

Abstract
Background & Aims
Differentiated pancreatic acinar cells expressing endogenous levels of mutant K-Ras do not spontaneously develop pancreatic ductal adenocarcinoma (PDAC). However, we hypothesized that acinar cells would develop PDAC in the presence of Ras activity levels mimicking those of human tumor cells.
Methods
We measured Ras activity in PDAC cells from mice and humans using a Raf pull-down assay. We compared the effects of acinar cell expression of mutant K-Ras at endogenous and elevated levels on Ras activity and on the development of PDAC.
Results
Ras activity was greatly elevated in PDAC cells compared to non-transformed cells expressing endogenous levels of mutant K-Ras. Expression of endogenous levels of mutant K-Ras in differentiated acinar cells resulted in moderately elevated Ras activity and in sparse murine pancreatic intraepithelial neoplasias (mPanINs) that did not spontaneously advance to PDAC unless the tumor suppressor p53 was simultaneous deleted. In contrast, expression of mutant K-Ras at higher levels generated Ras activity equal to that in PDAC. High Ras activity mimicking levels in PDAC led to acinar cell senescence and generated inflammation and fibrosis resembling the histological features of chronic pancreatitis (CP). With higher Ras activity in acinar cells abundant mPanINs formed and spontaneously progressed to both cystic papillary carcinoma (CPC) and metastatic PDAC.
Conclusions
There is an important relationship between Ras activity levels and the progression of PDAC. Sufficient Ras activity in pancreatic acinar induces several important pancreatic disease manifestations not previously reported and supports a potential direct linkage between CP, CPC and PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer death in the United States 1. Currently there are no effective non-surgical therapies for this disease. Therefore, considerable efforts have been made to develop animal models of PDAC useful for developing and testing new treatments. Recently mouse models have been described that faithfully recapitulate many important aspects of the human disease 2–4. These mouse models were developed by expression of activated mutants of K-Ras at endogenous levels from its native promoter in embryonic cells. These animals develop precursor lesions, mouse pancreatic intraepithelial neoplasia (mPanIN) 2, which progress over the course of 1–2 years into PDAC resembling the human disease. These models confirmed the potential role of K-Ras mutations as an initiator of PDAC and validated a model of PDAC progression starting from PanIN lesions and progressing to metastatic disease 2, 5.
It is well known that activating Ras mutations can induce either proliferation or senescence depending on the activity levels. In mammary gland, chronic low-level Ras induction results in tumor formation only after the spontaneous up-regulation of activated Ras and evasion of senescence checkpoints 6. In the pancreas of mice bearing targeted activating mutations at the endogenous K-Ras locus, a minor subset of cells display hyperproliferation and tumorigenesis 2, 4. In these models, the levels of Ras-GTP were significantly elevated in tumor lysates compared to non-tumor tissue expressing mutant K-Ras 7. These findings suggested that increased Ras activity may also play crucial roles in the process of pancreatic tumorigenesis.
In this study, we examined the effects of elevating Ras activity to different levels in pancreatic acinar cells in genetically engineered mice. We found that pancreatic acinar cells were transformed by endogenous levels of mutant K-Ras only in combination with genetic ablation of the tumor suppressor p53. Interestingly, Ras activity was greatly up-regulated in all PDAC cells examined whether mouse or human. Expression of high levels of mutant K-Ras in acinar cells using a transgenic approach led to levels of Ras activity similar to those found in PDAC cells, and caused mice to develop abundant mPanINs, cystic papillary carcinoma (CPC) and PDAC. High levels of Ras activity also led to wide-spread senescence of acinar cells and the development of profound inflammation and fibrosis resembling the histological features of human chronic pancreatitis (CP). Thus, a novel model expressing elevated levels of mutant K-Ras in pancreatic acinar cells developed CP, CPC and PDAC, providing the first evidence of a potential direct link between these diseases.
Results
Targeted Expression of Endogenous Levels of K-RasG12D in Pancreatic Acinar Cells Does Not Lead to PDAC Unless Accompanied by Loss of p53
To evaluate the effect of Ras activity specifically in pancreatic acinar cells, we utilized mice in which a tamoxifen-inducible Cre recombinase is expressed in differentiated acinar cells by a full-length mouse pancreatic elastase I gene promoter (Ela-CreERT)8. These mice exhibit tamoxifen-independent Cre activity after birth specifically in acinar cells with high levels of elastase expression, as has been fully described previously 8. Therefore, in the absence of tamoxifen Cre activity was restricted to well differentiated acinar cells. These mice were crossed with LSL-KRasG12D mice which express mutant K-RasG12D gene from the endogenous K-Ras promoter after Cre recombination9. Double-transgenic mice (LSL-KRas/Ela-CreERT) in the absence of tamoxifen developed typical mPanIN-1 lesions surrounded by limited focal fibrosis beginning within 2–3 months (Figure 1A). The mPanINs occurred infrequently in younger animals (~1 or 2 per section), but their number increased moderately over time (Figure 1B). These lesions were similar to those described when developmental promoters were used to activate expression of endogenous levels of mutant K-Ras 2. However, unlike what was observed using developmental promoters, these mPanIN-1 lesions did not progress to higher grade mPanINs or PDAC within 1.5 years of age (15/15). Yet, when the LSL-KRas/Ela-CreERT mice were further crossed with a conditional p53 deletion mice (LSL-KRas/p53f/f/Ela-CreERT), most of these mice developed PDAC within ~6 months (17/30) (Figure 1C). Therefore, acinar cells are readily transformed by endogenous expression of mutant K-Ras, but only after a second genetic hit.
Figure 1. Targeted expression of K-RasG12D to pancreatic acinar cells at endogenous levels induced non-progressing mPanIN-1 lesions unless p53 was deleted.
