

Abstract
Although activating mutations of FGFR3 are frequent in bladder tumors, little information is available on their specific effects in urothelial cells or the basis for the observed mutation spectrum. We investigated the phenotypic and signaling consequences of three FGFR3 mutations (S249C, Y375C, and K652E) in immortalized normal human urothelial cells (TERT-NHUC) and mouse fibroblasts (NIH-3T3). In TERT-NHUC, all mutant forms of FGFR3 induced phosphorylation of FRS2α and ERK1/2, but not AKT or SRC. PLCγ1 phosphorylation was only observed in TERT-NHUC expressing the common S249C and Y375C mutations, and not the rare K652E mutation. Cells expressing S249C and Y375C FGFR3 displayed an increased saturation density, related to increased proliferation and viability. This effect was significantly dependent on PLCγ1 signaling and undetectable in cells expressing K652E FGFR3, which failed to phosphorylate PLCγ1. In contrast to TERT-NHUC, expression of mutant FGFR3 in NIH-3T3 resulted in phosphorylation of Src and Akt. Additionally, all forms of mutant FGFR3 were able to phosphorylate Plcγ1 and induce morphological transformation, cell proliferation, and anchorage independent growth. Our results indicate that the effects of mutant FGFR3 are both cell type- and mutation-specific. Mutant FGFR3 may confer a selective advantage in the urothelium by overcoming normal contact inhibition of proliferation.
Keywords: fibroblast growth factor, FGFR3, bladder, urothelium, signaling
Introduction
Fibroblast growth factor receptor 3 (FGFR3) is one of four trans-membrane receptors that mediate the intracellular effects of fibroblast growth factors (FGFs) (Ornitz and Itoh, 2001). Structurally, FGFRs consist of an extracellular portion with three immunoglobulin-like domains, a trans-membrane segment, and an intracellular split tyrosine-kinase region. In normal physiological situations, signaling is triggered by the interaction of FGF with the extracellular domain of the receptor, which induces receptor dimerization and auto-phosphorylation. Activation of the receptor is followed by a phosphorylation cascade culminating in the activation of a number of signaling pathways, such as MAPK, PI3K and PKC (Dailey et al., 2005; Eswarakumar et al., 2005).
Germline missense mutations of FGFR3 resulting in its constitutive ligand-independent activation have been described in number of skeletal dysplasia syndromes (Eswarakumar et al., 2005). Functional and animal studies have shown that in chondrocytes, pathological activation of FGF signaling causes premature cell differentiation that halts limb growth and development. These effects are mediated by the activation of downstream targets with anti-proliferative and pro-apoptotic activity, such as the transcription factors STAT1, STAT3, and STAT5, and the cell cycle inhibitors p16, p18, p19, and p21, (Chen et al., 1999; Legeai-Mallet et al., 2004; Li et al., 1999; Sahni et al., 1999; Su et al., 1997).
Interestingly, somatic activating mutations of FGFR3 corresponding to the germline mutations found in skeletal dysplasia syndromes are observed in many urothelial carcinomas (UC) of the bladder (Cappellen et al., 1999; Sibley et al., 2001a; van Rhijn et al., 2002), suggesting that in a different cellular context FGFR3 may act as an oncogene. The FGFR3 mutations observed in UC are clustered in exons 7 (codons 248 and 249), 10 (codons 372, 373, 375, 382, and 393) and 15 (codon 652). S249C is the most common mutation in UC, occurring in more than 70% of mutant tumors, while Y375C is the second most common mutation with a frequency of around 22% (Tomlinson et al., 2007a). Both these mutations create unpaired cysteines in the proximal extracellular region, leading to the formation of disulfide bonds between adjacent receptors, thereby inducing ligand-independent dimerization and activation (Adar et al., 2002; d'Avis et al., 1998). Mutations within the kinase domain, such as K652E, are thought to induce a conformational change in the activation loop, resulting in constitutive auto-phosphorylation of the receptor (Webster et al., 1996). These mutations are only observed in a small proportion of cases. Currently, the reason for the differences in frequency of specific FGFR3 mutations in UC is unclear, although a role of smoking-related carcinogens in determining the occurrence or the type of FGFR3 mutations has been excluded (Wallerand et al., 2005).
