

Summary
Epithelial-mesenchymal interactions during lung development require extracellular signaling factors that facilitate branching morphogenesis. We show here that matrix metalloproteinases (MMPs) originating in the mesenchyme are necessary for epithelial branching and alveolization. We found that the delayed lung maturation characterized by abnormal branching and poor alveolization seen in mice deficient in epidermal growth factor receptor (Egfr−/−) is accompanied by aberrant expression of MMPs. By in situ zymography, the lungs from newborn Egfr−/− mice had low gelatinolytic activity compared with wildtype. Inhibition of MMPs in developing lungs in vivo or in vitro severely retarded morphogenesis. Egfr−/− mice had low expression of MT1-MMP/MMP14, which is a potent activator of gelatinase A/MMP2, in their lungs. Egf ligand increased MT1-MMP mRNA by tenfold in lung fibroblasts from wild type, but not from Egfr−/− mice. Extracts from lungs of Egfr−/− mice showed a tenfold reduction in active MMP-2, but only a slight decrease in proMMP-2 by zymography. At birth, MMP-2−/− mice had a lung phenotype characterized by abnormal lung alveolization which phenocopied that of Egfr−/− mice, albeit somewhat less severe. We conclude that proteolysis mediates epithelial/mesenchymal interactions during lung morphogenesis. From the phenotypes of the Egfr−/− mice, we identify MT1-MMP as a major downstream target of Egfr signaling in lung in vivo and in vitro. MT1-MMP is, in turn, necessary for activation of MMP-2, a mesenchymal enzyme that is required for normal lung morphogenesis.
Keywords: Matrix metalloproteases, MMP-2, Branching morphogenesis, Lung development, Egf signaling
Introduction
The extracellular matrix (ECM) contains instructive information for epithelial growth and differentiation, and is responsible for coordinating lung morphogenesis and growth (Matsui et al., 1996). Specific ECM components and their proteolytically cleaved fragments govern cell proliferation, differentiation and motility in organogenesis and tissue repair (Gustafsson and Fassler, 2000; Koshikawa et al., 2000; Roman, 1996). This is accomplished by the selective binding and timely release of growth factors, and generation of biologically active matrix fragments; these are processes that all depend on proteolysis (Werb, 1997). In recombination studies, factors from the distal embryonic lung ECM and mesenchyme induce proximal lung epithelium to undergo repeated invaginations reminiscent of future alveolar air spaces (Shannon et al., 1998). Furthermore, signals arising from specific regions of the mesenchyme and ECM program the epithelium to express distal epithelial-specific genes such as surfactant proteins (SP)-A, -B and -C (Shannon et al., 1998).
Morphogens expressed in the distal epithelium of the lung can signal mesenchyme differentiation (Motoyama et al., 1998; Park et al., 2000). Sonic hedgehog (Shh), which is widely expressed in the distal epithelium of the lung, disrupts normal epithelial to mesenchymal ratio by increasing proliferation of mesenchyme when expressed ectopically (Bellusci et al., 1997a). In its absence, or that of Gli2 and Gli3, which encode zinc-finger DNA binding proteins and act as transcription factors downstream of Shh, the esophagus fails to separate from trachea (Motoyama et al., 1998; Park et al., 2000). These data indicate that the mesenchyme, not the epithelium, is the most likely target of Shh signaling.
Secreted growth factors also play critical roles as signaling molecules in lung morphogenesis (Hogan, 1999; Warburton et al., 2000). The functional role of fibroblast growth factors (Fgfs) has been revealed by targeted expression of a dominant-negative Fgf receptor (Fgfr) in the primordial respiratory epithelium (Peters et al., 1994). Abrogation of Fgf signaling results in the complete absence of epithelial branching and differentiation in mouse lung (Peters et al., 1994). Deletion of Fgfr2(IIIb) results in viable mice with severe defects of lung (Arman et al., 1999). Fgf10 expressed in the mesenchyme at the earliest stage of lung development is required for migration and proliferation of primordial endoderm (Bellusci et al., 1997b; Sekine et al., 1999). In its absence, embryos lack lung buds (Min et al., 1998).
Epidermal growth factor (Egf) family members have been implicated in the development of the respiratory system in a diverse range of animal species from Drosophila to mammals (Hogan and Yingling, 1998; Kramer et al., 1999; Warburton et al., 2000). Moreover, defective signaling through the Egf receptor (Egfr) has been demonstrated in low birth-weight human infants at high risk for respiratory distress syndrome (Fondacci et al., 1994). Previous studies from this laboratory have established a role for Egf in lung development and epithelial maturation (Miettinen et al., 1995). Mice deficient in Egfr (Egfr−/−) die shortly after birth from respiratory failure (Miettinen et al., 1995; Miettinen et al., 1997). Furthermore, deficient alveolization and septation in these mice correlates with low expression of SP-C and thyroid-specific transcription factor (TTF-1) (Miettinen et al., 1997). It remains unclear as to what is the molecular basis for the defect in alveolization, and whether instructive signals originating from the mesenchyme or the epithelium are lacking.
While epithelial-mesenchymal interactions during lung development require secreted factors that allow branching morphogenesis in the lung, the nature of these factors remains poorly understood. Egf regulates expression and activation of ECM-degrading enzymes (van der Zee et al., 1998). Indeed, the cleft palate seen in some Egfr−/− mice may be due to decreased expression of MMPs (Miettinen et al., 1999). These findings suggest a plausible role for MMPs as downstream effector molecules of growth factor signaling in lung development. Thus, the alveolization defects in Egfr−/− mice may directly result from abnormal protease expression and/or function. In the present study we have tested the hypothesis that epithelial-mesenchymal interactions during lung development may be mediated through regulation of proteolysis. We sought to determine the factors originating in the mesenchyme that are necessary for prenatal epithelial branching, and postnatal completion of the alveolization process. Here we identify MMP-2 (gelatinase A) and MT1-MMP (MMP 14) as key proteases in lung development, and show that their activity occurs downstream of Egfr signaling.
Materials and Methods
Animals and isolation of embryonic lung fibroblasts
Timed-pregnant wildtype and Egfr+/− mice (Miettinen et al., 1995) (Swiss-Webster genetic background) were sacrificed at E10.5, E13.5, E18.5 and at postnatal days 1, 7 and 9. The plug date was considered day 0.5 of pregnancy. Embryos generated by crossing Egfr+/− mice were genotyped by Southern blot analysis as described previously (Miettinen et al., 1995), while newborn Egfr−/− mice were reliably identified by open-eye and wrinkled skin phenotypes. MMP-2−/−(Itoh et al., 1997) and littermate control mice (+/+ and +/−) from heterozygote matings were genotyped by standard zymography of serum.
Whole lungs from E12.5 Egfr+/− mice were dissected and embryos were genotyped by Southern blot analysis as described previously (Miettinen et al., 1995). Wild-type and Egfr−/− fibroblasts were separated from endoderm following a 30 minute treatment with 0.05% trypsin/EDTA (Gibco-BRL) and single cell suspensions were washed once in the medium (1:1 DMEM/F-12, 10% FCS, gentamycin 50 μg/ml) before seeding onto tissue culture-treated plates. Cells were grown to confluence and were deprived of serum factors 24 hours prior to stimulation with Fgf or Egf. All cells were used in experiments at passages 2–5.