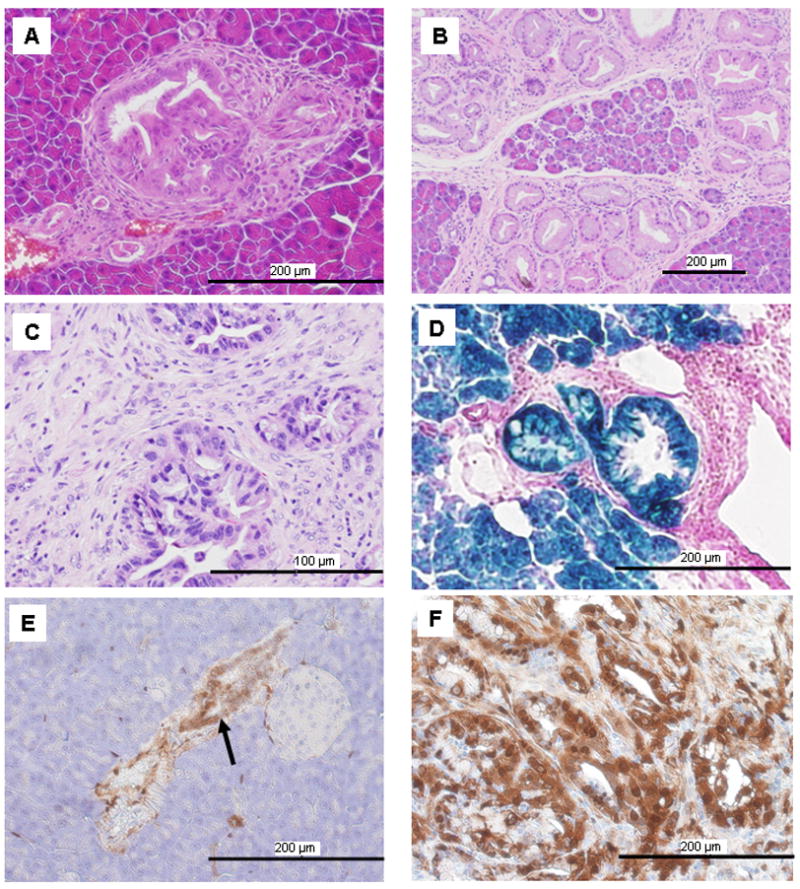
(A) mPanIN lesions developed in a 2-month-old LSL-KRas/Ela-CreERT mouse. (B) The number of mPanINs increased as the mice aged but did not progress (15 months old). (C) Mice (LSL-KRas/p53f/f/Ela-CreERT) which possessed endogenous expression of mutant K-Ras and p53 deletion readily developed PDAC (5 months old). (D). For lineage tracing, LSL-KRas/Ela-CreERT mice were further crossed with Rosa26 reporter mice such that cells of acinar lineage stained for β-galactosidase activity (blue). All mPanINs cells stained blue. (E). mPanIN cells (arrow) had elevated levels of p-Erk compared to surrounding acinar cells in a 4-month-old LSL-KRas/Ela-CreERT mouse detected using an anti-p-Erk antibody. (F). Cancer cells in PDAC developed from LSL-KRas/p53f/f/Ela-CreERT mice (5 months old) also showed strong p-Erk staining.
To determine the cellular origin of the mPanIN lesions in this model, these mice were further crossed with Rosa26R reporter mice (R26/LSL-KRas/Ela-CreERT). In these triple transgenic mice, mPanIN cells showed β-galactosidase activity, indicating the acinar lineage of mPanIN lesions (Figure 1D). We also observed elevated levels of p-Erk in mPanIN (Figure 1E) and PDAC cells (Figure 1F) compared to acinar cells which expressed endogenous levels of mutant K-Ras (Figure 1E). These data suggested that Ras activity was spontaneously up-regulated during PDAC development.
Elevated Ras Activity in PDAC Cells Can Be Mimicked by Transgenic Expression of Mutant K-Ras
We next examined the level of Ras activity in mouse models and human PDAC cell lines. Ras activity was moderately elevated in most of the acinar cells in pancreatic tissues expressing a single copy of mutant K-Ras (Figure 2A), similar to what was previously reported using a developmental promoter 2. In comparison, Ras activity was much more highly elevated in PDAC cell lines derived from tumors formed in LSL-KRas/p53f/f/Ela-CreERT mice (Figure 2A). Likewise, signaling molecules downstream from Ras, including p-Erk, were also more highly elevated in tumor cells than in non-transformed cells with endogenous levels of mutant K-Ras. Human PDAC cell lines with K-Ras mutations also exhibited very high Ras activity (Figure 2B). These data further support the hypothesis that Ras activity is spontaneously up-regulated during the development of PDAC, as has previously been suggested 2.
Figure 2. Transgenic expression of mutant K-Ras mimicked the elevated Ras activity levels found in pancreatic cancer cells.