Although FGFR3 mutations have also been found in multiple myeloma (Chesi et al., 2001) and carcinoma of the cervix (Cappellen et al., 1999), their frequency in these malignancies is much lower than in UC. Furthermore, except for benign skin tumors (Hafner et al., 2006), other solid epithelial tumors have been found negative for FGFR3 mutation (Karoui et al., 2001; Sibley et al., 2001b), pointing to a specific role in bladder carcinogenesis. Notably, FGFR3 mutations in UC are also found in flat urothelial hyperplasias (van Oers et al., 2006), implying that they may represent an early event during malignant transformation. The importance of FGFR3 signaling in bladder transformation is further emphasized by the effects of FGFR3 silencing in the bladder cancer cell lines MGHU3 and TCC97-7, which harbor the Y375C and S249C mutations respectively (Bernard-Pierrot et al., 2006; Tomlinson et al., 2007b). In both cases, decreased expression of mutant FGFR3 inhibited cell proliferation and anchorage-independent growth. Furthermore, a specific anti-FGFR3 antibody has been shown to inhibit proliferation of RT112, a bladder carcinoma cell line known to over-express wildtype FGFR3 (Martinez-Torrecuadrada et al., 2005).
Despite the overwhelming evidence linking FGFR3 mutation to bladder carcinogenesis, and its potential as a therapeutic target, most functional studies on FGFR3 have been performed either in chondrocytes or mouse fibroblasts. As FGFR3 activation appears to have diverse outcomes in different cellular contexts, it is crucial to investigate the signaling pathways mediated by mutant FGFR3 and their cellular consequences specifically in urothelial cells. In this study, we elucidate FGFR3 signaling in normal urothelial cells and show that mutant FGFR3 may confer a selective advantage by promoting proliferation and survival, in a partially PLCγ1-dependent manner. We also demonstrate a clear correlation between the frequency of different FGFR3 mutations in bladder tumors and the level of ligand-independence, signaling activation, and phenotypic consequences.
Results
Mutant forms of FGFR3 induce transformation of mouse fibroblasts
Initially, we assessed the ability of three mutant forms of FGFR3 that are found in UC (S249C, Y375C, and K652E) to transform NIH-3T3 cells. In these cells, expression of all FGFR3 mutations resulted in a transformed spindle-like morphology (Figure 1a), higher proliferation rate (p=0.004) (Figure 1b) and colony formation in soft agar (p=0.05) (Figure 1c). No effect on morphology, proliferation or anchorage-independence was observed for the ‘kinase dead’ control (S249C KD) (data not shown). All mutant forms of FGFR3 showed high levels of constitutive receptor phosphorylation (Figure 1d). Both the 130 and 120 kDa glycosylated forms of K652E FGFR3 were phosphorylated, while S249C and Y375C mutants showed activation only of the 130 kDa protein. Expression of all forms of mutant FGFR3 in NIH-3T3 increased phosphorylation of Frs2α, Plcγ1, and Src, and activated Erk1/2 and Akt (Figure 1d, Supplementary Figure 1).
Figure 1.

Effects of mutant FGFR3 in NIH-3T3 cells. (a) Morphology of cells expressing wildtype (WT), S249C, Y375C, and K652E FGFR3. Line bars indicate 100 μm. (b) Representative experiment showing the total cell numbers reached on day 7 by cells expressing WT or mutant FGFR3 and by control cells. (c) Fold difference in the number of colonies formed in soft agar by cells expressing WT or mutant FGFR3 and control cells. (d) Constitutive phosphorylation of mutant FGFR3 and ligand-independent activation of signaling cascades. Significant differences to control cells (transduced with an empty pFB vector) are indicated as ‘*’ (p<0.05) or ‘**’ (p<0.01)
Mutant FGFR3 increases saturation density in normal urothelial cells
Next we studied the effects of mutant FGFR3 in normal urothelial cells by expressing S249C, Y375C and K652E FGFR3 in TERT-NHUC. Expression levels were comparable to those observed in the bladder cancer cell line TCC97-7, derived from a low stage UC (data not shown). In contrast to the effects in NIH-3T3, expression of mutant FGFR3 in normal urothelial cells did not induce obvious morphological changes or anchorage-independent growth (data not shown). Furthermore, at subconfluence there was no difference in proliferation between cells expressing mutant FGFR3 and controls. However, TERT-NHUC expressing S249C and Y375C FGFR3 were characterized by an increased saturation density, and at confluence reached mean cell numbers 24% and 20% higher than controls (p≤0.05) (Figure 2a). Consistent with their increased saturation density, cells expressing mutant FGFR3 also acquired a different morphology at confluence. While controls produced a compact monolayer of flat cells, mutants displayed reduced cell-cell adhesion, formed ridges and acquired a multilayered appearance (Supplementary Figure 2).
Figure 2.

Effects of mutant FGFR3 in TERT-NHUC. (a) Saturation density of confluent cells. (b) Flow cytometric analysis of cell cycle profiles of confluent cells (day 12). (c) Representative experiment showing the number of viable cells expressing WT or mutant FGFR3, compared to control cells, at subconfluence (day 4) and confluence (day 12). (d) Expression levels of proteins involved in cell cycle and cell survival in confluent cultures (day 12); proteins were extracted from cells grown in full medium unless otherwise specified (DM, depleted medium). Significant differences to control cells (transduced with an empty pFB vector) are indicated as ‘*’ (p<0.05) or ‘**’ (p<0.01)
The saturation density of cells expressing K652E FGFR3 was similar to controls and cells expressing wildtype FGFR3 or S249C KD. Therefore, the magnitude of the phenotypic effect was of the order S249C>Y375C>K652E=Wildtype=Control, correlating with the relative frequencies at which these mutations are observed in bladder tumors.