In vivo MMP inhibition
Timed-pregnant mice were injected intraperitoneally with a broad, class-specific metalloproteinase inhibitor (GM6001) (Galardy et al., 1994) at 100 μg/g, or with carrier reagent (PBS), daily for 3 days starting at E10.5. Embryonic lungs were collected on E13.5, which corresponds to 1 day after initiation of the pseudoglandular stage (E12.5–16.5) of lung development. This concentration of GM6001 is shown to inhibit MMPs in animal models (Zheng et al., 2000) and at higher concentrations we did not see additional effects (data not shown). GM6001 is N-[(2R)-2-(hydroxyamidocarbonylmethyl)-4-methylpentanoyl]-L-tryptophan methylamide] (Galardy et al., 1994).
Lung organ culture and in vitro MMP inhibition
Wild-type E10.5 and E11.5 whole lungs were isolated and placed on top of polycarbonate membrane (VWR) floating on 1 ml of serum-free medium (1:1 DMEM/F-12, Transferrin 10 μg/ml, BSA 1 μg/ml, gentamycin 50 μg/ml) in 24-well culture plates. Lung organ cultures were treated with 20 μM of GM6001, a MMP inhibitor (Galardy et al., 1994) synthesized by AMS Scientific Inc., or control medium plus 1:1000 dilution of vehicle (DMSO), and placed in 5% CO2 at 37°C for 4 days. In order to obtain quality images of the lung, explants were removed from polycarbonate membranes, fixed in 4% paraformaldehyde (PFA), photographed, and the total number of branches determined for each organ. This concentration of GM6001 used here is the same as that used for tissue culture inhibition in our laboratory (Alexander et al., 2001; Chin and Werb, 1997); we observed no significant differences at higher concentrations of GM6001 (data not shown).
RNA isolation and RNAse protection assay (RPA)
We extracted total cellular RNA from lung tissue using RNAzol B (TEL-TEST, Inc.) according to the manufacturer’s protocol. RPA was performed on 10 μg of RNA using the In vitro Transcription Kit and the RPA Kit (Pharmingen) according to the manufacturer’s protocol. Briefly, a 1011 bp cDNA fragment from the protein-coding region of the mouse MT1-MMP gene was cloned into pT7Blue (Novagen) (Apte et al., 1997). After linearization of the plasmid with BamHI, T7 RNA polymerase was used to synthesize the 1011 bp antisense probe that was labeled with [α-32P]UTP. For synthesis of the MMP-2 antisense RNA probe, pSP65 was linearized with EcoRI, and SP6 RNA polymerase was used to generate and label the probe. A total of 1×106 cpm of radiolabeled probe was used to hybridize to 10 μg of total RNA extracted from the lungs. After the incubation, the hybridization mixture was treated with a mixture of RNAseA and RNAseT1. The protected hybridized RNA fragments were recovered by ethanol precipitation, resolved on 5% acrylamide, 8 M urea gels, exposed to Kodak film overnight, and the bands quantified using Quantity One software (Bio-Rad).
Histology
Mouse embryonic lungs were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin; sections were cut and standard histological techniques used for histology (hematoxylin and eosin, H and E). The trachea and lungs from 3-week-old mice were infused with 4% PFA at 20 cm water pressure overnight at 4°C before paraffin embedding.
In situ zymography
DQ™ gelatin, fluorescein conjugate (Molecular Probes) was reconstituted according to the manufacturer’s recommendations and diluted at 1:1 with 2% agar immediately before addition to cryostat sections of the lungs of newborn mice, followed by incubation at ambient temperature for up to 3 hours. This modification resulted in better localization of the gelatinase activity. Fluorescence intensity was monitored for 3 hours and lung sections from wild-type and Egfr−/− mice were photographed at equal time points. For inhibition of MMP activity, cryostat sections of the lung were incubated with 10 mM of 1,10-phenanthroline or control buffer for 1 hour prior to incubation with 1:1 dilution of DQ™ gelatin and 2% agar.
Gelatin gel zymography
Whole lung homogenates were lysed in 120 mM Tris-HCl buffer pH 8.7, 0.1% NP-40 and 5% glycerol. Insoluble aggregates and nuclei were removed by centrifugation and protein in the supernatant was quantified by standard protocols (Bradford). Samples (5 μg of protein) were added to non-denaturing loading buffer and separated in 10% SDS-polyacrylamide gels containing 0.02% gelatin. SDS was then removed by three 20 minute washes with 2.5% Triton X-100 before incubation for 24 hours at 37°C in the developing buffer (50 mM Tris-HCl pH 8, 5 mM CaCl2, 0.02% NaN3). Gels were then fixed and stained with 50% methanol and 10% acetic acid containing 0.3% w/v Coomassie Blue. MMP-2 and MMP-9 (gelatinase B) activity appeared as clear bands. Optical density of the clear bands was visualized using the Chemi-Doc gel documentation system (Bio-Rad) and quantified using Quantity One quantitation software (Bio-Rad). Results are shown as means and standard error of the mean. Statistical analysis were performed using the unpaired student’s t test, with significant differences considered as P<0.05.
In situ hybridization (ISH)
We used RNA probes prepared from plasmid vectors pSP65, pSP65 92b, pBSmo Sl-2, TRMII, p1011as, pSK T-1 and pTIMP-2 (Alexander et al., 1996; Apte et al., 1997) for MMP-2, gelatinase B, stromelysin-1, stromelysin-2, MT1-MMP, TIMP-1 and TIMP-2, respectively. The labeling of the probes and in situ hybridization (ISH) on prepared paraffin sections of lung tissue were carried out as previously described (Alexander et al., 1996; Apte et al., 1997; Chin and Werb, 1997). Briefly, the plasmids were linearized and [35S]UTP (1000 Ci/nmol, Amersham)-labeled probes were transcribed from the SP6 promoter using a transcription kit (Promega). The probes were fractionated with Sephadex G-50 (Pharmacia), precipitated with ethanol, mixed with hybridization mixture, and placed on the pretreated sections. The sections were incubated overnight at 55°C, washed in high stringency conditions, and dipped in autographic emulsion (Kodak NTB2). After exposure for 4–8 days, the emulsion was developed and the sections were counterstained with hematoxylin and mounted.
Statistics
All data are representative of at least three independent experiments with four to five mice in each in vivo experiment and are expressed as means±s.e.m. Significant differences (*), P≤0.05, are expressed relative to sham-treated control using the Student’s t-test. In vitro studies are representative of at least three independent experiments performed in triplicate, unless otherwise stated.