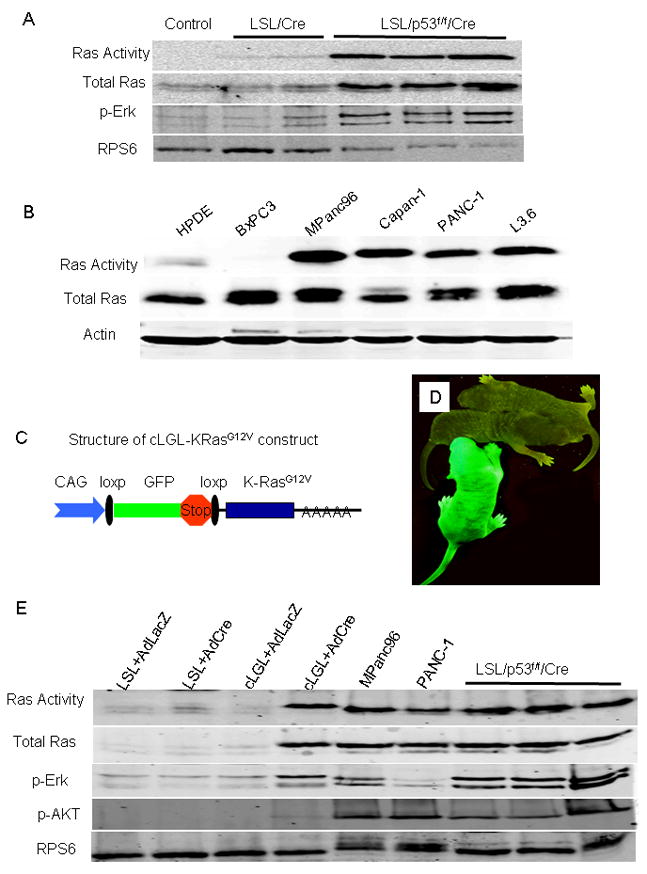
(A) Total Ras activity and p-Erk increased in PDAC cell lines isolated from LSL-KRas/p53f/f/Ela-CreERT (LSL/p53f/f/Cre) mice compared to untransformed pancreas tissue lysate from LSL-KRas/Ela-CreERT (LSL/Cre, 5 months old) and wild type control (control) mice. Deletion of p53 alone had no effect on Ras activity (data not shown). Ribosomal protein S6 (RPS6) was detected as a protein loading control. (B) Ras activity in human PDAC cells with K-Ras mutations (MPanc96, Capan-1, PANC-1 and L3.6) was significantly higher than that in immortalized human pancreatic duct cells (HPDE) and PDAC cells without K-Ras mutation (BxPC3). (C) Structure of the cLGL-KRasG12V ectopic expression transgene. K-RasG12V was engineered following a human CMV and chicken-β-actin chimeric promoter (CAG) and blocked by the proximal insertion of a loxp-green fluorescent protein (GFP)-stop-loxp cassette. (D) GFP was expressed in the whole body of cLGL-KRasG12V mouse (Green) but not in litter-mate controls (Gray). (E) Acini isolated from LSL-KRasG12D and cLGL-KRasG12V mice were infected either with a control adenovirus (AdLacZ) or with an adenovirus expressing Cre (AdCre). Ras activity was analyzed by Raf pull-down and compared with those in human PDAC cell lines or mouse PDAC cell lines isolated from LSL-KRas/p53f/f/Ela-CreERT (LSL/p53f/f/Cre) mice. Total Ras, p-Erk, p-AKT and RPS6 were measured in Western blots from sample aliquots before the pull-down assay.
To directly study the effects of higher Ras activity, we developed a new transgenic model in which K-RasG12V was engineered following a human CMV and chicken-β-actin chimeric promoter (CAG) 10 and blocked by the proximal insertion of a loxp-green fluorescent protein (GFP)-stop-loxp cassette (cLGL-KRasG12V; Figure 2C). Transgenic mice carrying this construct expressed GFP in the whole body (Figure 2D) before Cre recombination. The levels of Ras activity were compared between freshly prepared acinar cells isolated from LSL-KRasG12D and cLGL-KRasG12V mice in vitro by infecting the cells with a Cre-bearing adenovirus (AdCre). Under these conditions, the Ras activity measured in the acinar cells from the cLGL-KRasG12V mice after Cre-mediated recombination was higher than that from LSL-KRasG12D mice but was comparable to that observed in human and mouse PDAC cell lines (Figure 2E). Likewise, levels of p-Erk and p-AKT, indicators of Ras pathway activity, were also comparable between the cLGL-KRasG12V cell and human PDAC cells, but higher than those observed in the LSL-KRasG12D cells (Figure 2E). Thus, transgenic expression of mutant K-Ras achieved levels of Ras activity and down-stream signaling that was similar to those observed in mouse and human PDAC cells.
Higher Levels of Acinar Cell Ras Activity Induce Inflammation and Fibrosis Which Mimics the Histological Features of Human CP
To study the effects of a higher level of Ras activity in the pancreas, cLGL-KRasG12V mice were crossed with the Ela-CreERT mice and pups were allowed to develop in the absence of tamoxifen treatments. Five days after birth, the pancreata from double-transgenic mice showed few obvious differences in size, shape or color from littermate controls. Histologically these pancreata showed some signs of ADM, but were otherwise normal (Figure 3A). From 6 days, the pancreata from double-transgenic mice became firmer and bigger than those of the littermate controls. In adult mice, the pancreata became smaller than those of controls (Supplementary Figure 1). Histologically, changes in the pancreas in these mice as they aged involved a progression from normal pancreas to extensive fibrosis resembling CP (Figure 3A–3C). Chronic pancreatitis was characterized by loss of acinar cells, acinar-to-ductal metaplasia (ADM), leukocyte infiltration and replacement by stroma with collagen deposition (Supplementary Figure 1C). Cells observed within the stroma displayed pronounced smooth muscle actin (SMA) staining (Figure 3D), which suggested that they were activated stellate cells 11.
Figure 3. High levels of Ras activity in pancreatic acinar cells led to the development of CP.
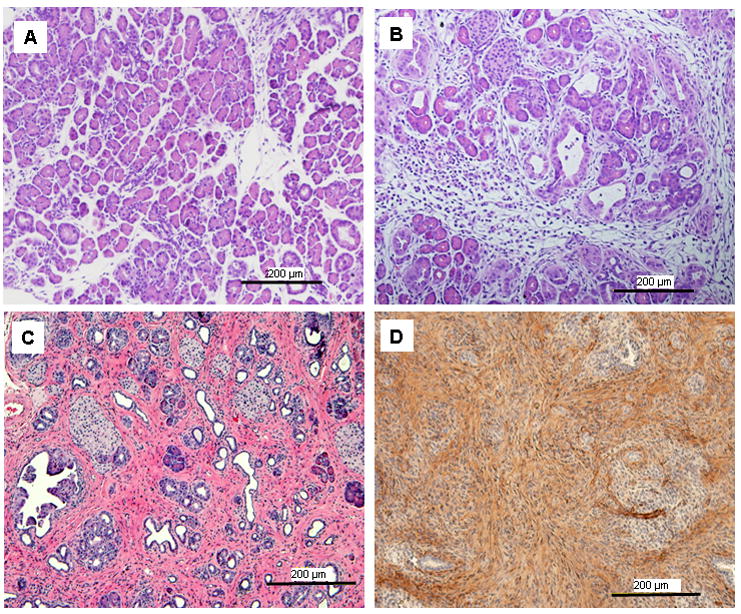
(A) At 5 days after birth, pancreata from cLGL-KRas/Ela-CreERT mice showed normal histology except for occasional sites of ADM. (B) At 9 days after birth, acinar cells were depleted from many areas with the development of fibrotic stroma. (C) In adult mice (2 months), the fibrosis expanded with abundant intercellular collagen deposition. (D) The stromal compartment of the pancreas (2 weeks old) showed abundant SMA expression.