Mutant FGFR3 promotes proliferation and viability of confluent TERT-NHUC
To investigate the cellular mechanisms that determine the higher saturation density associated with expression of mutant FGFR3, we analyzed both cell cycle profile and viability in subconfluent and confluent cells.
In controls, the number of cells in the G0/G1 phase of the cell cycle progressively increased at confluence, while the number of viable cells decreased. No significant differences in cell cycle profile or viability were observed between mutant and control cells in subconfluent cultures. However at confluence on day 12, only 59% of S249C were in G0/G1, compared with 68% of control and wildtype cells (p=0.004) (Figure 2b). A corresponding increase was detected in the proportion of S249C in S (8% vs 7%) (p=0.035) and G2/M phase (33% vs 25%) (p=0.004). Similar results were observed on days 8 and 10 (data not shown). The cell cycle profile of Y375C- and K652E-expressing cells was intermediate between S249C mutants and control cells (Figure 2b). No significant differences in cell cycle profile were measured between control cells and cells expressing S249C KD FGFR3 (data not shown).
Cells expressing S249C and Y375C FGFR3 were significantly more viable at confluence compared to controls or cells expressing wildtype FGFR3 (p≤0.001). On days 12 (Figure 2c) and 15 (data not shown), on average 45% of S249C cells were viable, compared with only 35% of control cells. Y375C showed an increase in viable cells on day 12 (41% vs. 35%) (Figure 2c) but no difference on day 15 (data not shown). In contrast to the other two mutants, the viability of confluent cells expressing K652E FGFR3 was similar to controls (p>0.05).
Mutant FGFR3 alters the expression of cell cycle and survival proteins in normal urothelial cells
Consistent with their altered cell cycle profile, confluent TERT-NHUC expressing mutant FGFR3 exhibited changes in cell cycle-related proteins (Figure 2d, Supplementary Figure 3a). Whilst in control cells the expression of the retinoblastoma protein (RB) was switched off upon reaching confluence, RB was expressed and phosphorylated in confluent cells expressing S249C FGFR3 and, to a lesser extent, in Y375C mutants. Furthermore, expression of CDK1 was increased in S249C and Y375C cells, and the difference was particularly pronounced when the cells were starved in depleted medium to block stimulation by other growth factors. The protein profile of cells expressing K652E FGFR3 was different from the other mutants, with low RB and intermediate CDK1 levels, consistent with their lower saturation density.
Three proteins involved in cell survival, MCL1, BCL-XL, and BCL2, were up-regulated in confluent cells expressing all types of mutant FGFR3 (Figure 2d, Supplementary Figure 3b). Surprisingly, there was no difference in the expression of these proteins between K652E and the other mutants, suggesting that differential expression of some other regulators of cell survival may be responsible for the lower viability of cells expressing K652E FGFR3.
Differential intensity of signaling is associated with distinct FGFR3 mutations
To investigate the reason for the differential behavior of cells expressing K652E FGFR3, we assessed the phosphorylation levels of FGFR3 mutant proteins and downstream effectors in urothelial cells. All mutant forms of FGFR3 were constitutively phosphorylated in the absence of ligand (Figure 3a). The level of activation of the K652E receptor was higher than the S249C and Y375C mutants (Figure 3a, Supplementary Figure 4a). Similar to results in NIH-3T3, K652E FGFR3 differed from the other mutants in that both the 130 KDa and 120 KDa forms were phosphorylated. Consistent with their constitutive activation, ligand-independent dimerization of S249C and Y375C FGFR3 was observed by western blotting under non-denaturing conditions (Figure 3b). Some differences were observed between the size of dimers formed by S249C and Y375C FGFR3, possibly as the result of differential glycosylation. K652E FGFR3 did not spontaneously form dimers, as this mutation does not produce unpaired cysteines.
Figure 3.

Constitutive activation of mutant FGFR3 and downstream signaling cascades in TERT-NHUC. (a) Constitutive phosphorylation of mutant FGFR3. (b) Western blot of cell lysates run under non-denaturing conditions showing ligand-independent dimerization of S249C and Y375C FGFR3. (c) Downstream signaling activated by mutant FGFR3 in confluent (day 12) and subconfluent (day 4) cells in full medium, and in subconfluent cells after 1 h in depleted medium.