Results
Egfr signaling controls lung development by regulating MMP activity during lung embryogenesis
Previous work from this laboratory has shown that mice deficient in Egfr signaling exhibit abnormal lung branching (Miettinen et al., 1995). Because this phenotype resembles that seen in embryos treated with MMP inhibitors, we hypothesized that Egfr signaling controls expression of morphogenic MMPs in the lung. To test this hypothesis, we first examined the proteolytic activity present in the developing lungs of Egfr−/− mice and their littermate controls. We used in situ zymography (Karelina et al., 2000) on unfixed frozen lung tissue to detect and localize active gelatin-degrading enzymes. We confirmed that airway branching phenotype was abnormal in Egfr−/− mice and revealed that Egfr−/− lungs exhibit low gelatinase activity compared with wild-type lungs (Fig. 1). This activity was caused by metalloproteases because preincubation of tissue with the zinc chelator 1,10-phenanthroline completely abolished proteolysis in wild-type lungs (Fig. 1). Furthermore, whole lung extracts from newborn Egfr−/− mice showed a partial decrease in proMMP-2, but a more robust decrease in activated MMP-2 (Fig. 2). Lung homogenates of E12.5 and 13.5 lungs also showed a significant decrease in MMP-2 activity (data not shown). This observation was specific for MMP-2, as there were no differences in MMP-9 activation seen by zymography (Fig. 2). The MMP-9 present in newborn lung homogenates was most likely due to alveolar macrophages, which are known to secrete MMP-9 (Vu and Werb, 2000). These results raised the question of which MMPs are dysregulated in Egfr−/− lungs and at what level of regulation.
Fig. 1.
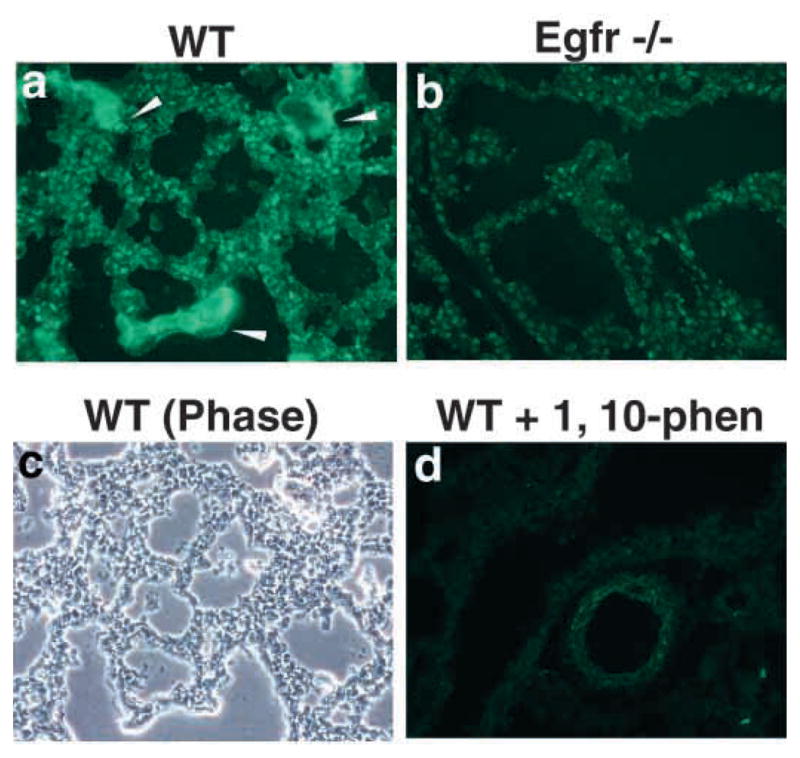
Analysis of activity of protease in lungs of Egfr−/− mice by in situ zymography. Cryostat sections of lungs from newborn wildtype (a,c,d) and Egfr−/− (b) after 3 hours of incubation with DQ gelatin. (a) Note gelatinolytic activity throughout the parenchyma and around the airway of wild-type lung (arrowhead), (d) which was inhibited in the presence of 1,10-phenanthroline. (c) Corresponding phase-contrast image from a.
Fig. 2.
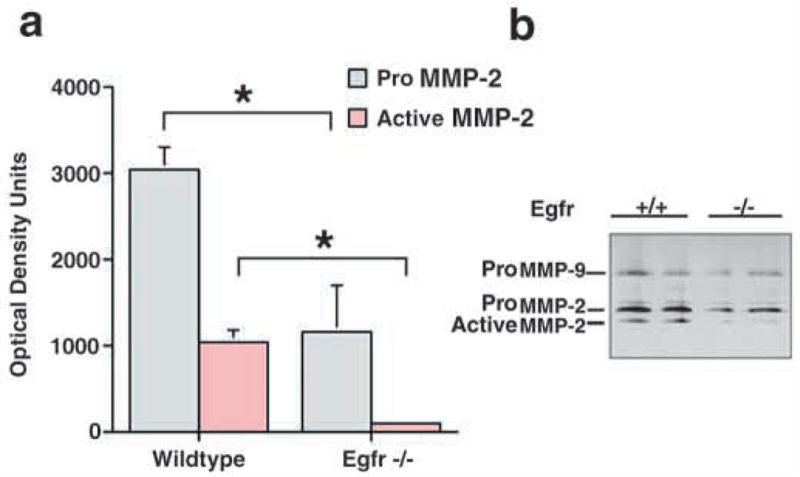
Inhibition of MMP-2 activation in Egfr−/− mice. Whole lung homogenates from newborn wildtype and Egfr−/− mice were analyzed by gelatin zymography. Quantification of zymograms (n=4) is shown in panel a, and the negative images of a representative zymogram in panel b. *P≤0.05 (Student’s t-test).
Inhibition of MMPs severely retards branching morphogenesis in the developing lung. The lung forms primary buds on the second day of embryonic stage E10.5. If MMPs are involved, then inhibition of MMPs should affect branching morphogenesis at this period of development. We injected timed-pregnant mice intraperitoneally with a broad, class-specific metalloproteinase inhibitor (GM6001) at 100 μg/g, or with carrier reagent (PBS), daily for three days starting at E10.5. Embryonic lungs were collected on E13.5, which corresponds to one day after the initiation of the pseudoglandular stage (E12.5–16.5) of lung development. Normal developing lung at E13.5 was characterized by elaboration of multiple epithelial branches into the hypercellular mesenchyme, shown by an arrow pointing to a freshly forming branch in Fig. 3c. Inhibition of MMPs resulted in severe retardation of branching in the developing lung, compared with sham-treated animals (Fig. 3). There was a lack of tertiary and fourth order branching of the endoderm that is normally seen by E13 in the developing lung. The cells forming the invaginating epithelium were also poorly organized and had lost their tight association with the mesenchyme. Furthermore, the mesenchyme in the embryos treated with GM6001 appeared poorly organized and hypoplastic compared with the sham-treated embryos. Slowing of lung branching in response to MMP inhibition was dose-dependent, as GM6001 given at 10 μg/g or 1 μg/g resulted in milder or no apparent branching defects, respectively (data not shown).
Fig. 3.