The loss of acinar cells that occurred in this model was associated with the expression of senescence-associated β-galactosidase (Figure 4A), with little evidence of apoptosis (Figure 4B). DEC1, another marker of senescence12, was dramatically increased early in this model (Figure 4C). Tumor suppressor genes p53, p15 and p16, which are known to be involved in oncogene induced senescence, were also elevated (Figure 4C and 4D). Senescent cells induced by oncogenic Ras in vitro have been reported to develop a senescence-associated secretory phenotype (SASP) which results in the secretion of inflammatory cytokines13. We found in this in vivo model that high levels of Ras activity induced increased expression on IL-1B, IL-6, Gro-α and GM-CSF (Figure 4F).
Figure 4. Elevated Ras activity in pancreatic acinar cells caused acinar cell senescence and stellate cell activation.
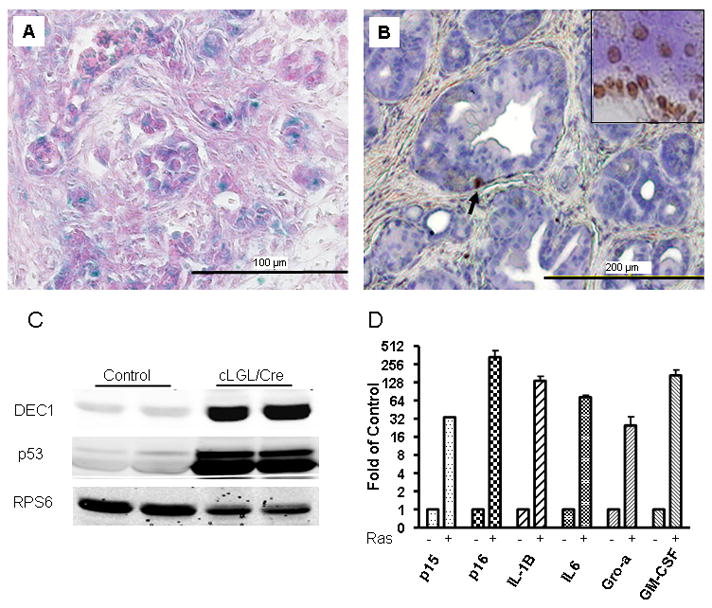
(A) In adult cLGL-KRas/Ela-CreERT mice (2 months old) many acinar cells stained positive for senescence-associated β-galactosidase. (B) Few pancreatic acinar cells showed apoptosis (arrow) as indicated by TUNEL assay (2 months old). DNase-treated tissue served as a positive control (insert). (C) DEC1 and p53 was up-regulated in cLGL-KRas/Ela-CreERT (cLGL/Cre) mice (2 months old) as measured by Western blot. D. Quantative RT-PCR showed that p15, p16, IL-1B, IL-6, Gro-α and GM-CSF were upregulated (shown in log2 scale) in cLGL-KRas/Ela-CreERT mice (1 month old) as compared with wild type littermate controls.
Elevated Levels of Ras Activity in Acinar Cells Results in Rapid Development of mPanIN, Cystic Papillary Carcinoma and PDAC
In the background of chronic pancreatitis, we observed the formation of multifocal ADM in the pancreas from all (100%) cLGL-KRasG12V/Ela-CreERT mice (Figure 5A). In younger mice, we observed primarily mPanIN-1 (Figure 5B) and occasionally mPanIN-2 lesions (Figure 5C). As the animals aged, there was an increase in the level of dysplasia and in the frequency of mPanIN-3 lesions (Figure 5D). Three out of 20 mice developed non-invasive cystic papillary carcinoma, which were composed of large cystic lesions (>1.0 mm) lined with highly dysplastic ductal epithelial cells forming finger-like papillae (Figure 5E). The frequencies of mPanIN lesions of different histological grades developed by different age groups are shown in Figure 5F. ADM and mPanINs in these mice exhibited typical characteristics similar to what were observed in other models (Supplementary Figure 2A–2F)2.
Figure 5. High levels of Ras activity caused acinar-to-ductal metaplasia and led to the formation of mPanINs and cystic papillary carcinoma.
