dependent manner (Fig. 1C,D). VPA- induced Bcl-2 protein increases appeared to be selective because the same treatment did not alter protein levels of a housing keeping gene, GAPDH (Fig. 1).
Figure 1.
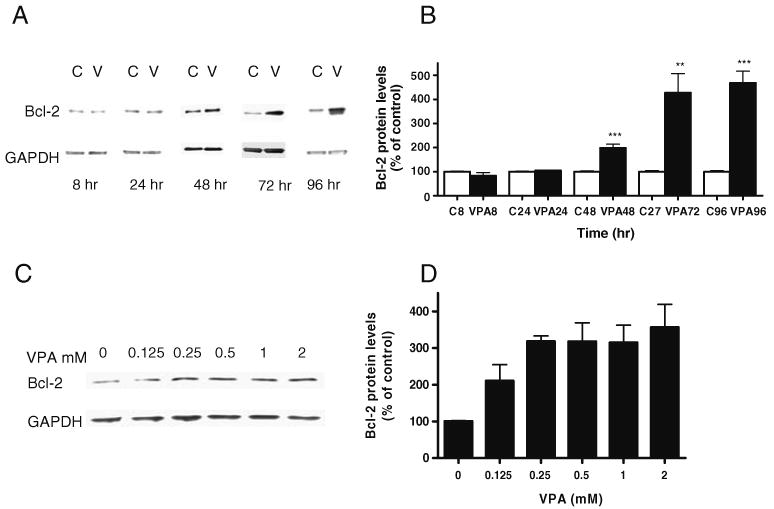
VPA increases bcl-2 protein levels in SH-SY5Y cells in a time- and concentration-dependent manner. SH-SY5Y cells were grown in DMEM medium supplemented with 10% FBS, antibiotics, and sodium pyridine to 80–90% confluence and treated thereafter for 8, 24, 48, 72, or 96 h with VPA (0.8 mM; VPA8, 24, 48, 72, or 96, respectively) or with vehicle control (C8, 24, 48, 72, or 96, respectively). A, C Cells were harvested and processed for Western blot analysis. B, D Densitometric results for Western blot analysis are displayed as percent of control, which was calculated as a mean of all of the controls. Data in the bar graph are means±SE from four sets of replicates. ***p<0.001; **p<0.01
Effects of VPA on Bcl-2 mRNA Levels
To investigate whether VPA-induced increases in Bcl-2 protein levels were, at least in part, due to enhanced bcl-2 gene expression, levels of bcl-2 mRNA were measured in cells prepared as in the Bcl-2 protein experiments. Treatment with VPA (0.8 mM) for 1, 2, or 3 days increased mRNA levels of bcl-2 by 120%, 175%, and 200%, respectively, compared to vehicle control-treated cells (Fig. 2). The effects appeared to be gene-specific because the same treatment did not alter the mRNA levels of the housekeeping gene, GADPH, used as the control (data not shown).
Figure 2.

VPA increases bcl-2 mRNA in a time-dependent manner. RT-PCR was utilized to quantitate bcl-2 mRNA levels from SH-SY5Y cells treated with 0.8 mM VPA for 1, 2, or 3 days (VPA1, 2, or 3, respectively) or with vehicle control (C1, 2, 3, respectively). Total cellular RNA was isolated from VPA-treated cells using Trizol reagent (Invitrogen) and purified using RNeasy mini kit (Qiagen). The RNA concentrations were determined in duplicates spectrophotometrically, and the quality of RNA was evaluated by visual inspection after electrophoresis of an aliquot of the RNA samples. Results are displayed as fold differences from respective controls for that day. Data in the bar graph are means±SE from three or more sets of replicates. ***p<0.001; **p<0.01, *p<0.05
Effects of VPA on the ERK and PI3K Pathways
Earlier studies have indicated that both ERK and PI3K pathways regulate bcl-2 gene expression (York et al. 2000) and that VPA activates both pathways (see “Introduction”). To evaluate whether these two sets of actions were related, the effects of VPA on these pathways were investigated in the same cell culture system in which VPA increased bcl-2 protein and mRNA levels (Figs. 1 and 2). Similar to our previous findings, treatment of SH-SY5Y cells with VPA for 2 days concentration-dependently increased activated phospho-ERK1/2 levels (Fig. 3A,B), but not total ERK1/2 levels. The same treatment also concentration-dependently increased activated phospho-Akt (ser473) levels and total Akt levels (Fig. 3C,D). The increases in activated phospho-Akt (ser473) levels appeared to be largely due to increases in total Akt levels (Fig. 3E). The mechanism by which VPA induces Akt protein will need to be investigated.
Figure 3.
