

Abstract
Based on the mild, thermal rearrangement of 1,2-dialkynylimidazoles to reactive carbene or diradical intermediates, a series of 1,2-dialkynylimidazoles were designed as potential irreversible p38 MAP kinase α-isoform (p38α) inhibitors. The synthesis of these dialkynylimidazoles and their kinase inhibition activity is reported. The 1-ethynyl-substituted dialkynylimidazole 14 is a potent (IC50 = 200 nM) and selective inhibitor of p38α. Moreover, compound 14 covalently modifies p38α as determined by ESI-MS after 12 h incubation at 37 °C. The unique kinase inhibition, covalent kinase adduct formation, and minimal CYP450 2D6 inhibition by compound 14 demonstrate that dialkynylimidazoles are a new, promising class of p38α inhibitors.
p38 MAP kinase (p38α) belongs to a family of serine/theronine kinases that serve as important mediators of inflammatory cytokines including tumor necrosis factor alpha (TNFα) and interleukin-1 beta (IL-1β).1,2 Elevated levels of the pro-inflammatory cytokines are associated with a number of diseases, such as toxic shock syndrome, rheumatoid arthritis, osteoarthritis, diabetes and inflammatory bowel disease.3 Therefore, inhibition of p38α is considered to be a potential therapeutic strategy.4 A number of p38α inhibitors have been synthesized and characterized.5 Although these compounds show good inhibition of p38α, many also inhibit other protein kinases with similar or greater potency.6
There has been a growing interest in irreversible inhibitors of protein kinases,7 and a number of these drugs are in clinical trials.8 Advantages of irreversible kinase inhibition include increased selectivity,9 duration,10 and therapeutic utility, especially against kinases that are resistant to competitive, ATP-binding pocket-targeting drugs.11 Additionally, irreversible inhibitors and related selective, covalent kinase modifying small molecules are of interest as probes for chemical genetics studies.12 While certain natural products and ATP analogs irreversibly inhibit kinases,13 none are selective towards p38α. Thus, there is a need to develop selective and irreversible inhibitors that target p38α. We have discovered a novel thermal cyclization and rearrangement of 1,2-dialkynylimidazoles (DAIms) (Scheme 1). Mild thermolysis of DAIms in the presence of chlorinated solvents or HCl leads to the isolation of imidazo[1,2-a]pyridine (ImPy) products, which may result from trapping of an initially-formed diradical intermediate via aza-Bergman cyclization.13
Scheme 1.

Thermal cyclization and rearrangement of 1,2-dialkynylimidazoles
Thermolysis under neutral conditions in non-halogenated solvents affords products derived from trapping cyclopentapyrazine (CyPP) carbene intermediates by H-atom abstraction, C–H bond insertion, and alkene addition reactions.14–16 The CyPP carbene is proposed to be derived from an intermediate cyclic cumulene that results from collapse of the diradical.14 Non-covalent association between DAIms and a kinase may facilitate the rate-determining aza-Bergman cyclization.
The formation of reactive diradical and carbene intermediates under mild conditions from DAIms has led us to propose that DAIms can be designed to undergo kinase binding-induced cyclization and covalent inactivation of specific kinase targets. Specifically, the structural similarity between DAIms and the known p38α inhibitors such as SB-20358017 and RWJ-6765718 (Figure 1) has inspired the design and inhibition studies of p38α-targeting DAIms described here.
Figure 1.
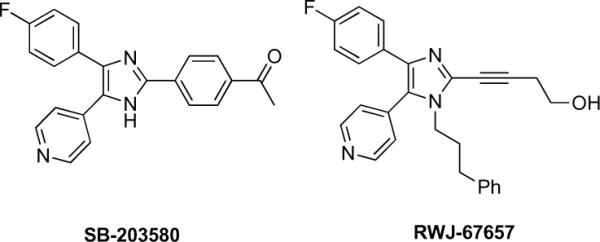
Examples of 4,5-diarylimidazole p38a inhibitors.
An initial route to kinase-targeting dialkynylimidazoles is shown in Scheme 2. The known 4(5)-(4-fluorophenyl)-5(4)-(4-pyridyl)imidazole 119 was protected with trityl group. Interestingly, this reaction only afforded one regioisomer, which was assigned as the 5-(4-fluorophenyl)-4-(4-pyridyl)imidazole 2 based on COSY and NOESY NMR. Compound 2 was deprotonated with n-BuLi at 0 °C, and quenched with I2 to give the 2-iodo-imidazole 3, which was deprotected in aqueous TFA to afford 4. Coupling of the lithium anion of imidazole 4, formed by deprotonation with LHMDS, with phenyl(phenylethynyl)-iodonium tosylate20 afforded a 15 % yield of a 1:1 mixture of the regioisomeric N-alkynyl-2-iodoimidazoles 5 and 6. The separated regioisomers were subjected to Sonogashra coupling with various terminal acetylene partners to provide the regioisomeric dialkynylimidazoles 7 and 8. The regiochemical assignments within this series were made based on the X-ray crystal structure of 7b shown in Figure 2.21
Scheme 2.