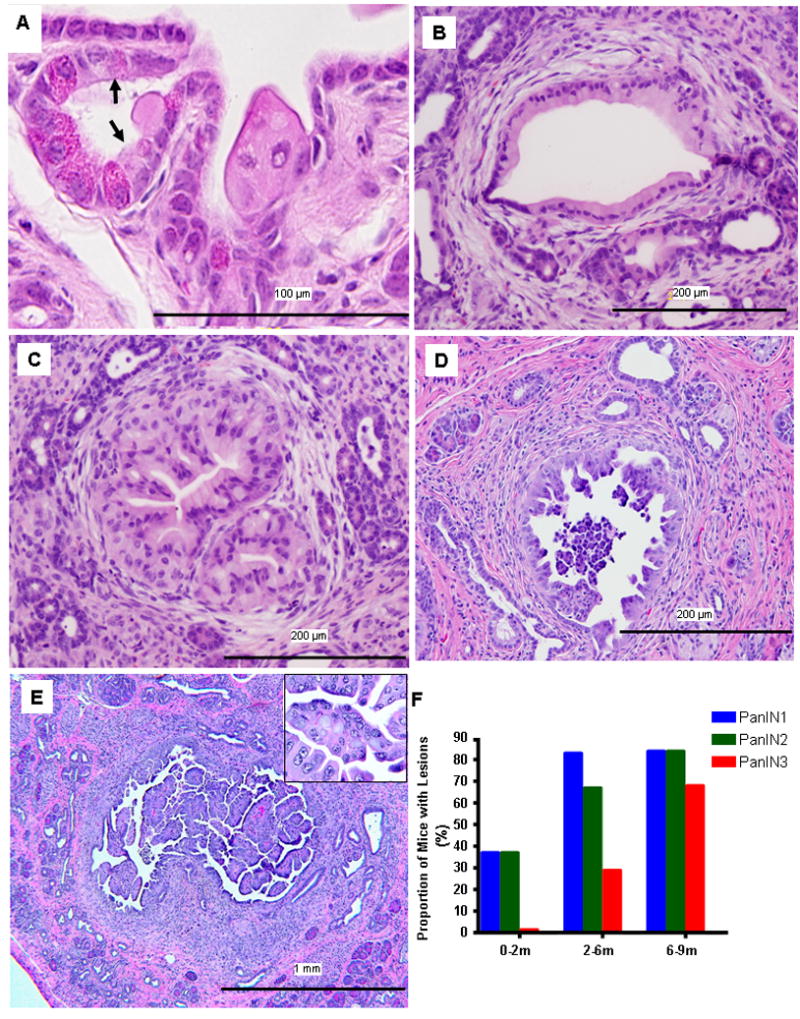
(A) In cLGL-KRas/Ela-CreERT mice (2 months old) acinar-to-ductal metaplasia (arrows) were observed. (B–E) Transgenic expression of mutant K-Ras induced typical mPanINs which progressed over time including mPanIN-1 (B, 2 months), mPanIN-2 (C, 4 months), and mPanIN-3 (D, 6 months) lesions and cystic papillary carcinoma (E, 7 months). (F) The grade of the mPanIN lesions present in the pancreas of transgenic animals progressed with age. Each group consisted of ~20 animals.
At 4 months of age, one mouse developed PDAC (Figure 6A) with metastasis to the liver (Supplementary Figure 2G) and lungs (data not shown). By 6 months of age, 29% (7/24) of mice developed PDAC. At 9 months, the incidence for PDAC increased to 50% (10/20). Metastatic PDAC cells were also identified in peripancreatic lymph nodes (Figure 6B). Some mice showed extensive tumor invasion into the diaphragm (Figure 6C). All tumors were adenocarcinomas composed of neoplastic glands, similar to those seen in human PDAC. No staining of the acinar cell marker, amylase, was detected by immunohistochemistry (Supplementary Figure 2H). The tumors were moderately to poorly differentiated, with 10–40% desmoplastic tumor stroma. Negative staining for GFP supported that the PDAC cells originated from pancreatic acinar cells (Figure 6D).
Figure 6. Invasive and metastatic PDAC developed spontaneously from pancreatic acinar cells with levels of Ras activity that mimicked those of human PDAC.
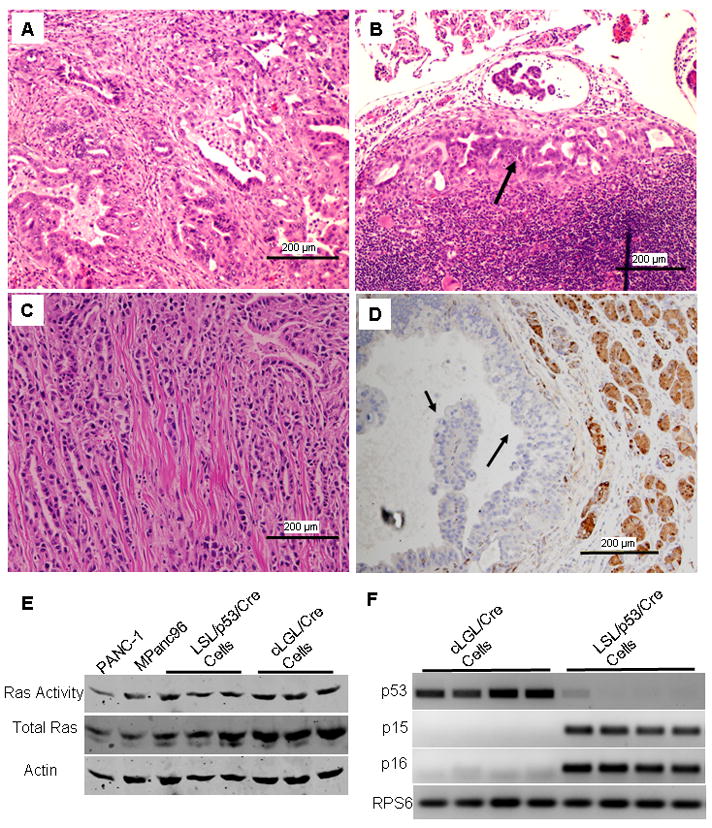
(A) PDAC developed in cLGL-KRas/Ela-CreERT mice (4 months). (B) Metastatic cancer cells were observed in the lymph nodes. (C) Some PDAC invaded the diaphragm. (D) GFP expression was lost in cancer cells, suggesting they were of pancreatic acinar origin. (E) Ras activity was measured in three independent cancer cell lines developed from LSL-KRas/p53f/f/Ela-CreERT (LSL/p53/Cre Cells) mice and cLGL-KRas/Ela-CreERT (cLGL/Cre Cells) and compared with those found in human PDAC cells. (F) RT-PCR measurement showed that cancer cells from cLGL-KRas/Ela-CreERT (cLGL/Cre Cells) mice lost expression of p15 and p16 but retained p53. In contrast, cancer cells from LSL-KRas/p53f/f/Ela-CreERT (LSL/p53/Cre Cells) mice lost p53 expression but retained intact p15 and p16.