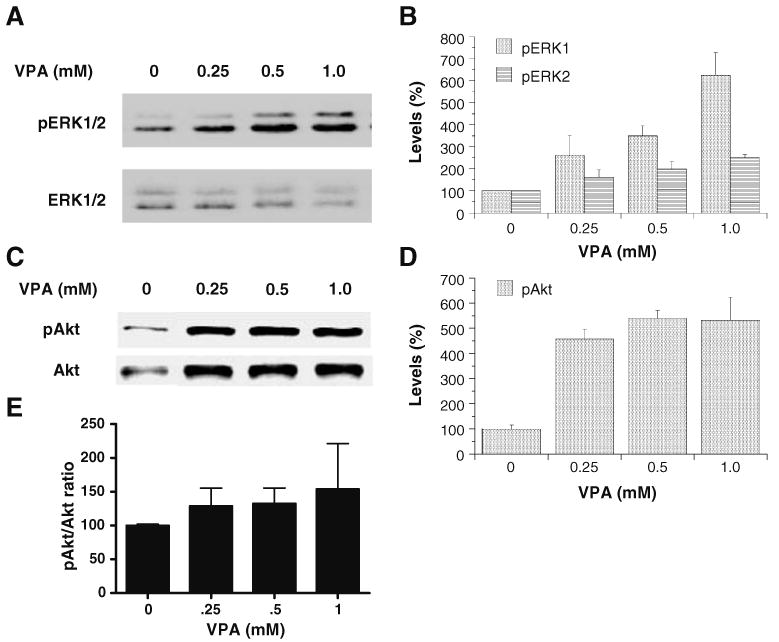
VPA enhances phospho-ERK1/2 and phospho-Akt levels in SH-SY5Y cells in a concentration-dependent manner. SH-SY5Y cells were grown as previously described and treated with VPA (0.8 mM) for 48 h, collected and processed for Western blot analysis, and probed with either phospho-ERK1/2 (Thr202/Tyr204) or phospho-Akt (Ser473) antibody (A, C). Densitometric results for Western blot analysis are displayed as percent of control, which was calculated as a mean of all of the controls (B, D). The phospho-Akt/Akt ratios were also increased by VPA treatment in a concentration-dependent manner (E). Data in the bar graph are means±SE from four sets of replicates
p90 ribosomal S6 kinase (RSK) is a converging point for the ERK and PI3K pathways. The RSK family of serine–threonine kinases includes four highly homologous isoforms with dual functional kinase domains, one in the N terminus that phosphorylates RSK substrates and one in the C terminus that activates the N terminus kinase domain (Frodin and Gammeltoft 1999). Full activation of RSK1 requires phosphorylation by both ERK and PI3K pathway constituents. ERK phosphorylates the C-terminal domain at thr573, allowing for C-terminal-induced phosphorylation of ser380 and autophosphorylation of thr359 and ser363 in the hinge region which further allows for phosphorylation of the N terminus at ser221 by phosphoinositide-dependent kinase 1 (PDK1; Richards et al. 1999). Thus, RSK occupies a pivotal position within the cellular milieu that is responsible for cell survival and may be critical itself in coordinating ERK and PI3K pathway activities for orchestration of neural plasticity. RSK phosphorylates CREB and increases CREB-induced transcriptional activity (Richards et al. 1999). Treatment of SH-SY5Y cells with VPA for 2 days concentration-dependently increased activated phospho-RSK1 levels (Fig. 4A,B), but not total RSK1 levels. The same treatment also concentration-dependently increased activated phospho-CREB levels, but not total CREB levels (Fig. 4C,D). These data indicate that VPA activates common downstream targets of the ERK and PI3K pathways and suggest that VPA also increases CREB-mediated gene expression through these pathways.
Figure 4.