Reagents and conditions: (a) Et3N, Ph3CCl, CH2Cl2 (58 %); (b) i) n-BuLi, ii) I2, THF, 0 °C (60 %); (c) TFA, H2O (83 %); (d) LHMDS, PhI+CCPhTsO−; (e) RCCH, Pd(PPh3)4, CuI, Et3N.
Figure 2.
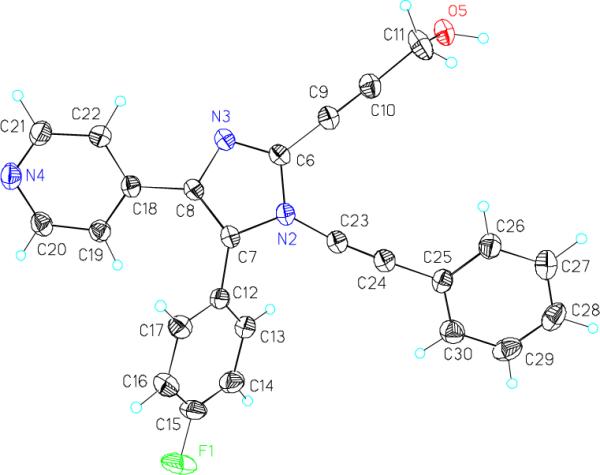
X-ray crystal structure of dialkynylimidazole 7b.
Although providing access to select kinase-targeting dialkynylimidazoles, the synthetic route shown in Scheme 2 suffers from a number of limitations associated with the alkynyliodonium coupling reaction. Only the phenylethynyl and TMS-ethynyl iodonium reagents could be employed in this coupling,22 and even in these cases, the yields are poor and mixtures of regioisomers are produced.
An improved synthetic route to these dialkynylimidazoles employing the recently reported copper-catalyzed N-alkynylation of imidazoles with bromoalkynes was devised (Scheme 3). Treating 4-fluorophenylimidazole 923 with TIPS-protected bromo-acetylene in the presence of catalytic CuI and 2-acetyl-cyclohexanone as ligand affords a 9:1 mixture of regioisomeric alkynylimidazoles 10b and 10a, respectively, in 79 % yield.22 Iodination of the 2-position of 10b affords the 2-iodoimidazole 11, which undergoes Sonogashira coupling with O-TIPS-protected homopropargyl alcohol to give the dialkynylimidazole 12 in 73 % yield.24 Deprotonation of 12 with n-BuLi followed by iodine quench affords the 5-iodoimidazole 13 in 74 % yield. A final Suzuki-Miyaura coupling of the 5-iodo imidazole 13 with pyridine-4-boronic acid followed by TBAF deprotection gives **the dialkynyl-imidazole 14.25 Mild thermolysis of 14 at 80° C under acidic conditions in the presence of chloride afforded 15, the product of HCl addition to the diradical, in 50 % yield.
Scheme 3.

Reagents and conditions: (a) BrCCTIPS, CuI, AcC, Cs2CO3, dioxane, 50 °C overnight followed by reflux for 4 h (79 %, 1:9 10a/10b); (b) i) n-BuLi, ii) I2, THF, −78 °C (91 %); (c) TIPSOCH2CH2CCH, Pd(PPh3)4, CuI, Et3N (73 %); (d) i) n-BuLi, ii) I2, THF, −78 °C (74 %); (e) pyridine 4-boronic acid, Pd(PPh3)4, K2CO3 (41 %); (f) TBAF, THF, −78 °C (89 %); (g) Me4NCl, TfOH, DMF, 80 °C, 5 days (50 %).
All of these 1,2-dialkynylimidazoles were assayed against p38α MAPK at a fixed time-point of 60 min (Table 1).26 Compounds 7a–c and 8a–c display modest inhibition at 10 μM concentration. In this series there is little difference in activity between the 1-alkynyl-5-fluorophenyl regioisomers 7a–c and the 1-alkynyl-5-pyridylisomers 8a–c, in contrast to reported 1-substituted pyridylimidazole p38α inhibitors.28 Interestingly, the 1-ethynyl-substituted analog 14 is a potent inhibitor of p38α. Compound 14 completely inhibits p38α at 10 μM (Table 1), and has an IC50 for p38α of 200 nM.27 In comparison, the IC50 of 14 against p38 (5.4 μM) is >25-fold higher. Dialkyny-limidazole 14 was also assayed at concentration of 20 μM against a panel of 53 additional human kinases. Only one kinase, (MAPK4/HGK) was strongly inhibited (>90% inhibition at 20 μM, IC50 = 4.2 μM), while six additional kinases were moderately inhibited (between 50–90% inhibition, see Supporting Information). The cyclized 15 also inhibited p38α (IC50 = 370 nM).