Signaling was examined in cells at various degrees of confluence, in full and depleted medium (Figure 3c, Supplementary Figure 4b). In all conditions tested, no differences were observed between mutant and control cells regarding the levels of activated p38, JNK, SRC and AKT (data not shown). Constitutive phosphorylation of FRS2α was detected in mutant FGFR3-expressing cells in all conditions. Increased phosphorylation of ERK1/2 was observed in mutant-expressing cells at subconfluence in depleted medium and at confluence in full medium. However, in exponentially growing cells in full medium, ERK1/2 was equally activated in all cell types, presumably due to stimulation via other growth factor receptors. Similarly, increased PLCγ1 phosphorylation in FGFR3 mutant cells was only observed after confluence or starvation. Interestingly, in contrast to our findings in NIH-3T3 cells, in TERT-NHUC PLCγ1 phosphorylation was detected exclusively in cells expressing S249C and Y375C FGFR3, but not K652E.
Overall, these results suggest that although all mutant forms of FGFR3 are constitutively phosphorylated in urothelial cells, they differ in their ability to activate downstream signaling cascades, particularly with regard to PLCγ1 phosphorylation.
Differential ligand dependence of mutant forms of FGFR3
We next examined whether cells expressing wildtype and mutant FGFR3 were responsive to FGF1 stimulation in terms of receptor activation (Figure 4a, Supplementary Figure 5) and signaling (Figure 4b, Supplementary Figure 5). Phosphorylation of wildtype FGFR3 could be induced by a short incubation with FGF1 (Figure 4a). In contrast, the strong constitutive phosphorylation of S249C FGFR3 was unaffected by treatment with FGF1, suggesting complete ligand-independence. On the other hand, Y375C and K652E FGFR3 showed increased levels of phosphorylation in response to ligand stimulation (Figure 4a).
Figure 4.
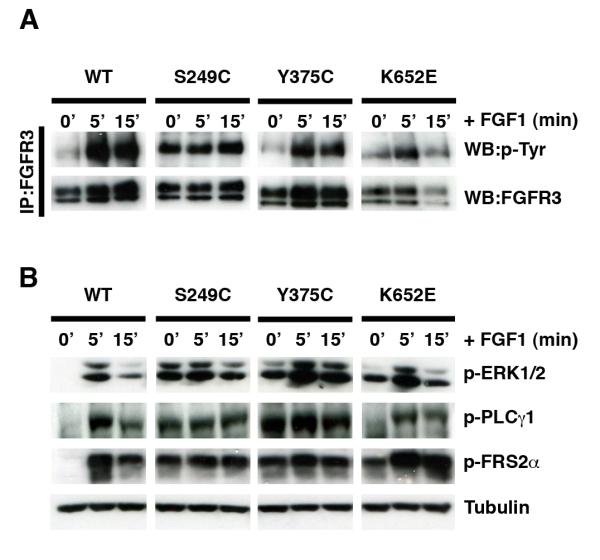
Ligand independence of different forms of mutant FGFR3 expressed in TERT-NHUC. Receptor phosphorylation (a) and signaling activation (b) in starved subconfluent cells, before and after stimulation with FGF1.
As expected, FGF1 induced phosphorylation of FRS2α, ERK1/2 and PLCγ1 in normal urothelial cells over-expressing wildtype FGFR3 (Figure 4b). Consistent with their complete ligand-independence, FGF1 treatment failed to stimulate signaling further in cells expressing the S249C mutation. However, cells expressing the Y375C mutation, responded to FGF1 treatment with a small increase in ERK1/2 activation, while phosphorylation levels of FRS2α and PLCγ1 remained unchanged. Cells expressing the K652E mutated protein displayed the most pronounced response to ligand, with increased phosphorylation of PLCγ1, ERK1/2 and FRS2α (Figure 4b). Overall, therefore, the degree of ligand-independence of the different mutant FGFR3 was in the order S249C>Y375C>K652E.
PLCγ1 phosphorylation contributes to FGFR3-mediated increase in saturation density
To clarify whether the lack of constitutive PLCγ1 phosphorylation may explain the different phenotypic behavior associated with the K652E mutation, we used a construct encoding a S249C FGFR3 protein with a mutated PLCγ1 binding site (S249C+Y762F). This mutant protein was able to constitutively activate FRS2α and ERK1/2, but failed to phosphorylate PLCγ1 (Figure 5a). When expressed in NIH-3T3, S249C+Y762F FGFR3 was associated with morphological transformation (Figure 5b), increased proliferation (p=0.004) (Figure 5c), and increased colony formation in soft agar (p=0.05) (Figure 5d), compared to control cells, indicating that PLCγ1 signaling is not essential for FGFR3-induced transformation in this cell type.
Figure 5.