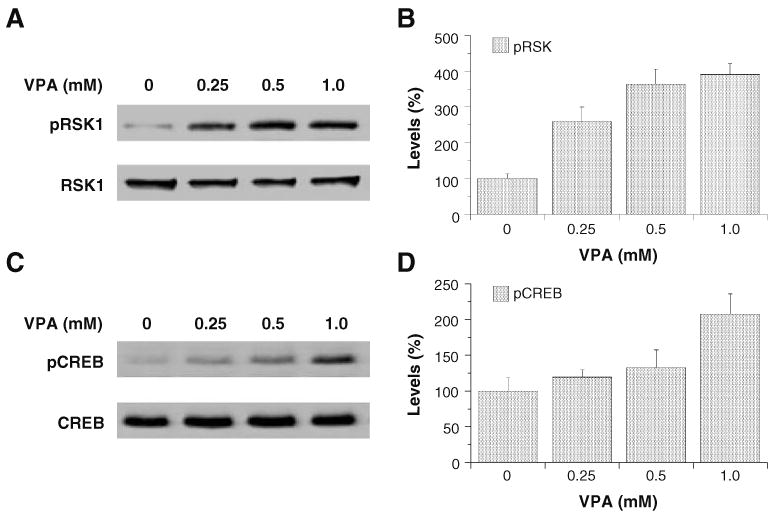
VPA enhances phospho-RSK1 and phospho-CREB levels in SH-SY5Y cells in a dose-dependent manner. SH-SY5Y c1ells were grown as previously described and treated with VPA (0.8 mM) for 48 h, collected and processed for Western blot analysis, and probed with either phospho-RSK1 (Thr359/Ser363) or phospho-CREB (Ser133) antibody (A, C). Densitometric results for Western blot analysis are displayed as percent of control, which was calculated as a mean of all of the controls (B, D). Data in the bar graph are means±SE from four sets of replicates
Effects of VPA on the Activity of a Human bcl-2 Promoter–Reporter Gene Construct
Given that the effects of VPA on bcl-2 mRNA levels could be due to either promoter activation enhancement, mRNA stabilization, or both, the effect of VPA on luciferase expression driven by a human bcl-2 promoter was examined. Because the pTATA-Luc construct contained its own promoter, in addition to the bcl-2 promoter we cloned into the plasmid construct, we compared luciferase activities in pTATA-Luc transfected SH-SY5Y cells with and without the bcl-2 promoter construct. Cells transfected with the bcl-2 promoterless construct displayed barely detectable luciferase activities, whereas cells transfected with the bcl-2 promoter displayed significantly greater luciferase expression, indicating that the enhancer elements in the bcl-2 gene promoter fragment cloned into the pTATA-Luc construct played a major role in luciferase expression in our experimental system (data not shown). Furthermore, VPA treatment concentration-dependently increased luciferase expression levels in SH-SY5Y cells transfected with the bcl-2 promoter construct (Fig. 5). A point mutation of the CRE consensus site of the bcl-2 construct resulted in a significant reduction in, but not abolishment of, VPA-induced luciferase expression in our cell culture system (Fig. 5), indicating that CREB transcription factors are involved in VPA-induced bcl-2 promoter activation and that a CREB-independent mechanism is likely involved.
Figure 5.

VPA enhances activity of a bcl-2 promoter construct in SH-SY5Y cells in a dose-dependent manner and is partially dependent on a viable CRE site within the bcl-2 promoter. A luciferase reporter gene construct driven by a promoter containing a human bcl-2 gene promoter fragment was transfected into SH-SY5Y cells which were treated with various doses of VPA for 48 h to produce a dose–response curve for the promoter activity. A luciferase assay system was used to assess the promoter activity. Additionally, a single base pair mutation was made in the CRE site of the human bcl-2 promoter fragment and transfected into SH-SY5Y cells which were treated with the same doses of VPA for 48 h to produce a dose–response curve for the promoter activity. Data in the bar graph are means±SE from three or more sets of replicates. A two-way ANOVA of the data indicates that there is a highly significant group effect (WT vs mutant, F=57, p<0.0001), a highly significant concentration effect (F=117.3, p< 0.0001), and a highly significant interaction effect between group and concentration effects (F=14.88, p<0.0001)
Roles of ERK and PI3K Pathways in the Activation of a Human bcl-2 Promoter–Reporter Gene Construct and in VPA's Enhancement of this Promoter's Activity
Consistent with predictions from previous data, incubations of transfected SH-SY5Y cells with MEK inhibitor PD98059 and/or PI3K inhibitor LY294002 significantly reduced luciferase gene expression driven by the bcl-2 promoter (Fig. 6A), suggesting that both the ERK and PI3K pathways are sufficient to control bcl-2 promoter activities. Incubation with PD98059 or LY294002 markedly attenuated VPA induction of this promoter (Fig. 6B), implying that both pathways are required for and may function synergistically or cooperatively to induce VPA's enhancement of bcl-2 promoter activity.
Figure 6.