Table 1.
In vitro activity of 1,2-dialkynylimidazoles against p38α
| Compound | p38α % inhibition (@ 10 μM)a |
|---|---|
| 7a | 19 |
| 8a | 28 |
| 7b | 63 |
| 8b | 83 |
| 7c | 53 |
| 8c | 75 |
| 14 | 100 |
Tests were carried out in duplicate.
Dialkynylimidazole 14 (100 μM) was incubated with non-phosphoryated p38α (5 μM) at 37 °C in 50 mM HEPES, 10 mM MgCl2, 2 mM DTT, 1 mM EGTA, pH 7.5 for 12 h, followed by extensive dialysis, and the sample was analyzed by ESI- MS. A new peak in the mass spectrum at m/z = 41896, which corresponds to addition of a single molecule of 14 (MW = 331) to p38α, was observed (~25 % adduct) (Figure 3). Under identical conditions but with 1 mM DTT present, the adduct was the predominant species observed (Supporting Information).
Figure 3.

(a) ESI-MS spectrum of unphosphorylated p38α incubated for 12 h at 37 °C; (b) ESI-MS spectrum of unphosphorylated p38α incubated with dialkynylimidazole 14 for 12 h at 37 °C, followed by extensive dialysis.
A common concern for pyridinylimidazole MAPK inhibitors such as RWJ 67657 and SB-203580 is their inhibition of cytochrome P450 (CYP450) enzymes, which may be linked to hepatotoxicity.29 Interestingly, the dialkynylimidazole 14 displays a much lower level of inhibition of CYP450 2D6 (4% inhibition at 10 μM) compared to SB-203580 (78% inhibition at 10 μM).
In summary, novel p38α-targeting dialkynylimidazoles were designed, synthesized and evaluated. Although 1-phenethynyl-substituted dialkynylimidazoles 7a–c and 8a–c are only modest inhibitors of p38α, the 1-ethynyl-substituted dialkynylimidazole 14 is a potent and selective inhibitor. Commensurate with the increased facility of rearrangement of 1-ethynyl-substituted dialkynylimidazols relative to 1-phenethynyl analogues,16 compound 14 forms a covalent adduct with p38α. However, the conditions for p38α adduct formation (12 h at 37 °C) are much milder than those required for cyclization/trapping of 14 to afford 15 (5 days at 80 °C), indicating that the kinase may facilitate the cyclization of 14. Further studies on the site and mechanism of this covalent modification of p38α by 1-ethynyl-substituted dialkynylimidazoles are on-going. The unique kinase inhibition, covalent kinase adduct formation, and minimal CYP450 2D6 inhibition by compound 14 demonstrate that dialkynylimidazoles are a new, promising class of p38α inhibitors.
Supplementary Material
Acknowledgments
We thank Dr. Herng-Hsiang Lo for ESI-MS analysis; and Dr. Vincent Lynch for X-ray structure determination. This research was supported by grants from the Robert Welch Foundation (F-1298 to SMK, F-1390 to KND), the Texas Higher Education Coordinating Board (to SMK), and the NIH (GM59802 to KND).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. Nature. 1994;372:739. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 2.Lee JC, Young PR. J. Leuk. Biol. 1996;59:152–7. doi: 10.1002/jlb.59.2.152. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann M, Brennan FM, Maini RN. Annu. Rev. Immunol. 1996;14:397. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 4.Lee JC, Kassis S, Kumar S, Badger A, Adams JL. Pharmacol. Ther. 1999;82:389. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]
- 5.Pettus LH, Wurz RP. Curr. Top. Med. Chem. 2008;8:1452. doi: 10.2174/156802608786264245. [DOI] [PubMed] [Google Scholar]
- 6.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. Biochem. J. 2007;408:297. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Denny WA. Pharmacol. Therap. 2002;93:253–261. doi: 10.1016/s0163-7258(02)00194-8. [DOI] [PubMed] [Google Scholar]; (b) Rastelli G, Rosenfeld R, Reid R, Santi DV. J. Struct. Biol. 2008;164:18. doi: 10.1016/j.jsb.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Mukherji D, Spicer J. Expert Opin. Invest. Drugs. 2009;18:293. doi: 10.1517/13543780902762843. [DOI] [PubMed] [Google Scholar]