MMP function is required for normal branching morphogenesis. Timed-pregnant mice were treated with 100 mg/kg of GM6001 (b,d) or vehicle control (PBS) (a,c) starting at E10.5 for three days. (a,b) Equivalent sections from left lung of embryos treated by sham inhibition (a) or with MMP inhibitor (b), then stained with H and E. Dark boxes indicate the region of epithelial mesenchymal contact presented at higher magnification in c and d. The arrow in c indicates a newly forming branch. Arrowheads in b and d point to loose or poorly organized mesenchyme. Bars, 200 μm (a,b); 50 μm (c,d). Li, liver; Lu, lung; H, heart.
We next examined whether blockage of MMPs specifically inhibits branching in the developing lungs. Isolated embryonic lungs in culture are capable of branching ex vivo for several days in the absence of serum factors (Shannon et al., 1998). Inhibition of metalloproteinases using soluble GM6001 in the absence of serum resulted in retardation of branching by two-to threefold compared with the control (Fig. 4A,B). In addition, cells in the explant cultures treated with GM6001 were viable and displayed normal branching when returned to control media (data not shown).
Fig. 4.

(A) MMPs regulate lung bud branching in lung organ cultures. Numbers above the panels represent the number of days in culture. E11.5 lung buds were isolated from timed-pregnancies and placed in serum-free media in the presence or absence of MMP inhibitor (GM6001). Following four days growth in vitro, lung buds show retardation of branching in the presence of MMP inhibitor as compared to control. (B) Analysis of branching inhibition. Quantification by counting the number of newly formed secondary and tertiary branches in E10.5 (n=4) and E11.5 (n=3) whole lungs following four days of culture. *P≤0.05 (Student’s t-test).
MMP-2 and MT1-MMP are expressed during lung development
We next sought to determine which MMPs are expressed in embryonic lung. We examined expression of stromelysin-1 (MMP-3), stromelysin-2 (MMP-10), gelatinase A (MMP-2), gelatinase B (MMP-9), MT1-MMP (MMP-14), and their natural inhibitors, tissue inhibitor of metalloproteinases 1 (TIMP-1) and TIMP-2 in three well characterized stages of embryonic lung development (Warburton et al., 2000): embryonic stage/primary budding (9.5–11.5 days post-coitum; E9.5–11.5), pseudoglandular (E12.5–16.5), and canalicular (E17–18) stages by in situ hybridization (ISH). Expression of stromelysin-1 (MMP-3), stromelysin-2 (MMP-10), gelatinase B (MMP-9) or TIMP-1 mRNA were not detected in the epithelium or the mesenchyme at any stages (Fig. 5, absence of white grains; and data not shown). By contrast, we found abundant expression of mRNA for gelatinase A (MMP-2), MT1-MMP (MMP-14) and TIMP-2 in the embryonic lung at E11.5, E13.5, E18.5 (Fig. 5, arrows pointing to white grains; and data not shown). MMP-2 expression was confined to cells of mesenchymal origin (fibroblasts, cartilage, and endothelium) and was completely absent from lung epithelium (Fig. 6e,f and inset, absence of white grains). By contrast, MT1-MMP and TIMP-2 were expressed in both the epithelium and the mesenchyme, although the epithelium appeared to express lower message levels than the mesenchyme (Fig. 6a–d and insets). Incubation with mouse sense probes for MMP-2, MT1-MMP and TIMP-2 performed under parallel conditions showed no tissue binding (data not shown).
Fig. 5.
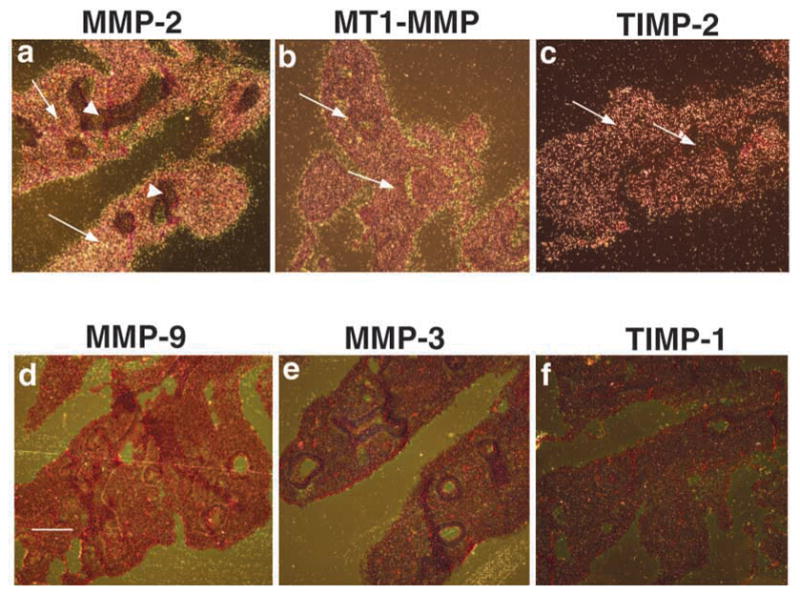
Constitutive expression of MMP-2, MT1-MMP and TIMP-2 mRNA during lung development. Lung sections from normal fetuses were taken at E12.5. For ISH analysis the following antisense probes were used: (a) MMP-2, (b) MT1-MMP, (c) TIMP-2, (d) MMP-9, (e) MMP-3, (f) TIMP-1. Note the presence of message in (a,b,c) and its absence in (d,e,f). Arrows point to the presence and arrowheads to the absence of message. Bar, 200 μm (a–f).
Fig. 6.
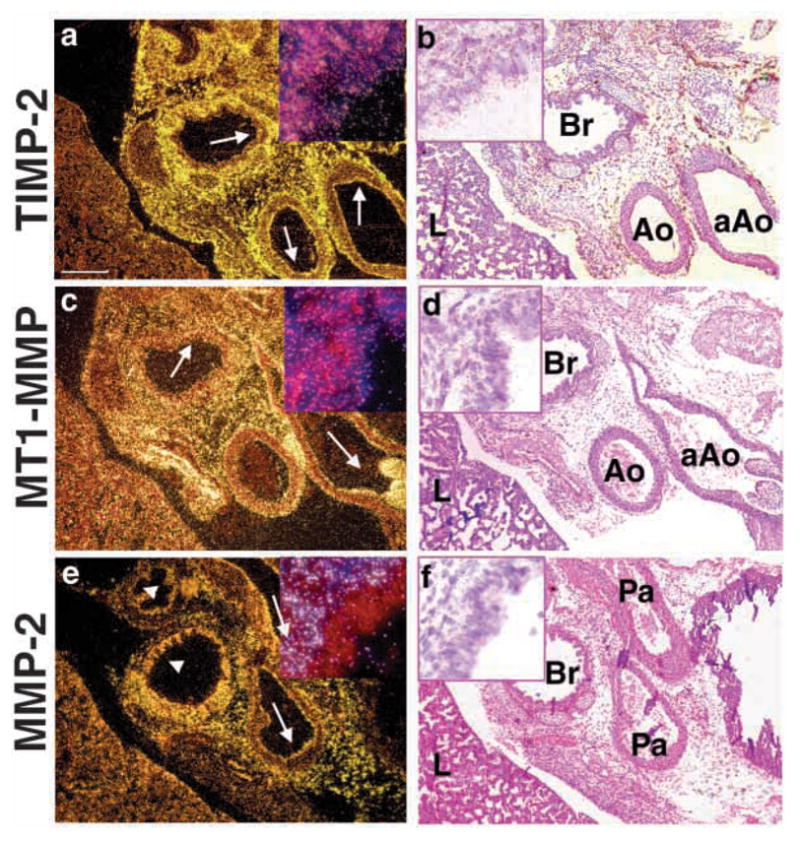
Localization of MMP-2 mRNA in the mesenchyme. Embryos from normal fetuses were taken at E18.5. For ISH analysis the following antisense probes were used: (a) TIMP-2, (c) MT1-MMP, (e) MMP-2. (b,d,f) H and E counterstain of darkfield images are shown. Note localization of TIMP-2 and MT1-MMP to the epithelia and mesenchyme (arrows), and absence of MMP-2 in the epithelia (arrowheads). Inset in each panel is 60× magnification of bronchial region from the corresponding image. aAo, ascending aorta; Ao, aorta; Br, bronchus; L, lung; Pa, pulmonary artery. Bar, 200 μm (a–f).