Chemical inhibition of either the ERK or PI3K pathway drastically reduces activity of a human bcl-2 promoter–reporter gene construct and inhibition of both pathways virtually abolishes the activity. A SH-SY5Y cells were grown as before, transfected with the promoter construct, and treated with PD98059 (50 μM) or LY294002 (10 μM) or both for 48 h and subsequently collected and processed for luciferase assay. B The same experiment was set up again with VPA (0.8 mM final concentration) added to half of the cells from each treatment condition 2 h after inhibitor treatment. Cells were collected and a luciferase assay system was again used to assess the promoter activity. Data in the bar graph are means±SE from three or more sets of replicates. ***p<0.001, **p<0.01 from DMSO controls; ###p< 0.001, ##p<0.01 from DMSO/VPA
Role of RSK1 in VPA-Induced Activity of a Human bcl-2 Promoter–Reporter Gene Construct
RSK phosphorylates and activates CREB through which it can induce bcl-2 promoter activity. An RNAi strategy was used to specifically block RSK1 to further evaluate the role of RSK1 as a converging point for the ERK and PI3K pathways on bcl-2 promoter activity. Transfections of RSK1-specific plasmid vector-mediated shRNAs (12 and 23), but not the mutated shRNA (23mut) construct, in SH-SY5Y cells down-regulated RSK1 protein levels (Fig. 7A,B). RSK1 shRNA (sequence 23) and RSK inhibitor Ro 31-8220 reduced activities of the bcl-2 promoter (Fig. 7C) and also attenuated valproate-induced activities of this promoter (Fig. 7D).
Figure 7.

RSK1 knockdown attenuates activity of a human bcl-2 promoter–reporter gene construct. Several RSK1-specific shRNAs, designed using online tools, as well as a LacZ control that coded for β-galactosidase, were cloned into a U6 promoter-driven vector and tested for RSK1 knockdown efficiency. Additionally, a 1-bp mutation of the most efficient shRNA (sequence 23) was designed (23mut) to rescue or reverse the knockdown and to show that the knockdown was not due to off-target effects. A, B SH-SY5Y cells were grown as before and transfected with the shRNA constructs for 2 days before cells were collected and processed for RSK1-specific immunoblotting and GAPDH as an off-target control. C Sequence 23 RSK1-specific shRNA and the LacZ control shRNAs were transfected into SH-SY5Y cells as above. Cells were collected 2 days later and processed for bcl-2 promoter activity. RSK1-specific shRNA significantly reduced promoter activity (D). The previous experiment was set up again with VPA (0.8 mM final concentration) added 2 h after the shRNA transfections. Cells were collected and processed as before. RSK1-specific shRNA significantly reduced VPA-induced enhancement of promoter activity compared to that of the LacZ shRNA. Data in the bar graph are means±SE from three or more sets of replicates. ***p< 0.001, **p<0.01 from control; ###p<0.001 from Control/VPA
Effects of ERK Pathway, PI3K Pathway, and RSK1 Inhibitions on Bcl-2 Protein Levels
In our experiments, ERK pathway, PI3K pathway, and RSK1 inhibitions altered baseline and VPA-induced increases in activities of the bcl-2 promoter. We, therefore, further tested whether these promoter effects resulted in changes in protein levels. Chemical inhibitions of the ERK and PI3K pathways, as well as RSK1 knockdown, reduced Bcl-2 protein levels and also attenuated Bcl-2 levels in inhibited cells that were treated with valproate (Fig. 8).
Figure 8.
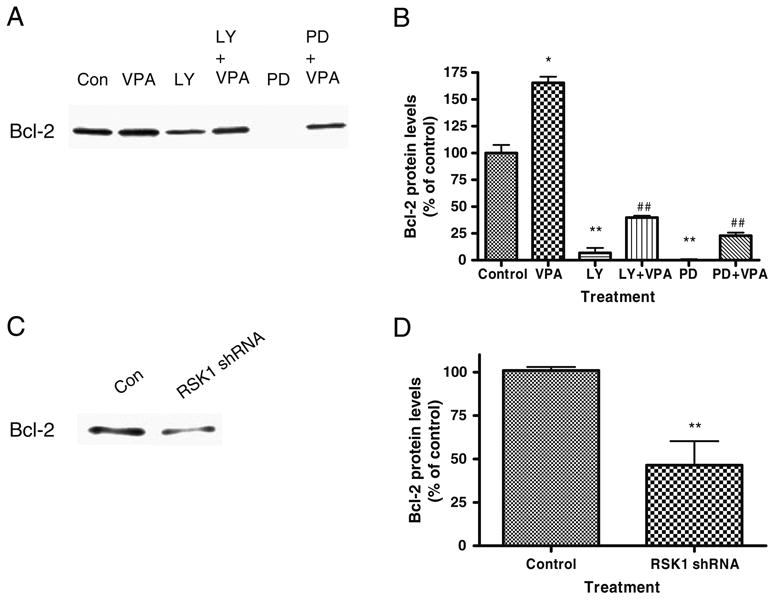
Chemical inhibition of the ERK or PI3K pathway or both or RSK1 knockdown reduces Bcl-2 protein levels. SH-SY5Y cells were grown as before and treated with PD98059 (50 μM) or LY294002 (10 μM) or DMSO vehicle control with or without VPA (0.8 mM) for 3 days. A Cells were collected and processed for immunoblot analysis with anti-Bcl-2 or GAPDH load control. Densitometric measurements were obtained as before and represent means±SE of four independent experiments. C and D SH-SY5Y cells were grown to confluence and transfected with the (sequence 23) RSK1-specific shRNA construct for 2 days. Cells were collected and harvested for immunoblot analysis with anti-Bcl-2. Data in the bar graph are means±SE from three or more sets of replicates. **p<0.01, *p<0.05, ##p<0.01 compared to VPA treatment alone
Discussion