- 9.Cohen MS, Zhang C, Shokat KM, Taunton J. Science. 2005;308:1318. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Tsou HR, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, Rabindran SK, Shen R, Shi XQ, Wang YF, Upeslacis J, Wissner A. J. Med. Chem. 2005;48:1107. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]; (b) Tummino PJ, Copeland RA. Biochemistry. 2008;47:5481. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]; (c) Smith AJT, Zhang XY, Leach AG, Houk KN. J. Med. Chem. 2009;52:225. doi: 10.1021/jm800498e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalczyk Anja, Klueter Sabine, Rode Haridas B., Simard Jeffrey R., Gruetter Christian, Rabiller Matthias, Rauh Daniel. Bioorg. Med. Chem. 2008;16:3482. doi: 10.1016/j.bmc.2008.02.053. [DOI] [PubMed] [Google Scholar]
- 12.Blair JA, Rauh D, Kung C, Yun CH, Fan QW, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM. Nat. Chem. Biol. 2007;3:229. doi: 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- 13.(a) Khandekar SS, Feng BB, Yi T, Chen S, Laping N, Bramson N. J Biomol. Screen. 2005;10:447. doi: 10.1177/1087057105274846. [DOI] [PubMed] [Google Scholar]; (b) Barluenga S, Dakas PY, Boulifa M, Moulin E, Winssinger N. C. R. Chim. 2008;11:1306. [Google Scholar]
- 14.Nadipuram AK, Kerwin SM. Tetrahedron. 2006;62:3798. [Google Scholar]
- 15.Nadipuram AK, David WM, Kumar D, Kerwin SM. Org. Lett. 2002;4:4543. doi: 10.1021/ol027100p. [DOI] [PubMed] [Google Scholar]
- 16.Kerwin SM, Nadipuram A. Synlett. 2004:1404. [Google Scholar]
- 17.Gallagher TF, Fier-Thompson SM, Garigipati RS, Sorenson ME, Smietana JM, Lee D, Bender PE, Lee JC, Laydon JT, Griswold DE, Chabot-Fletcher MC, Breton JJ, Adams JL. Bioorg. Med. Chem. Lett. 1995;5:1171–1176. [Google Scholar]
- 18.Wadsworth SA, Cavender DE, Beers SA, Lalan P, Schafer PH, Malloy EA, Wu W, Fahmy B, Olini GC, Davis JE, Pellegrino-Gensey JL, Wachter MP, Siekierka JJ. J. Phamacol. Exp. Therap. 1999;291:680. [PubMed] [Google Scholar]
- 19.Boehm JC, Smietana JM, Sorenson ME, Garigipati RS, Gallagher TF, Sheldrake PL, Bradbeer J, Badger AM, Laydon JT, Lee JC, Hillegass LM, Griswold DE, Breton JJ, ChabotFletcher MC, Adams JL. J. Med. Chem. 1996;39:3929. doi: 10.1021/jm960415o. [DOI] [PubMed] [Google Scholar]
- 20.Koser GF, Rebrovic L, Wettach RH. J. Org. Chem. 1981;46:4324. [Google Scholar]
- 21.Crystallographic data for compound 7b have been deposited with the Cambridge Crystallographic Data Centre as CCDC740499. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44 (0)1223-336033 or deposit@ccdc.cam.ac.uk].
- 22.Laroche C, Li J, Freyer MW, Kerwin SM. J. Org. Chem. 2008;73:6462. doi: 10.1021/jo801118q. [DOI] [PubMed] [Google Scholar]
- 23.Ho K-K, Auld DS, Bohnstedt AC, Conti P, Dokter W, Erickson S, Feng D, Inglese J, Kingsbury C, Kultgen SG, Liu R-Q, Masterson CM, Ohlmeyer M, Rong Y, Rooseboom M, Roughton A, Samama P, Smit M-J, Son E, Van der Louw J, Vogel G, Webb M, Wijkmans J, You M. Bioorg. Med. Chem. Lett. 2006;16:2724. doi: 10.1016/j.bmcl.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 24.Laroche C, Kerwin SM. Tetrahedron Lett. 2009;50:5194. [Google Scholar]
- 25.See Supporting Information for characterization data for all compounds.
- 26.All kinase inhibition studies were performed by Invitrogen using the Z'-lyte™ assay at [ATP] = Km[app] and protein substrate concentration of 20 μM.
- 27.The inhibition due to 14 in these assays is primarily due to non-covalent inhibition: preincubation of the kinase with 14 for 60 min prior to the addition of ATP did not change the IC50 value.
- 28.Laufer S, Wagner G, Kotschenreuther D. Angew. Chem. Int. Ed. 2002;41:2290. doi: 10.1002/1521-3773(20020703)41:13<2290::AID-ANIE2290>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 29.Adams JL, Boehm JC, Kassis S, Gorycki PD, Webb EF, Hall R, Sorenson M, Lee JC, Ayrton A, Griswold DE, Gallagher TF. Bioorg. Med. Chem. Lett. 1998;8:3111. doi: 10.1016/s0960-894x(98)00549-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.