To further evaluate the levels of Ras activity in PDAC cells developed from different models, we measured Ras activity in cancer cell lines developed from LSL-KRas/p53f/f/Ela-CreERT and cLGL-KRas/Ela-CreERT mice as wells as human PDAC cell lines (Figure 6E). We observed comparable levels of Ras activity in each of these cancer cell lines, suggesting that Ras activity progressed to peak levels in PDAC cells even when the cancers were initiated with endogenous levels of mutant K-Ras expression. All four independent cancer cells from cLGL-KRas/Ela-CreERT mice lost expression of p15 and p16 but retained p53 (Figure 6F). These p53 were verified as wild-type by DNA sequencing. Conversely, cancer cells from LSL-KRas/p53f/f/Ela-CreERT mice lacked p53 expression as expected, but retained intact p15 and p16 (Figure 6F).
Discussion
In this study, we investigated the importance of Ras activity levels in the development of PDAC. We demonstrated that the level of Ras activity in PDAC cells, whether mouse or human, is highly elevated. We confirmed that adult pancreatic acinar cells are largely refractory to endogenous levels of mutant K-Ras 4, 14, 15 and showed for the first time that this resistance can be readily overcome by deletion of p53. Furthermore, we observed that levels of Ras activity that mimicked the levels found in PDAC cells had profound effects on acinar cells that have not been reported previously, including the spontaneous development of CP, CPC and PDAC. Therefore, our data indicate that the levels of Ras activity are of great importance in the development of pancreatic diseases.
It was previously reported that levels of Ras-GTP were significantly elevated in bulk tumor tissues from mice expressing endogenous levels of K-Ras, although it was unclear if the elevated activity reflected increased signaling in cancer cells or an alteration in the balance of cell types in the tumors 7. In the current study, we identified the cancer cells themselves as the source of this high Ras activity. We also observed that both mouse and human PDAC cells possessed greatly elevated Ras activity compared to untransformed cells expressing endogenous levels of mutant K-Ras. When these high levels of Ras activity were directly mimicked by transgenic expression of mutant K-Ras in pancreatic acinar cells, we observed the development of senescence, diffuse CP, ADM, multifocal mPanINs and a high incidence of PDAC at a relatively early age. These data support the concept that a threshold of Ras activity is important for the induction of cellular transformation and that this level can be achieved spontaneously in models with endogenous levels of mutant K-Ras expression 6 or directly by expression of higher levels of mutant K-Ras.
The mechanisms responsible for the stochastic events resulting in the elevation of Ras activity in cells expressing mutant K-Ras from the endogenous promoter are currently unknown. It may be partly explained by the common phenomenon of amplification of the mutant K-Ras allele 7. However, other alterations in signaling pathways may also contribute to the increased Ras activity independently or in addition to K-Ras mutation. For example, it has also been observed that combining low levels of expression of mutant K-Ras with other stimuli known to increase Ras activity such as transforming growth factor-α (TGFα) 16 or cholecystokinin (CCK)4 leads to much more profound phenotypes. This supports the concept that up-regulated Ras pathway activity by extrinsic stimuli may also play an important role in transformation.
The current study confirmed previous observations that low endogenous levels of Ras activity are insufficient to transform fully differentiated pancreatic acinar cells 4, 14, 17. However, it also shows for the first time that combination of p53 deletion with endogenous levels of mutant K-Ras in acinar cells readily leads to PDAC. Furthermore, we observed that adult acinar cells were spontaneously transformed by elevated levels of mutant K-Ras expression that resulted in levels of Ras activity similar to those observed in PDAC cells. Taken together, these data support that pancreatic acinar cells can be a cell origin of PDAC providing elevated Ras activity and loss of tumor suppressor genes occurs.
In two previous studies in which endogenous mutant K-Ras expression were initiated by elastase4 or pdx1 promoters18, chronic pancreatitis induced by secretagogue administration were found to accelerate PDAC formation. This study differs from previous ones in that higher levels of Ras activity drove both chronic pancreatitis and spontaneous PDAC in the absence of additional treatments. Thus, Ras activity above a threshold is sufficient for both inflammatory and transforming responses. Of note, the treatments commonly utilized to induce pancreatitis also activate Ras19. Therefore, one possible explanation for the previous observations is that the treatments used to generate inflammation themselves elevated the level of Ras activity above a threshold leading to the observed effects.
These data support a model in which there are two significant barriers to PDAC development, as have been previously proposed in mammary cancer 6, 20. One barrier is the elevation of levels of Ras activity beyond a critical threshold. This barrier can be overcome either by increased extrinsic Ras activators, such as co-expression of TGF-α 21 or administration of CCK 4, 18, or, as in the current study, by elevated expression of mutant K-Ras. Yet, even with high levels of Ras activity there remains a latency of several months before tumors form. Furthermore, elevated Ras activity primarily drives cellular senescence rather that tumorigenesis. Thus, another barrier, the loss of tumor suppressors, is required for evasion of oncogene-induced senescence and full transformation. As shown here and other studies, deletion or mutation of p53 22, p16 7 or p19 23 can dramatically accelerate tumor formation. These two barriers are not independent. Loss of tumor suppressors is permissive for cells with spontaneously up-regulated Ras activity to evade oncogene-induced senescence. It also appears that the spontaneous loss of tumor suppressors is accelerated by high levels of Ras activity, likely due to the associated increase in genetic instability 24.