Egfr−/− lungs lack activated MMP-2 due to a deficiency in MT1-MMP gene expression
We next looked at the expression pattern of MMP-2, and MT1-MMP mRNAs by in situ hybridization. Interestingly, there was abundant expression of MMP-2 and TIMP-2, but little to no expression of MT1-MMP in Egfr−/− mice (Fig. 7; and data not shown). ProMMP-2 is proteolytically cleaved by MT1-MMP to yield active MMP-2 (Holmbeck et al., 1999). Because it is widely accepted that ISH is not a quantitative method for assessing mRNA in the tissue, we chose to use an RNAse protection assay (RPA) to quantify the mRNA level in 1- and 9-day-old lungs from wild-type and Egfr−/− mice. The RPA confirmed the ISH data obtained from embryonic lung showing that, while lungs from 1-day-old postnatal Egfr−/− mice express MMP-2 mRNA at levels similar to their littermate controls, much less MT1-MMP mRNA was detected in lungs of Egfr−/− mice (Fig. 8a). There was also a decrease in MMP-2 mRNA in 9-day-old Egfr−/− lungs compared with the wildtype. Egfr−/− mice rarely live beyond the first day of life and the occasional mice that live to a few weeks do not thrive, suffering from severe emaciation and dehydration. Thus, not surprisingly, we found less MMP-2 mRNA from these 9- (Fig. 8b) or 7-day-old (data not shown) Egfr−/− mice compared with the littermate controls. These data are consistent with our in situ data (Fig. 7; and data not shown). Thus, Egfr signaling is essential in lung organogenesis and for proper activation of MMP-2 through a mechanism that involves the expression of MT1-MMP.
Fig. 7.
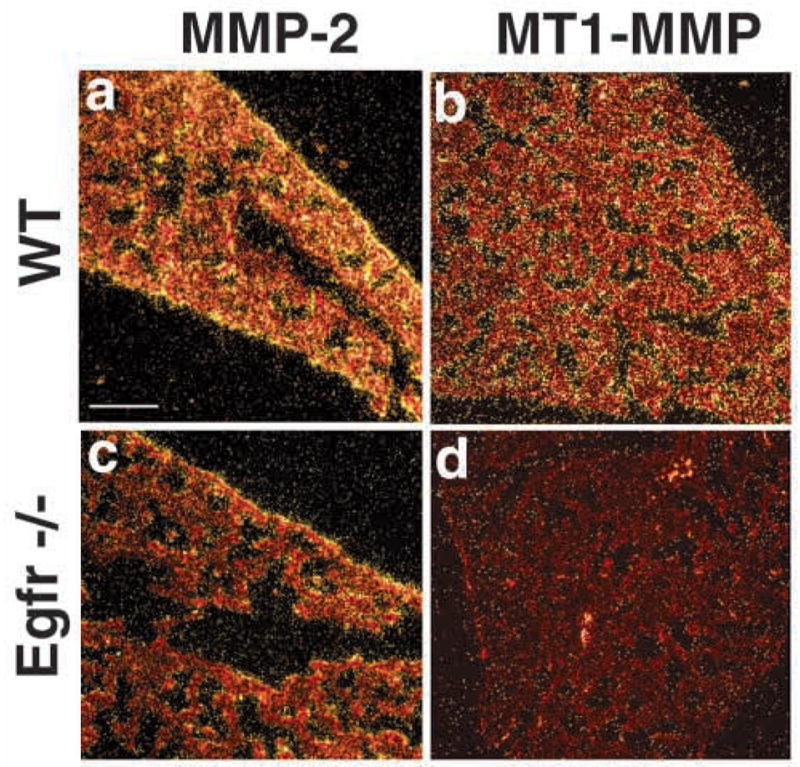
Decreased expression of MT1-MMP, but not MMP-2, mRNA in Egfr−/− mice. Lung from 18.5 dpc (E18.5) wildtype (a,b) or Egfr−/−(c,d) used to detect MMP-2 (a,c) and MT1-MMP (b,d) mRNA by in situ hybridization. Note the abundant presence of MMP-2 and MT1-MMP mRNA in a,b,c, and absence of MT1-MMP in d. Bar, 100 μm (a–d).
Fig. 8.

Quantification of MT1-MMP and MMP-2 mRNA expression by RPA. (a) There was little to no detectable protected MT1-MMP mRNA in the 1- or 9-day-old Egfr−/− lungs, while protected mRNA fragments were detected in the control littermate. (b) By contrast, MMP-2 mRNA was protected from degradation using the MMP-2 RNA probe with RNA from wild-type and Egfr−/− mice. (c) Egf regulates MT1-MMP expression in lung fibroblasts. Fibroblasts were isolated from E12 lungs produced by mating of Egfr+/− mice. Egfr−/− and wild-type lung fibroblasts were deprived of serum factors overnight, followed by stimulation with 100 ng of murine Egf or Fgf-2 for 16 hours. Total RNA was then isolated and used for quantification of MT1-MMP mRNA by RPA. Major protected bands were quantified and the amount of increase over baseline are as shown.
Egfr signaling directly regulates MT1-MMP gene expression in embryonic lung fibroblasts
We next asked whether Egfr signaling directly and/or specifically regulates MT1-MMP gene expression in the embryonic lung mesenchyme. E12 lung fibroblasts from wild-type, but not Egfr−/− mice stimulated with Egf for 16 hours upregulated MT1-MMP mRNA tenfold (Fig. 8c). Furthermore, Egf regulation of MT1-MMP was specific, since Fgf-2 failed to increase MT1-MMP message in the wild-type embryonic fibroblasts (Fig. 8c). RPA studies represent at least three independent experiments.