The results of the present study suggest that VPA enhances Bcl-2 expression through multiple molecular mechanisms. One of these mechanisms is associated with concerted activations of the ERK and PI3K pathways that converge at RSK to stimulate CREB phosphorylation of a consensus CRE site of the major promoter of bcl-2. At the protein level, VPA concentration- and time-dependently increased Bcl-2 protein levels (Fig. 1). These protein increases were associated with increases in bcl-2 mRNA levels (Fig. 2) and with activation of ERK and PI3K pathways (Fig. 3) as well as increases in activated phospho-RSK1 and phospho-CREB levels (Fig. 4). Both ERK and PI3K pathways are known to regulate RSK and, through RSK-induced transactivational activity of CREB, regulate bcl-2 transcription (Fig. 9). Thus, the protein and mRNA data suggest that VPA activates the bcl-2 promoter in part by enhancing CREB transcriptional activity through activation of ERK and/or PI3K pathways. The follow-up experiments showed that VPA augmented activity of the human bcl-2 promoter–reporter gene construct and that a point mutation within the CRE site attenuated VPA-induced promoter activation enhancement (Fig. 5), confirming that VPA increases Bcl-2 protein levels partially through a transcriptional mechanism involving transcription factors interacting with the CRE site. Blockages of ERK and PI3K pathways and knockdown of RSK1 attenuated the promoter activity in the absence or presence of VPA (Figs. 6 and 7), supporting the substantial roles that both pathways and RSK, a converging point for these pathways, play in the regulation of bcl-2 expression. Their significances in bcl-2 regulation were further demonstrated by experiments showing that chemical inhibition of ERK or PI3K pathway or RSK1 knockdown reduced Bcl-2 protein levels in the absence or presence of VPA (Fig. 8). However, blocking either of these two pathways or RSK did not completely abolish induction of bcl-2 by VPA, suggesting that additional mechanisms are involved in VPA-mediated induction of Bcl-2.
Figure 9.

VPA regulates Bcl-2 in part by activating both the ERK and PI3K pathways. Lines with arrowheads indicate activation of designated target. Lines with filled-in circles indicate inhibition of designated target
The cascade-associated effects of VPA on bcl-2 transcriptional regulation and protein levels are similar to those produced by neurotrophic factors, yet the temporal profiles of the effects are different. Neurotrophins such as brain-derived nerve growth factor (BDNF) and nerve growth factor (NGF) activate both ERK and PI3K pathways and stimulate Bcl-2 expression through a mechanism involving CREB (Almeida et al. 2005). The present study indicates that VPA behaves like a neurotrophic factor in producing a cascade-associated effect on Bcl-2 regulation. Neurotrophin-induced ERK and PI3K pathway activations are characterized by rapid onsets and short lasting durations (minutes to hours), while VPA-induced ERK activation is characterized by delayed onsets and persistent durations (days; Hao et al. 2004). In concert with this temporal profile, the effects of VPA on Bcl-2 expression are also characterized by a pattern of delayed onset and long-lasting duration. Although CREB profoundly affects regulation of Bcl-2 expression by NGF, a study using a series of deletion constructs of a bcl-2 promoter has shown that NGF activates the bcl-2 promoter independent of the promoter's CRE site (Liu et al. 1999). Interestingly, it has been found that neurotrophin 3 (NT-3)-induced increases in Bcl-2 protein levels in oligodendrocyte progenitors is dependent on the promoter's CRE site, indicating a direct role for CREB in regulating bcl-2 gene activity in response to NT-3 (Saini et al. 2004). Recently, it was reported that B cells from mutant CRE-bcl-2 mice exhibit significantly reduced expression of Bcl-2 (Xiang et al. 2006). The present study found that although CRE mutation reduced VPA-induced bcl-2 promoter activation, VPA was still able to induce promoter activity, suggesting that additional transcriptional mechanisms are involved in induction of the bcl-2 promoter by both neurotrophins and VPA. Along this line, RSK is one of several known CREB kinases including protein kinase (PKC), cAMP-dependent kinase (PKA), mitogen and stress-activated kinase (MSK) 1, mitogen-activated protein kinase-activated protein kinase, (MAPKAP) 2 and 3 and calcium-calmodulin kinases (CaMK) II and IV. In cultured rat striatal neurons, it was found that PMA-stimulated PKC activation induces CREB phosphorylation in a dose-dependent manner that was reversed by PKC inhibitors, chelerythrine and Go6983, by CaMK inhibitor KN93 and by ERK inhibitors PD98059 and U0126 (Mao et al. 2007), suggesting that a network of signal transduction pathways converge at various downstream targets to affect CRE-associated transcription. Nevertheless, NGF and BDNF have been shown to promote cell survival by stimulating expression of bcl-2 by their ability to phosphorylate ser133CREB by RSK, a recognized CREB kinase (Mayr and Montminy 2001). Furthermore, overexpression of dominant negative CREB induces cell death that can be rescued by overexpression of Bcl-2 (Mayr and Montminy 2001).