Another major finding in this study was that Ras activity at levels similar to those observed in PDAC cells drove the development an inflammatory phenotype similar to the histological features of human chronic pancreatitis. This observation raises an issue concerning the commonly observed presence of K-Ras mutations in CP. It has been well recognized that K-Ras mutations are present in nearly all PDAC 25, 26. However, it is less well appreciated that K-Ras mutations are also found in nearly 30–40% of CP patients 27. The biological significance of K-Ras mutations in CP has not been understood. One popular hypothesis is that prolonged inflammatory conditions of CP increase the probability of gene mutations, as has been reported28. However, K-Ras mutations have also been observed in hyperplastic ducts within normal pancreas29. Therefore, it is likely that K-Ras mutations can occur prior to the development of CP. In the current study, we observed that elevation of Ras activity in pancreatic acinar cells led directly to CP. Thus, our study provides evidence of a potential functional relationship between K-Ras mutations and CP in which mutant K-Ras is a cause rather than a secondary effect of CP. The present study also suggests that the activity level, rather than the presence of mutations, is the critical parameter. It is intriguing to speculate that the inflammatory response observed in this study may have been associated with the induction of senescent cells by high levels of Ras activity. Senescent cells are known to secrete myriad factors associated with inflammation 13. This animal model should be useful for future studies to elucidate the senescence-associated secretory phenotype and the development of fibrosis in vivo. It is noted that not all pancreases of CP patients possess K-Ras mutations. However, it is known that the Ras signaling pathway is activated by several stimuli known to cause CP in mouse models including CCK treatments19, expression of Cox-230, and expression of TGFα 31. Thus, we speculate that increases of Ras activity in pancreatic acinar cells brought about by various treatments may mediate the development of fibrosis and inflammation even in the absence of K-Ras mutations. Whether these concepts apply to the human disease will require further investigation.
We also observed that acinar cells gave rise to CPC in the mouse model with higher levels of Ras activity. This is the first evidence that acinar cells can give rise to this pathological condition. In mouse models, cystic papillary neoplasms are defined as large (>1 mm) cystic structures composed of usually papillary, noninvasive epithelial proliferations with varying degrees of cellular atypia. These lesions resemble human intraductal papillary mucinous neoplasms (IPMN) 4, 32 which are considered an important precursor lesion to PDAC33. In a recent study in transgenic mice, it was observed that combination of expression of mutant K-Ras with over-expression of TGFα were able to lead to the development of structures resembling IPMN19. In that study, it was suggested that the CPCs did not originate from pancreatic acinar cells based on the observation that the cells in the CPCs did not express acinar specific genes. However, in the current study, we observed that acinar cells rapidly lost differentiated gene expression upon metaplasia such that it is difficult to know the cell of origin without lineage tracing.
In summary, elevated Ras activity plays crucial roles in the development of pancreatic cancer and provides a possible direct linkage between CP, CPC and PDAC. Based on our observations and taking into account observations from other models, we propose a scheme to explain the role of Ras activity in pancreatic disease progression (Figure 7). According to this mechanistic view, two key obstacles to the formation of PDAC are achievement of a high level of Ras activity and the loss of tumor suppressors. High levels of Ras activity also generate an inflammatory response resembling CP that likely accelerates genetic changes. The animal model described in this study will provide a powerful tool for further investigations of the mechanisms underlying chronic pancreatitis, pancreatic cancer and the transition between these diseases.
Figure 7. Ras activity levels control the development of pancreatic diseases.
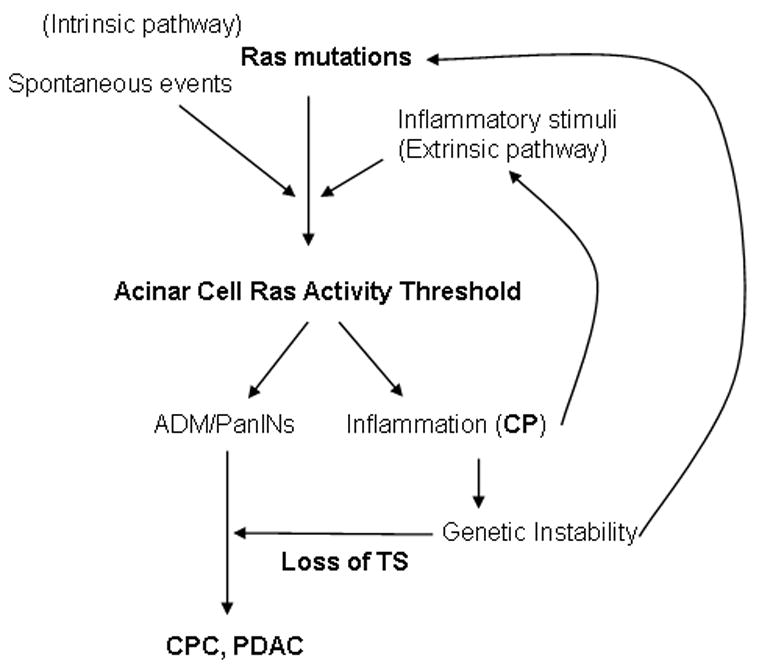
For the initiation of pancreatic diseases, Ras activity levels must be elevated beyond a threshold. This can be accomplished either by high levels of extrinsic Ras activators (extrinsic pathway) or by intrinsic alterations of Ras activity including activating mutations of K-Ras (Intrinsic pathway), or by a combination of these two pathways. Once reached, levels of Ras activity above a threshold cause acinar cells to undergo ADM and to form PanINs. High levels of Ras activity also lead to the generation of inflammation resembling chronic pancreatitis (CP). Inflammation induced genetic instability likely increases the probability of genetic alterations including mutations in K-Ras and loss of tumor suppressors (TS) which are required for the development of PDAC.
Materials and Methods
Detailed materials and methods are described in the Supplementary Materials.
Genetically Engineered Mice
cLGL-KRasG12V transgenic mice were produced by pronuclear microinjection at the M. D. Anderson Cancer Center Genetically Engineered Mouse Facility. LSL-KRasG12D and p53f/f conditional deletion mice were obtained from the Mouse Models for Human Cancer Consortium Repository (Rockville, MD.) 9, 34. All animal experiments were approved by the Institutional Animal Care and Use Committees of the University of Texas MD Anderson Cancer Center.
Ras Activity Assay
Total Ras activity was measured in cells lysed with beads coated with Raf1-RBD (Upstate Biotechnology, Lake Placid, NY).