Newborn lungs from MMP-2−/− mice phenocopy abnormal lung branching in Egfr−/− mice
Our data point to a role of Egfr signaling for proper activation and expression of MMP-2 during lung development. To determine whether regulation of MMP-2 function is necessary for lung morphogenesis, we examined lungs from the mice lacking MMP-2 (Itoh et al., 1997). Although these mice are born apparently normal (Itoh et al., 1997), upon careful examination we observed that 15% of newborn MMP-2−/− mice gasped for breath and in all mice the lungs had abnormally large alveolar spaces, fewer septations and thinner interstitial tissue than in control littermates (Fig. 9), which is similar to, but slightly less severe than, the phenotype seen in Egfr−/− lungs.
Fig. 9.
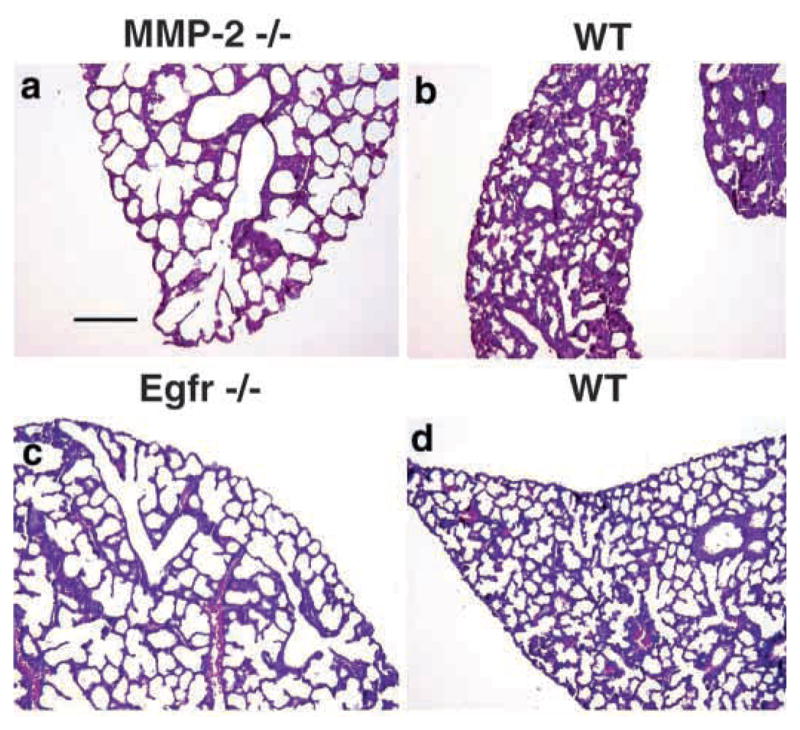
Lungs from newborn MMP-2−/− mice phenocopy branching defects in Egfr−/− mice. HandE stained sections from lungs of newborn (a) MMP-2−/−, (c) Egfr−/−, (b,d) their wild type littermate controls, respectively. Note that the distal airway phenotype of MMP-2−/− mice resembles that of the Egfr−/− mice. Bar, 200 μm (a–d).
Discussion
MMPs contribute to lung branching morphogenesis and alveolization
In the present study we have shown that signaling through Egfr and MMPs contributes to embryonic and postnatal lung development by acting in the same pathway. Furthermore signaling through the Egfr is necessary and sufficient for up-regulation of MT1-MMP in mouse lung fibroblasts. We have observed that MMP-2 and MT1-MMP are expressed constitutively in the embryonic lung, and play a crucial role in lung development. In support of our findings, other studies have shown expression of MMP-2 and MT1-MMP in rabbit and murine embryonic lungs (Fukuda et al., 2000; Reponen et al., 1992). Interestingly, despite reports of MMP-2 expression in rabbit epithelial cells (Fukuda et al., 2000), we found complete absence of MMP-2 mRNA by ISH or immunohistochemistry (data not shown) in the embryonic lung endoderm. MMP-2 was primarily expressed in the mesenchymal cells, and was completely absent from the proximal and distal airway epithelium. By contrast, MT1- MMP and TIMP-2 were expressed constitutively in both the embryonic mesenchyme and epithelial cells. Since MT1-MMP and TIMP-2 are required for MMP-2 activation (Caterina et al., 2000; Holmbeck et al., 1999), and are co-expressed during mouse embryogenesis (Apte et al., 1997) this would result in activation of MMP-2 both in the mesenchymal compartment and on the surface of epithelial cells. This is exactly where we observed gelatinase activity in situ.
Tubular structures such as lung follow a highly stereotyped and detailed process of invasion into the mesenchyme to form the final 3D structure of the organ. Interestingly, exogenous addition of collagenase to organ cultures inhibits lung branching (Ganser et al., 1991). However, there is no evidence that collagenase is expressed at this time in morphogenesis. Instead, our in vivo inhibition studies, in which we treated pregnant mice with GM6001, point to a positive role for MMPs in epithelial cell branching, because embryonic lungs failed to undergo secondary and tertiary branching.
What is the functional role of ECM-degrading enzymes during branching morphogenesis in vivo? We found that branch formation required functional MMPs. The simplest hypothesis is that MMPs are required for degradation of an ECM protein that is a necessary component for migration of epithelial cells during invagination. During development lung ECM is in a constant state of turnover. One role of MMP-2 could be to cleave ECM proteins such as elastin, laminin-5 or collagen types IV or VII, producing bioactive fragments, which would then induce migration of epithelial cells in vitro (Karelina et al., 2000; Koshikawa et al., 2000; Salo et al., 1999).
MT1-MMP is the downstream target of Egfr signaling
In this study we provide strong evidence for the role of MMPs in branching morphogenesis during embryonic lung development. Lung buds in mice are first formed from invagination of a single epithelial endoderm tube into the splanchnic mesoderm at E9.5 (Hogan, 1999; Warburton et al., 2000). The 3D growth of the primary lung buds may be controlled by the Shh and its downstream transcriptional molecules Gli2 and Gli3 (Bellusci et al., 1997a), whereas the pattern of secondary branch formation is most likely controlled by growth factors (Hogan, 1999; Warburton et al., 2000) and proteases as shown by our studies. Egfr and many of its ligands are abundantly expressed in the developing mouse lung, suggesting an autocrine and/or paracrine interaction in the developing mouse embryo (Warburton et al., 1992). Embryonic lung synthesizes several Egfr ligands and responds to Egf, Tgf-α and amphiregulin by precocious branching (Schuger et al., 1996; Warburton et al., 1992).
Most MMPs (e.g. MMP-2) are secreted as inactive zymogens. Therefore, their activators (e.g. MT1-MMP) may ultimately control ECM degradation during morphogenesis (Caterina et al., 2000). Our data point to MT1-MMP and its activation of MMP-2 as a major downstream target of signaling by this receptor tyrosine kinase that results in branching morphogenesis in lung development. We provide direct evidence for the role of MMP-2 during lung branching morphogenesis and show that MT1-MMP, a potent activator for the same enzyme is upregulated by an Egfr ligand in culture. Although expression of MMP-2 mRNA and protein was only slightly decreased in mice with a null mutation in Egfr, its activation was greatly compromised. However, MT1-MMP, which is required for cell surface activation of MMP-2, is downregulated in the absence of Egfr signaling. Since proMMP-2 can also be activated by other enzymes, it is not surprising that we found a small amount of active MMP-2 in the whole lung homogenates.