Lithium, like VPA, has been shown to regulate gene transcription. Treatment of rat cerebellar granule cells with therapeutically relevant concentrations increases AP-1 and CRE binding activities that are persistent for 7 days (Ozaki and Chuang 1997). Furthermore, it has been demonstrated that chronic lithium treatment enhances glutamate-induced increases in MEK activity and that PD98059 prevents lithium-induced increases in pCREB levels in glutamate-treated neurons (Kopnisky et al. 2003). We have shown that chronic treatment of VPA and lithium in rodent chow increases levels of pERK1/2, pRSK1, pCREB, and BDNF in rat frontal cortex and hippocampus (Einat et al. 2003) as well as immunoreactivity of Bcl-2 in the frontal cortex (Chen et al. 1999b) and VPA-induced immunoreactivity of pERK1/2 in rat prefrontal cortex (Hao et al. 2004).
Several studies indicate that BDNF itself is a target of VPA (Chen et al. 2006). Treatment with VPA increases BDNF mRNA and protein levels in cultured cells and in specific brain regions of rodents (Fukumoto et al. 2001; Einat et al. 2003). As suggested in some studies, mood stabilizers induce autocrine or paracrine processes that contribute to BDNF protective and neurotrophic effects through stimulation of receptor tyrosine kinases and thereby activate the ERK and PI3K pathways (Chen et al. 2006). It has been demonstrated that cultured cortical progenitor cells express BDNF and NT-3 and that their receptors TrkB and TrkC constitute an autocrine-paracrine neurotrophin loop that activates downstream intracellular effectors PI3K and MEK (Barnabe-Heider and Miller 2003). Interestingly, NGF-induced activation of the ERK pathway is known to preferentially stimulate BDNF promoter IV in PC12 cells independent of NGF-activated signaling pathways involving PKA and PKC (Park et al. 2006). Thus, the autocrine–paracrine processes that regulate VPA's effects on BDNF are also involved in regulating Bcl-2 expression and are mediated through ERK and PI3K pathways.
Several other known targets of VPA may account for additional mechanisms through which VPA regulates Bcl-2 levels. VPA is considered to be a direct inhibitor of HDAC, hyperacetylating core histones H3 and H4 (Phiel et al. 2001). Hyperacetylated histones are associated with relaxed chromatin conformation and derepression of transcriptional activity. VPA-induced HDAC inhibition accounts for expression of NeuroD, a critical inducer of neural progenitor cell differentiation (Hsieh et al. 2004). A previous study reported that VPA mediates gene expression driven by a CMV promoter through HDAC inhibition (Phiel et al. 2001). GSK-3 is phosphorylated by both RSK and Akt, resulting in the inactivation of its catalytic functions (De Mesquita et al. 2001). GSK-3 phophorylates CREB, which attenuates CREB transcriptional activity (Leinninger et al. 2004). VPA directly or indirectly inactivates GSK-3 which would predictably contribute to VPA induction of the bcl-2 promoter. Lithium inhibits inositol monophosphatase, resulting in, as suggested, inositol depletion. Inositol depletion in the CNS is a well-known theory explaining the clinical action of at least one mood stabilizer, lithium (Berridge et al. 1989). Recent studies suggest that VPA also induces inositol depletion through inhibition of its de novo biosynthesis (Shaltiel et al. 2004).
In summary, results of the present study demonstrate that VPA, like neurotrophins, activates both ERK and PI3K pathways and that these pathway activations contribute to the effects of VPA on bcl-2 gene transcription, Bcl-2 protein levels, and bcl-2 cellular functions. Given the recent pathological findings in BD patients (see “Introduction”), VPA's long-lasting neurotrophin-like effect on Bcl-2 may play a critical role in the treatment of BD and in the prevention of mood episodes which are currently under investigation in our laboratory.