Western Blot Analysis
To evaluate the levels of Ras, actin, RPS6, total Erk, p-Erk, p-AKT, p53 and DEC1 expression, cell or tissue lysates were separated by SDS-PAGE and immunodetection was done as described in Supplementary Methods.
Activation of K-Ras Expression in Primary Pancreatic Acinar Cells with Cre-bearing Adenovirus in vitro
Primary pancreatic acinar cells were prepared and incubated with adenoviruses at a titer which was previously shown to be nearly 100% efficient and incubated at 37 °C overnight.
Histology and Immunohistochemistry
Mice pancreata were fixed in 10% formalin and embedded in paraffin. H&E staining was performed in the M. D. Anderson Cancer Center Histology Core. Immunohistochemistry was done using RTU Vectastain Elite ABC Universal kit (Vector Laboratories, Burlingame, CA).
β-Galactosidase Activity Assays
Frozen sections were fixed in 2% formaldehyde/0.2% glutaraldehyde in phosphate-buffered saline for 5 min and incubated with X-gal-containing reaction mixture provided by the manufacturer (Millipore, Billerica, MA).
TUNEL Assay
To evaluate apoptotic cells we used the in situ Cell Death Detection kit according to the manufactures suggestions (Roche, Indianapolis, IN).
Periodic Acid-Schiff (PAS) and Sirius Red Staining
The mucin and collagen expression were stained with Periodic Acid-Schiff (PAS) reagents and Sirius Red Staining (Sigma, St. Louis, MO).
RT-PCR and Quantative RT-PCR
Total RNA preparation and quantitative RT-PCR were performed as we previously described 35.
Supplementary Material
Acknowledgments
This research was supported by NIH DK052067, 5R21DK068414, M.D. Anderson Support Core grant CA16672, M.D. Anderson Pancreatic Specialized Programs of Research Excellence (SPORE) grant P20 CA101936, and by the Lockton Endowment.
Footnotes
Conflict of Interest: No conflict of interest to disclose for all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 3.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
- 4.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Hruban RH, Adsay NV, bores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Kloppel G, Longnecker DS, Luttges J, Offerhaus GJ. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 7.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji B, Song J, Tsou L, Bi Y, Gaiser S, Mortensen R, Logsdon C. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis. 2008;46:390–395. doi: 10.1002/dvg.20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 11.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian Y, Zhang J, Yan B, Chen X. DEC1, a basic helix-loop-helix transcription factor and a novel target gene of the p53 family, mediates p53-dependent premature senescence. J Biol Chem. 2008;283:2896–2905. doi: 10.1074/jbc.M708624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, Maitra A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siveke JT, Einwachter H, Sipos B, Lubeseder-Martellato C, Kloppel G, Schmid RM. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007;12:266–279. doi: 10.1016/j.ccr.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan RD, Zheng CF, Guan KL, Williams JA. Activation of MAP kinase kinase (MEK) and Ras by cholecystokinin in rat pancreatic acini. American Journal of Physiology. 1995;268:G1060–G1065. doi: 10.1152/ajpgi.1995.268.6.G1060. [DOI] [PubMed] [Google Scholar]
- 20.Ferbeyre G. Barriers to Ras transformation. Nat Cell Biol. 2007;9:483–485. doi: 10.1038/ncb0507-483. [DOI] [PubMed] [Google Scholar]
- 21.Greten FR, Wagner M, Weber CK, Zechner U, Adler G, Schmid RM. TGF alpha transgenic mice. A model of pancreatic cancer development. Pancreatology. 2001;1:363–368. doi: 10.1159/000055835. [DOI] [PubMed] [Google Scholar]
- 22.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 23.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, Hanahan D, Redston MS, Chin L, DePinho RA. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopnin PB, Agapova LS, Kopnin BP, Chumakov PM. Repression of sestrin family genes contributes to oncogenic Ras-induced reactive oxygen species up-regulation and genetic instability. Cancer Res. 2007;67:4671–4678. doi: 10.1158/0008-5472.CAN-06-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c- K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 26.Hruban RH, van Mansfeld AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN, Kensler TW, Bose KK, Cameron JL, Bos JL. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant- enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol. 1993;143:545–554. [PMC free article] [PubMed] [Google Scholar]
- 27.Luttges J, Diederichs A, Menke MA, Vogel I, Kremer B, Kloppel G. Ductal lesions in patients with chronic pancreatitis show K-ras mutations in a frequency similar to that in the normal pancreas and lack nuclear immunoreactivity for p53. Cancer. 2000;88:2495–2504. doi: 10.1002/1097-0142(20000601)88:11<2495::aid-cncr10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 28.Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology. 1996;110:227–231. doi: 10.1053/gast.1996.v110.pm8536861. [DOI] [PubMed] [Google Scholar]
- 30.Muller-Decker K, Furstenberger G, Annan N, Kucher D, Pohl-Arnold A, Steinbauer B, Esposito I, Chiblak S, Friess H, Schirmacher P, Berger I. Preinvasive duct-derived neoplasms in pancreas of keratin 5-promoter cyclooxygenase-2 transgenic mice. Gastroenterology. 2006;130:2165–2178. doi: 10.1053/j.gastro.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 31.Wagner M, Greten FR, Weber CK, Koschnick S, Mattfeldt T, Deppert W, Kern H, Adler G, Schmid RM. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev. 2001;15:286–293. doi: 10.1101/gad.184701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein A, Klimstra DS, Kloppel G, Lauwers GY, Longnecker DS, Luttges J, Maitra A, Offerhaus GJ, Perez-Gallego L, Redston M, Tuveson DA. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 33.Hruban RH, Maitra A, Kern SE, Goggins M. Precursors to pancreatic cancer. Gastroenterol Clin North Am. 2007;36:831–49. vi. doi: 10.1016/j.gtc.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonkers J, Meuwissen R, van der GH, Peterse H, van d V, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 35.Ji B, Bi Y, Simeone D, Mortensen RM, Logsdon CD. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology. 2001;121:1380–1390. doi: 10.1053/gast.2001.29557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.