Contribution of Plcγ1 phosphorylation to FGFR3-mediated transformation in NIH-3T3. (a) Activation of signaling cascades in cells expressing S249C FGFR3 or S249C with a mutation in the PLCγ1 binding site (S249C+Y762F). (b) Morphology of NIH-3T3 cells expressing S249C+Y762F FGFR3 and control cells. Line bars indicate 100 μm. (c) Representative experiment showing total cell numbers reached on day 7 by NIH-3T3 cells expressing S249C and S249C+Y762F FGFR3, compared with controls (d) Fold difference in the number of colonies >200 μm formed in soft agar by NIH-3T3 cells expressing S249C and S249C+Y762F FGFR3, compared with control cells. Significant differences to control cells (transduced with an empty pFB vector) are indicated as ‘*’ (p<0.05) or ‘**’ (p<0.01)
In TERT-NHUC, abolishing PLCγ1 phosphorylation significantly reduced the increase in saturation density associated with S249C FGFR3 (13% vs. 24%, p=0.05) (Figure 6a), suggesting that PLCγ1 signaling contributes to this phenotype. Confluent TERT-NHUC expressing the S249C+Y762F mutant showed an increase in the number of cells in G0/G1 phase (63% vs. 59%) (p=0.006), a decrease in the number of cells in G2/M (27% vs 33%) (p=0.004) (Figure 6b), and a lower viability (39% vs. 45%, p=0.047) (Figure 6c) compared with S249C cells. These results confirm our hypothesis that lack of PLCγ1 phosphorylation may contribute to the lower viability and saturation density exhibited by cells expressing the K652E FGFR3 mutation. However, cells expressing S249C+Y762F FGFR3 achieved higher cell numbers at confluence compared with K652E and control cells (p=0.05) (Figures 2a and 6a), suggesting that other signaling pathways also modulate this phenotype and may be differentially activated between K652E and the other mutants.
Figure 6.
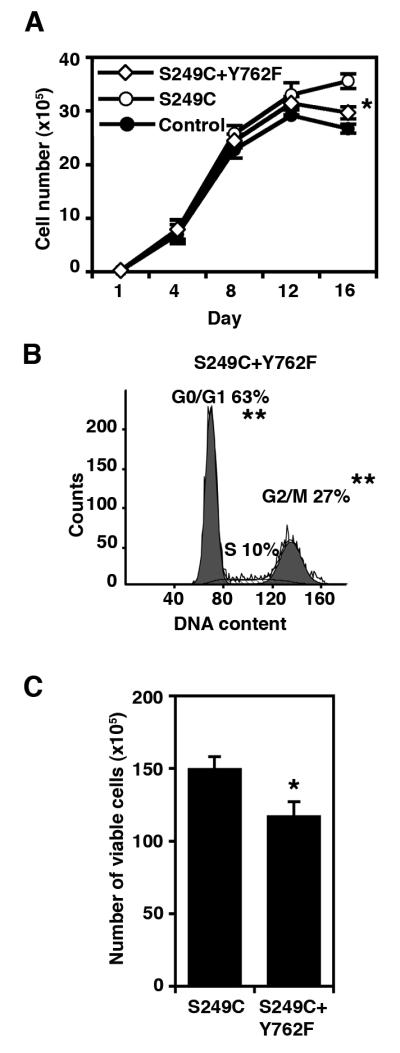
Contribution of PLCγ1 phosphorylation to saturation density in TERT-NHUC. (a) Growth curves showing saturation density achieved by cells expressing S249C+Y762F FGFR3, compared with cells expressing S249C FGFR3 and control cells. (b) Flow cytometric analysis of cell cycle profiles of confluent cells (day 12) expressing S249C+Y762F FGFR3. (c) Representative experiment showing the number of viable cells at confluence (day 12) in cultures expressing S249C+Y762F FGFR3 compared with cells expressing S249C FGFR3. Significant differences to S249C cells are indicated as ‘*’ (p<0.05) or ‘**’ (p<0.01)
Discussion
FGFR3 is the predominant FGF receptor in the urothelium, where its expression levels are up to 20-fold higher than other FGFRs (Tomlinson et al., 2005). A number of studies have underlined a link between bladder carcinogenesis and dysregulated, ligand-independent signaling due to activating mutations of FGFR3. Despite its important physiological and pathological roles in the urothelium, however, most information regarding the effects of mutant FGFR3 derives from functional investigations performed in other tissues or cell types. This is the first study to specifically investigate the signaling and phenotypic consequences of mutant FGFR3 in normal urothelial cells.
Our results show that in urothelial cells mutant FGFR3 initiates a sequence of phosphorylation events mediated by FRS2α and PLCγ1, leading to the specific activation of the ERK1/2 signaling pathway, without simultaneous phosphorylation of JNK or p38. In contrast to findings in NIH-3T3, no differences in the activation of Src and Akt were detected in TERT-NHUC expressing mutant FGFR3. Previous studies have also described differences in FGFR3 signaling between different cell types. For example, whilst the STAT1 signaling pathway mediates the effects of FGFR3 in chondrocytes, STAT1 is not activated by FGFR3 signaling in NIH-3T3 (Sahni et al., 1999), or multiple myeloma cell lines (Ronchetti et al., 2001). Similarly we have not detected STAT1 phosphorylation in normal urothelial cells expressing mutant FGFR3 (unpublished). The diversity of FGFR3 signaling in different cellular contexts may be due to tissue-specificity in the expression of modulators or mediators of signaling, and underlines the crucial importance of investigating the effect of FGFR3 mutation specifically in the target cells of interest.