MMP-2 null mice phenocopy the Egfr−/− lung phenotype
If the major target of signaling through the Egfr is activation of MMP-2, then MMP-2−/− mice should have the same phenotype as Egfr−/− mice. Mice with null mutations of MMP-2 and Egfr showed strikingly similar abnormal distal airway branching and abnormal tissue architecture by histology. This raises the question of what substrates of MMP-2 are involved in airway development. MMP-2 cleaves many proteins in the interstitial space, basement membrane and on cell surfaces (Werb, 1997). Elastin and collagen remodeling are specific to lung and are necessary for alveolar development (Wright et al., 1999). Since active MMP-2 in the lung is a potent elastinolytic enzyme (Shipley et al., 1996), its absence in both Egfr−/− and MMP-2−/− lungs is expected to result in significant disturbance in alveolization postnatally. Indeed, in support of this hypothesis, we found severely impaired alveolization concomitant with abnormal elastin deposition in 15% of the MMP-2−/− mice (F.K. and K.R., unpublished). Thus elastin is one of the in vivo targets of MMP-2 in lung that is controlled by Egf signaling. These data provide further evidence for the coordinated signaling through Egfr and MMP-2 activation in lung branching morphogenesis.
However, does Egfr originally activate pathways other than MMP-2 activation? Although MMP-2−/− mice exhibited only mild respiratory compromise, these mice may have a spectrum of lung diseases that may only be manifest clinically after a pathological insult (Shapiro, 2000). By contrast, mice deficient in Egfr signaling die shortly after birth from respiratory failure and lack epithelial cell maturity (Miettinen et al., 1995). This suggests that there are additional downstream targets of Egfr signaling other than MMPs. Thus, while some features in branching morphogenesis are shared between these mice, lack of MMP-2 activation alone does not produce the lethal respiratory failure in Egfr−/− mice. It is unlikely that this is due to a direct role of MT1-MMP, because MT1-MMP null mice do not manifest respiratory failure (Holmbeck et al., 1999). More likely, these affects are due to the functions of Egfr signaling as a survival factor (Gibson et al., 1999; Sibilia et al., 2000).
Our data indicate that MMPs in vivo are downstream effectors of growth factors mediating growth and differentiation. However, the epithelial basement membrane and the ECM of the mesenchymal stroma must be degraded for proper branching and invagination of the epithelial cells to proceed. MMP-2 is expressed constitutively throughout the mesenchyme, although it may be regulated locally at the activation level by the Egfr through expression of MT1-MMP on both epithelial and mesenchyme. Although we suspect abnormal elastin deposition, we have not elucidated the full component of protein targets of these MMPs in the lung in vivo.
Membrane-bound MMP-2, in close association with its activator MT1-MMP, plays an important role, not only in branching of the epithelium, but also in the angiogenesis and cell invasion that are required for lung development (Haas et al., 1998; Haas et al., 1999). Although growth retardation in MT1-MMP−/− mice (Holmbeck et al., 1999) and poor branching morphogenesis are seen in MMP-2−/− mice, they do not exhibit a phenotype as severe as the in vivo inhibition of MMPs. This is reminiscent of previous in vitro studies showing that inhibition of MMPs can block endothelial tube formation and correlate MMP-2 activity with sprouting of endothelial tubes from pre-existing microvessel segments (Haas and Madri, 1999). Furthermore, microvascular endothelial cells upregulate expression of MMP-2 and MT1-MMP when grown on 3D type I collagen matrices (Haas et al., 1998). Whether other members of the MMP family act during lung morphogenesis in the absence of MMP-2 activation, or whether coordination of epithelial and vascular development is essential, are subjects for further study.
Acknowledgments
The authors thank Kim Kampman and Haifa Al-Khatib for assistance with lung techniques and Jeffrey Rosen for thoughtful discussions. We thank S. Itohara for providing breeding pairs of the MMP-2 null mice. This work was supported by a mentored clinician-scientist award (K08 HL03732) to F.K., and by grants from the Human Frontier Science Program (RG005/1999M), the National Institutes of Health (HD26732 and CA57621) and the Sandler Family Asthma Fund to Z.W.
References
- Alexander CM, Hansell EJ, Behrendtsen O, Flannery ML, Kishnani NS, Hawkes SP, Werb Z. Expression and function of matrix metalloproteinases and their inhibitors at the maternal-embryonic boundary during mouse embryo implantation. Development. 1996;122:1723–1736. doi: 10.1242/dev.122.6.1723. [DOI] [PubMed] [Google Scholar]
- Alexander CM, Selvarajan S, Mudgett J, Werb Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. J Cell Biol. 2001;152:693–703. doi: 10.1083/jcb.152.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte SS, Fukai N, Beier DR, Olsen BR. The matrix metalloproteinase-14 (MMP-14) gene is structurally distinct from other MMP genes and is co-expressed with the TIMP-2 gene during mouse embryogenesis. J Biol Chem. 1997;272:25511–25517. doi: 10.1074/jbc.272.41.25511. [DOI] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Gorivodsky M, Lonai P. Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc Natl Acad Sci USA. 1999;96:11895–11899. doi: 10.1073/pnas.96.21.11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development. 1997a;124:53–63. doi: 10.1242/dev.124.1.53. [DOI] [PubMed] [Google Scholar]
- Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997b;124:4867–4878. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- Caterina JJ, Yamada S, Caterina NC, Longenecker G, Holmback K, Shi J, Yermovsky AE, Engler JA, Birkedal-Hansen H. Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2 (Timp-2) gene alters proMMP-2 activation. J Biol Chem. 2000;275:26416–26422. doi: 10.1074/jbc.M001271200. [DOI] [PubMed] [Google Scholar]
- Chin JR, Werb Z. Matrix metalloproteinases regulate morphogenesis, migration and remodeling of epithelium, tongue skeletal muscle and cartilage in the mandibular arch. Development. 1997;124:1519–1530. doi: 10.1242/dev.124.8.1519. [DOI] [PubMed] [Google Scholar]
- Fondacci C, Alsat E, Gabriel R, Blot P, Nessmann C, Evain-Brion D. Alterations of human placental epidermal growth factor receptor in intrauterine growth retardation. J Clin Invest. 1994;93:1149–1155. doi: 10.1172/JCI117067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Ishizaki M, Okada Y, Seiki M, Yamanaka N. Matrix metalloproteinases and tissue inhibitor of metalloproteinase-2 in fetal rabbit lung. Am J Phys Lung Cell Mol Phys. 2000;279:L555–L561. doi: 10.1152/ajplung.2000.279.3.L555. [DOI] [PubMed] [Google Scholar]