Acknowledgments
This work was supported by the NIMH intramural program (Manji and Chen) and a NARSAD research award (Chen).
Abbreviations
- Bcl-2
B-cell lymphoma 2
- BD
bipolar (mood) disorder
- CRE
cyclic adenosine monophosphate response element
- CREB
cyclic adenosine monophosphate response element binding protein
- ERK
extracellular signal-regulated kinase
- GSK-3
glycogen synthase kinase-3
- HDAC
histone deacetylase
- IMPase
inositol monophosphatase
- PI3K
phosphatidylinositol 3-kinase
- RSK
p90 ribosomal S6 kinase
- VPA
valproate
References
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM, et al. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death and Differentiation. 2005;12:1329–1343. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- Atack JR, Prior AM, Fletcher SR, Quirk K, McKernan R, Ragan CI. Effects of L-690,488, a prodrug of the bisphosphonate inositol monophosphatase inhibitor L-690,330, on phosphatidylinositol cycle markers. The Journal of Pharmacology and Experimental Therapeutics. 1994;270:70–76. [PubMed] [Google Scholar]
- Barnabe-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. The Journal of Neuroscience. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmaker RH. Bipolar disorder. The New England Journal of Medicine. 2004;351:476–486. doi: 10.1056/NEJMra035354. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: A unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997;385:434–439. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. Journal of Neurochemistry. 1999a;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, et al. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. Journal of Neurochemistry. 1999b;72:879–882. doi: 10.1046/j.1471-4159.1999.720879.x. [DOI] [PubMed] [Google Scholar]
- Chen PS, Peng GS, Li G, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Molecular Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- Chuang DM. The antiapoptotic actions of mood stabilizers: Molecular mechanisms and therapeutic potentials. Annals of the New York Academy of Sciences. 2005;1053:195–204. doi: 10.1196/annals.1344.018. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. Journal of Neurochemistry. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- De Mesquita DD, Zhan Q, Crossley L, Badwey JA. p90-RSK and Akt may promote rapid phosphorylation/inactivation of glycogen synthase kinase 3 in chemoattractant-stimulated neutrophils. FEBS Letters. 2001;502:84–88. doi: 10.1016/S0014-5793(01)02669-2. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/S0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Di Daniel E, Cheng L, Maycox PR, Mudge AW. The common inositol-reversible effect of mood stabilizers on neurons does not involve GSK3 inhibition, myo-inositol-1-phosphate synthase or the sodium-dependent myo-inositol transporters. Molecular and Cellular Neuroscience. 2006;32:27–36. doi: 10.1016/j.mcn.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging studies of mood disorders. Biological Psychiatry. 2000;48:813–829. doi: 10.1016/S0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Pandey GN. ERK MAP kinase signaling in post-mortem brain of suicide subjects: Differential regulation of upstream Raf kinases Raf-1 and B-Raf. Molecular Psychiatry. 2006;11:86–98. doi: 10.1038/sj.mp.4001744. [DOI] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, et al. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. The Journal of Neuroscience. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. Journal of Neurochemistry. 1999;73:1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Molecular and Cellular Endocrinology. 1999;151:65–77. doi: 10.1016/S0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology. 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Janiri L. Sodium- and magnesium-valproate in vivo modulate glutamatergic and GABAergic synapses in the medial prefrontal cortex. Psychopharmacology. 2006;185:255–262. doi: 10.1007/s00213-006-0317-3. [DOI] [PubMed] [Google Scholar]
- Hao Y, Creson T, Zhang L, Li P, Du F, Yuan P, et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. The Journal of Neuroscience. 2004;24:6590–6599. doi: 10.1523/JNEUROSCI.5747-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proceedings of the National Academy of Sciences of the USA. 2004;101:16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2¢-deoxycytidine. Breast Cancer Research and Treatment. 2003;81:177–186. doi: 10.1023/A:1026146524737. [DOI] [PubMed] [Google Scholar]
- King TD, Gandy JC, Bijur GN. The protein phosphatase-1/inhibitor-2 complex differentially regulates GSK3 dephosphorylation and increases sarcoplasmic/endoplasmic reticulum calcium ATPase 2 levels. Experimental Cell Research. 2006;312:3693–3700. doi: 10.1016/j.yexcr.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopnisky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang DM. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. 2003;116:425–435. doi: 10.1016/S0306-4522(02)00573-0. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Biebl M, Wilhelm D, Li M, Friedlander RM, Winkler J. Increased generation of granule cells in adult Bcl-2-overexpressing mice: A role for cell death during continued hippocampal neurogenesis. The European Journal of Neuroscience. 2005;22:1907–1915. doi: 10.1111/j.1460-9568.2005.04377.x. [DOI] [PubMed] [Google Scholar]
- Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H, et al. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. Journal of Neurochemistry. 2004;91:238–251. doi: 10.1111/j.1471-4159.2004.02725.x. [DOI] [PubMed] [Google Scholar]
- Lai JS, Zhao C, Warsh JJ, Li PP. Cytoprotection by lithium and valproate varies between cell types and cellular stresses. European Journal of Pharmacology. 2006;539:18–26. doi: 10.1016/j.ejphar.2006.03.076. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Backus C, Uhler MD, Lentz SI, Feldman EL. Phosphatidylinositol 3-kinase and Akt effectors mediate insulin-like growth factor-I neuroprotection in dorsal root ganglia neurons. The FASEB Journal. 2004;18:1544–1546. doi: 10.1096/fj.04-1581fje. [DOI] [PubMed] [Google Scholar]
- Liu YZ, Boxer LM, Latchman DS. Activation of the Bcl-2 promoter by nerve growth factor is mediated by the p42/p44 MAPK cascade. Nucleic Acids Research. 1999;27:2086–2090. doi: 10.1093/nar/27.10.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao LM, Tang Q, Wang JQ. Protein kinase C-regulated cAMP response element-binding protein phosphorylation in cultured rat striatal neurons. Brain Research Bulletin. 2007;72:302–308. doi: 10.1016/j.brainresbull.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature reviews. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Suhan T, Michaelis UR, et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death and Differentiation. 2006;13:446–453. doi: 10.1038/sj.cdd.4401759. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Chuang DM. Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain. Journal of Neurochemistry. 1997;69:2336–2344. doi: 10.1046/j.1471-4159.1997.69062336.x. [DOI] [PubMed] [Google Scholar]
- Park SY, Lee JY, Choi JY, Park MJ, Kim DS. Nerve growth factor activates brain-derived neurotrophic factor promoter IV via extracellular signal-regulated protein kinase 1/2 in PC12 cells. Molecules and Cells. 2006;21:237–243. [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. The Journal of Biological Chemistry. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Phrolov K, Applebaum J, Levine J, Miodovnick H, Belmaker RH. Single-dose intravenous valproate in acute mania. The Journal of Clinical Psychiatry. 2004;65:68–70. doi: 10.4088/jcp.v65n0111. [DOI] [PubMed] [Google Scholar]
- Rajkowska G. Cell pathology in bipolar disorder. Bipolar Disorders. 2002;4:105–116. doi: 10.1034/j.1399-5618.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Richards SA, Fu J, Romanelli A, Shimamura A, Blenis J. Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Current Biology. 1999;9:810–820. doi: 10.1016/S0960-9822(99)80364-9. [DOI] [PubMed] [Google Scholar]
- Saini HS, Gorse KM, Boxer LM, Sato-Bigbee C. Neurotrophin-3 and a CREB-mediated signaling pathway regulate Bcl-2 expression in oligodendrocyte progenitor cells. Journal of Neurochemistry. 2004;89:951–961. doi: 10.1111/j.1471-4159.2004.02365.x. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Roth KA. Regulation of neuronal cell death and neurodegeneration by members of the Bcl-2 family: Therapeutic implications. Current Drug Targets CNS and Neurological Disorders. 2005;4:25–39. doi: 10.2174/1568007053005127. [DOI] [PubMed] [Google Scholar]
- Shaltiel G, Shamir A, Shapiro J, et al. Valproate decreases inositol biosynthesis. Biological Psychiatry. 2004;56:868–874. doi: 10.1016/j.biopsych.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vaden DL, Ju S, Ding D, Geiger JH, Greenberg ML. Genetic perturbation of glycolysis results in inhibition of de novo inositol biosynthesis. The Journal of Biological Chemistry. 2005;280:41805–41810. doi: 10.1074/jbc.M505181200. [DOI] [PubMed] [Google Scholar]
- Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T, et al. Benefit of valproic acid in suppressing disease progression of ALS model mice. The European Journal of Neuroscience. 2004;20:3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Molecular and Cellular Biology. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Wang J, Boxer LM. Role of the cyclic AMP response element in the bcl-2 promoter in the regulation of endogenous Bcl-2 expression and apoptosis in murine B cells. Molecular and Cellular Biology. 2006;26:8599–8606. doi: 10.1128/MCB.01062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Molecular and Cellular Biology. 1998;18:1946–1955. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Molecular and Cellular Biology. 2000;20:8069–8083. doi: 10.1128/MCB.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G. The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. The Journal of Biological Chemistry. 2001;276:31674–31683. doi: 10.1074/jbc.M104309200. [DOI] [PubMed] [Google Scholar]