We have also shown that, as a result of FGFR3 mutation, normal urothelial cells reach higher cell numbers at confluence, due to a combination of increased proliferation and viability, and exhibit consistent changes in a number of proteins involved in cell cycle control and survival. Expression of RB, for example, was switched off in confluent control cells, an event consistent with G1 arrest and confluence-induced differentiation in some epithelial cell types (Guy et al., 2001). In contrast, mutant cells retained RB expression and the protein was hyper-phosphorylated. Furthermore, the cell cycle regulator CDK1, which modulates RB phosphorylation (Santamaria et al., 2007; Yamamoto et al., 1995), was up-regulated in cells expressing mutant FGFR3. Complete loss of RB function is associated with both p53-dependent and -independent apoptosis, creating a selective pressure for the acquisition of an anti-apoptotic phenotype (Harbour and Dean, 2000). Indeed, a number of pro-survival factors were found to be up-regulated in cells expressing mutant FGFR3, including BCL2, Bcl-XL, and MCL1. Thus our observations suggest that at confluence TERT-NHUC cease dividing and, as result of RB downregulation, either differentiate or die. In contrast, cells expressing mutant FGFR3 survive and continue proliferating by phosphorylating RB and inducing mechanisms to escape apoptosis. Some of these events have been observed in bladder tumors in vivo. High expression of Cdk1 and hyperphosphorylated Rb has been described in mouse SV40 T-induced bladder tumors (Garcia-Espana et al., 2005) and expression of BCL2 and Bcl-XL is observed in a large proportion of human bladder carcinomas (Korkolopoulou et al., 2002). Since our observations indicate that these proteins may represent downstream effectors of FGFR3 signaling in urothelial cells, it will be interesting to assess their expression in bladder tumors in relation to FGFR3 mutation status.
Interestingly, we found that the ability of mutant FGFR3-expressing cells to reach a higher saturation density was partially dependent on PLCγ1 phosphorylation, as this was reduced in cells expressing S249C FGFR3 with a mutation disrupting the PLCγ1-binding site. This finding is consistent with a previous report showing that abolishing PLCγ1 signaling attenuates, but does not completely eliminate, the ability of mutant FGFR3 to transform Ba/F3 hematopoietic cells (Chen et al., 2005), indicating that transformation in this cell type requires both PLCγ-dependent and -independent mechanisms, possibly relating to MAPK activation. Although mutant FGFR3 has not as yet been implicated in dysregulation of contact inhibition of cell proliferation and movement, PLCγ1 activation could potentially modulate cytoskeletal reorganization and cell adhesion through PKC signaling and release of calcium from the endoplasmic reticulum (Wells and Grandis, 2003). Furthermore, in some cell types FGF can signal through non-canonical pathways involving a number of cell-cell and cell-matrix adhesion molecules (Murakami et al., 2008).
As expected for a normal cell type, TERT-NHUC require further genetic alterations for the acquisition of a completely transformed phenotype, and FGFR3 mutation alone was not sufficient to induce anchorage independent growth in soft agar or tumor formation in nude mice (unpublished). However, the observed proliferative and survival advantage under confluent conditions in culture suggests that FGFR3 mutation as an initiating or early genetic event could contribute to clonal expansion or the development of hyperplasia within the urothelium in vivo.
Interestingly, the majority of UC with FGFR3 mutations also show an up to 20-fold increase in the levels of FGFR3 protein and over-expression of the wildtype receptor is found in 42% of bladder tumors without detectable FGFR3 mutation, indicating that this may be an additional mechanism for FGFR3 dysregulation (Tomlinson et al., 2007a). In this study, FGFR3 expression in transduced cells was comparable to levels in the bladder tumor cell line TCC97-7 and representative of the range of over-expression observed in bladder tumors (Tomlinson et al., 2007b). It is currently unclear whether during malignant transformation FGFR3 up-regulation precedes or follows its mutation. In this study, no proliferative or survival advantage was observed in cells over-expressing wildtype receptor, although phosphorylation and signaling could be induced by FGF1 stimulation, suggesting that over-expression of wildtype FGFR3 may be advantageous in those tumors with high levels of endogenous or exogenous ligand.