- Galardy RE, Grobelny D, Foellmer HG, Fernandez LA. Inhibition of angiogenesis by the matrix metalloprotease inhibitor N-[2R-2-(hydroxamidocarbonymethyl)-4-methylpentanoyl)]-L-tryptophan methylamide. Cancer Res. 1994;54:4715–4718. [PubMed] [Google Scholar]
- Ganser GL, Stricklin GP, Matrisian LM. EGF and TGF alpha influence in vitro lung development by the induction of matrix-degrading metalloproteinases. Int J Dev Biol. 1991;35:453–461. [PubMed] [Google Scholar]
- Gibson S, Tu S, Oyer R, Anderson SM, Johnson GL. Epidermal growth factor protects epithelial cells against Fas-induced apoptosis. Requirement for Akt activation. J Biol Chem. 1999;274:17612–17618. doi: 10.1074/jbc.274.25.17612. [DOI] [PubMed] [Google Scholar]
- Gustafsson E, Fassler R. Insights into extracellular matrix functions from mutant mouse models. Exp Cell Res. 2000;261:52–68. doi: 10.1006/excr.2000.5042. [DOI] [PubMed] [Google Scholar]
- Haas TL, Davis SJ, Madri JA. Three-dimensional type I collagen lattices induce coordinate expression of matrix metalloproteinases MT1-MMP and MMP-2 in microvascular endothelial cells. J Biol Chem. 1998;273:3604–3610. doi: 10.1074/jbc.273.6.3604. [DOI] [PubMed] [Google Scholar]
- Haas TL, Madri JA. Extracellular matrix-driven matrix metalloproteinase production in endothelial cells: implications for angiogenesis. Trends Cardiovasc Med. 1999;9:70–77. doi: 10.1016/s1050-1738(99)00014-6. [DOI] [PubMed] [Google Scholar]
- Haas TL, Stitelman D, Davis SJ, Apte SS, Madri JA. Egr-1 mediates extracellular matrix-driven transcription of membrane type 1 matrix metalloproteinase in endothelium. J Biol Chem. 1999;274:22679–22685. doi: 10.1074/jbc.274.32.22679. [DOI] [PubMed] [Google Scholar]
- Hogan BL. Morphogenesis. Cell. 1999;96:225–233. doi: 10.1016/s0092-8674(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Yingling JM. Epithelial/mesenchymal interactions and branching morphogenesis of the lung. Curr Opin Genet Dev. 1998;8:481–486. doi: 10.1016/s0959-437x(98)80121-4. [DOI] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- Karelina TV, Bannikov GA, Eisen AZ. Basement membrane zone remodeling during appendageal development in human fetal skin. The absence of type VII collagen is associated with gelatinase-A (MMP2) activity. J Invest Dermatol. 2000;114:371–375. doi: 10.1046/j.1523-1747.2000.00886.x. [DOI] [PubMed] [Google Scholar]
- Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol. 2000;148:615–624. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer S, Okabe M, Hacohen N, Krasnow MA, Hiromi Y. Sprouty: a common antagonist of FGF and EGF signaling pathways in Drosophila. Development. 1999;126:2515–2525. doi: 10.1242/dev.126.11.2515. [DOI] [PubMed] [Google Scholar]
- Matsui R, Thurlbeck WM, Shehata EI, Sekhon HS. Two different patterns of airway branching regulated by different components of the extracellular matrix in vitro. Exp Lung Res. 1996;22:593–611. doi: 10.3109/01902149609070032. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Warburton D, Bu D, Zhao JS, Berger JE, Minoo P, Koivisto T, Allen L, Dobbs L, Werb Z, Derynck R. Impaired lung branching morphogenesis in the absence of functional EGF receptor. Dev Biol. 1997;186:224–236. doi: 10.1006/dbio.1997.8593. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R, Werb Z. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat Genet. 1999;22:69–73. doi: 10.1038/8773. [DOI] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12:3156–3161. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama J, Liu J, Mo R, Ding Q, Post M, Hui CC. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet. 1998;20:54–57. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L. Targeted expression of a dominant negative FGF receptor blocks branching morphogenesis and epithelial differentiation of the mouse lung. EMBO J. 1994;13:3296–3301. doi: 10.1002/j.1460-2075.1994.tb06631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reponen P, Sahlberg C, Huhtala P, Hurskainen T, Thesleff I, Tryggvason K. Molecular cloning of murine 72-kDa type IV collagenase and its expression during mouse development. J Biol Chem. 1992;267:7856–7862. [PubMed] [Google Scholar]
- Roman J. Extracellular matrix and lung inflammation. Immunol Res. 1996;15:163–178. doi: 10.1007/BF02918505. [DOI] [PubMed] [Google Scholar]
- Salo S, Haakana H, Kontusaari S, Hujanen E, Kallunki T, Tryggvason K. Laminin-5 promotes adhesion and migration of epithelial cells: identification of a migration-related element in the gamma2 chain gene (LAMC2) with activity in transgenic mice. Matrix Biol. 1999;18:197–210. doi: 10.1016/s0945-053x(99)00012-8. [DOI] [PubMed] [Google Scholar]
- Schuger L, Johnson GR, Gilbride K, Plowman GD, Mandel R. Amphiregulin in lung branching morphogenesis: interaction with heparan sulfate proteoglycan modulates cell proliferation. Development. 1996;122:1759–1767. doi: 10.1242/dev.122.6.1759. [DOI] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Shannon JM, Nielsen LD, Gebb SA, Randell SH. Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev Dyn. 1998;212:482–494. doi: 10.1002/(SICI)1097-0177(199808)212:4<482::AID-AJA2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Shapiro SD. Animal models for COPD. Chest. 2000;117:223S–227S. doi: 10.1378/chest.117.5_suppl_1.223s. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Doyle GA, Fliszar CJ, Ye QZ, Johnson LL, Shapiro SD, Welgus HG, Senior RM. The structural basis for the elastolytic activity of the 92-kDa and 72-kDa gelatinases. Role of the fibronectin type II-like repeats. J Biol Chem. 1996;271:4335–4341. doi: 10.1074/jbc.271.8.4335. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, Schlessinger J, Wagner EF. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell. 2000;102:211–220. doi: 10.1016/s0092-8674(00)00026-x. [DOI] [PubMed] [Google Scholar]
- van der Zee E, Jansen I, Hoeben K, Beertsen W, Everts V. EGF and IL-1 alpha modulate the release of collagenase, gelatinase and TIMP-1 as well as the release of calcium by rabbit calvarial bone explants. J Periodontal Res. 1998;33:65–72. doi: 10.1111/j.1600-0765.1998.tb02293.x. [DOI] [PubMed] [Google Scholar]
- Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- Warburton D, Seth R, Shum L, Horcher PG, Hall FL, Werb Z, Slavkin HC. Epigenetic role of epidermal growth factor expression and signalling in embryonic mouse lung morphogenesis. Dev Biol. 1992;149:123–133. doi: 10.1016/0012-1606(92)90269-m. [DOI] [PubMed] [Google Scholar]
- Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV. The molecular basis of lung morphogenesis. Mech Dev. 2000;92:55–81. doi: 10.1016/s0925-4773(99)00325-1. [DOI] [PubMed] [Google Scholar]
- Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Wright C, Strauss S, Toole K, Burt AD, Robson SC. Composition of the pulmonary interstitium during normal development of the human fetus. Pediatr Dev Pathol. 1999;2:424–431. doi: 10.1007/s100249900145. [DOI] [PubMed] [Google Scholar]
- Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Chapman HA, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]