Notably, our data indicate a clear hierarchy for specific mutations in relation to ligand-independence, signalling and phenotypic effect, that reflects the frequency at which they are detected in bladder tumors (S249C>Y375C>K652E), thus suggesting that the spectrum of FGFR3 mutations observed in UC may relate to selection for their potency. In contrast, all forms of mutant FGFR3 were equally transforming in NIH-3T3, underlining the danger of extrapolating to urothelial cells conclusions drawn from functional studies in other cell lines. Cell type- and mutation-specific differences in the effects of mutant FGFR3 are also emphasized by previous studies. In NIH-3T3, in line with our results, all types of mutant FGFR3 were highly phosphorylated and able to induce transformation (Agazie et al., 2003; Bernard-Pierrot et al., 2006; Chesi et al., 2001; Hart et al., 2001; Ronchetti et al., 2001; Webster and Donoghue, 1997). In other cell lines, however, the Y375C mutant was only moderately phosphorylated and partially ligand-dependent (Adar et al., 2002; Bonaventure et al., 2007; Harada et al., 2007), which is consistent with our observations in TERT-NHUC. In HEK293T, the K650E mutation was highly phosphorylated, completely ligand-independent and able to activate PLCγ1 and STAT1 (Gibbs and Legeai-Mallet, 2007; Harada et al., 2007; Su et al., 1997), while in multiple myeloma cell lines it showed only weak phosphorylation, was partially ligand-dependent and unable to activate STAT1 signaling even after FGF stimulation (Ronchetti et al., 2001). An earlier functional study in BaF3 cells also indicated partial ligand-dependence of the K650E mutant (Naski et al., 1996). To our knowledge, the degree of ligand-independence of the S249C mutant has not been examined previously, although the adjacent mutation R248C was shown to be fully activated in BaF3 cells (Naski et al., 1996) but only moderately phosphorylated in HEK293 cells (Bonaventure et al., 2007; Gibbs and Legeai-Mallet, 2007).
As K652E FGFR3 displayed a high level of constitutive phosphorylation when expressed in normal urothelial cells, the reasons for the less intense signaling and phenotypic effect is not completely clear. Differential localization of K652E FGFR3 and signaling from distinct cellular compartments could explain some of these differences. Interestingly, in both NIH-3T3 and TERT-NHUC we detected phosphorylation of the 120 KDa form of K652E, but not of S249C or Y375C FGFR3. This observation is consistent with previous reports showing that the immature form of K650E FGFR3 protein may be activated and may signal from the endoplasmic reticulum rather than the cell membrane (Lievens and Liboi, 2003). However, in TERT-NHUC K652E was able to activate FRS2, an observation consistent with membrane rather then cytoplasmic localization (Lievens et al., 2006). Another possible explanation of the differences between K652E and the other mutants is that dimerization of the receptor, which does not occur with the K652E mutation, may be essential for the physical interaction with some downstream signaling effector or for trans-phosphorylation of some tyrosine residues, such as Y762. This may explain why in urothelial cells K652E was capable of phosphorylating PLCγ1 only after stimulation with FGF1.
In conclusion, we have elucidated the signaling pathways activated by mutant FGFR3 in normal urothelial cells and identified putative downstream targets. We have demonstrated that mutant FGFR3 confers both proliferative and survival advantages to urothelial cells, and that this effect is partially PLCγ1-dependent. We have also demonstrated a clear correlation between the degree of ligand-independence and constitutive signaling of specific mutant FGFR3 proteins that is directly related to the frequency of these mutations in bladder cancer, implying a proportional selective advantage for cells carrying these mutations.
Materials and Methods
Cell culture
Telomerase-immortalized normal human urothelial cells (TERT-NHUC) (Chapman et al., 2006) were cultured in Keratinocyte Growth Medium 2 (KGM2) (Promocell GmbH., Heidelberg, Germany) supplemented with provided growth factors, 0.09 mM CaCl2, and 30 ng/ml cholera toxin (Sigma-Aldrich Company Ltd., Poole, UK). For FGF1 stimulation experiments, cells were treated with 20 ng/ml recombinant human FGF1 and 10 μg/ml heparin (R&D Systems Europe Ltd., Abingdon, UK) after 1 h incubation in depleted medium (KGM2 without added growth factors). NIH-3T3 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich) with 10% FCS (Invitrogen Ltd, Paisley, UK) and 2 mM L-glutamine. Both cell lines were cultured in a humidified atmosphere at 37°C in 5% CO2.
Expression vectors and transduction of cell lines
Site-directed mutagenesis on FGFR3 IIIb cDNA was used to create the S249C, S249C+K510A (‘kinase-dead’, S249C KD) (Monsonego-Ornan et al., 2002), S249C+Y762F, Y375C, and K652E mutations. The presence of the correct mutation was verified by sequencing and wildtype and mutant FGFR3 were cloned into a retroviral expression vector (pFB; Stratagene, La Jolla, CA) containing a hygromycin resistance cassette (Tomlinson et al., 2005). The expression vectors were transfected into Phoenix A cells using siPORT XP-1 (Ambion, Huntingdon, UK). TERT-NHUC and NIH-3T3 cells were incubated with retroviral supernatant containing 8 μg/ml polybrene and selected with hygromycin 48 h after transduction.
Phenotypic assays
For all phenotypic assays, 1-3×103 cells/cm2 were seeded in triplicate and each experiment was repeated at least three times. All experiments were carried out shortly after transduction (<30 population doublings). To investigate anchorage-independent growth, cells were cultured in medium containing 0.3% agarose with weekly feeding. Colonies bigger than 200 μm were counted on day 28, after staining with 8 mM p-iodonitrotetrazolium violet (Sigma-Aldrich). For growth curves, cells were counted with a Z2 Coulter™ analyzer (Beckman Coulter Ltd., High Wycombe, UK). The Guava EasyCyte™ Plus System 3 (Guava Technologies, Inc., Stamford, UK) was used to examine cell viability with the ViaCount® dye-exclusion test (Guava Technologies), and cell cycle profile with propidium iodide staining. Cell cycle profiles were analysed using ModFit LT 2.0 software (Verity Software House, Topsham, US).
Western blotting
Protein extraction and quantification was carried out as described (Tomlinson et al., 2005). Heat-denatured proteins were separated by 7.5-12.5% SDS-PAGE, transferred to Hybond-C membrane (GE Healthcare Life Sciences, Buckinghamshire, UK), and incubated with 1:1000 primary antibodies. For detection of FGFR3 dimers, the denaturation step was omitted. Primary antibodies used were: anti-FGFR3 clone B9, anti-ERK1/2, anti-phospho-ERK1/2 (Tyr 204), anti-RB, and anti-CDK1 from Insight Biotechnology Ltd. (Wembley, UK), anti-phospho-PLCγ1 (Tyr 783), anti-PLCγ1, anti-phospho-FRS2α (Tyr 436), anti-phospho-SRC family (Tyr 416), anti-phospho-p38 MAPK (Thr 180/Tyr 182), anti-phospho-RB (Ser 807/811), anti-phospho-AKT (Ser 473), anti-MCL1, anti-BCL2 and anti-BCL-XL from New England Biolabs Ltd. (Hitchin, UK). Bound primary antibodies were detected using HRP-conjugated secondary antibody and the ECL Plus kit (GE Healthcare Life Sciences). Blots were reprobed with anti-actin clone AC-40 (Sigma-Aldrich Company Ltd) or anti-tubulin alpha (MorphoSys UK Ltd., Kidlington, UK) as loading control.
Immunoprecipitation
For detection of phosphorylated FGFR3, protein lysates were incubated overnight at 4°C with 2 μl of rabbit anti-FGFR3 cytoplasmic antibody (Sigma-Aldrich Company Ltd), followed by 2 h incubation at 4°C with protein A-sepharose® beads (Sigma-Aldrich Company Ltd). Eluted proteins were separated by 7.5% SDS-PAGE. Phosphorylated FGFR3 was detected using 1:1000 anti-phosphotyrosine antibody clone 4G10 (Millipore, Hampshire, UK) in 5% BSA (Sigma-Aldrich).
Statistical analysis
Significant differences between cell types were assessed using the Mann-Whitney test, with the SPSS 12.0 statistical analysis software (SPSS Inc., Chicago, US). A p≤ 0.05 was accepted as significant.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Dr D Podolsky for donating the FGFR3 IIIb cDNA, and Dr R Agami for the pFB expression vector. This work was funded by a grant from Cancer Research UK (C6228/A5433).
Footnotes
Conflict of interests
The authors declare no conflict of interest.
References
- Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J Bone Miner Res. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- Agazie YM, Movilla N, Ischenko I, Hayman MJ. The phosphotyrosine phosphatase SHP2 is a critical mediator of transformation induced by the oncogenic fibroblast growth factor receptor 3. Oncogene. 2003;22:6909–6918. doi: 10.1038/sj.onc.1206798. [DOI] [PubMed] [Google Scholar]
- Bernard-Pierrot I, Brams A, Dunois-Larde C, Caillault A, Diez de Medina SG, Cappellen D, et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis. 2006;27:740–747. doi: 10.1093/carcin/bgi290. [DOI] [PubMed] [Google Scholar]
- Bonaventure J, Horne WC, Baron R. The localization of FGFR3 mutations causing thanatophoric dysplasia type I differentially affects phosphorylation, processing and ubiquitylation of the receptor. Febs J. 2007;274:3078–3093. doi: 10.1111/j.1742-4658.2007.05835.x. [DOI] [PubMed] [Google Scholar]
- Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- Chapman EJ, Hurst CD, Pitt E, Chambers P, Aveyard JS, Knowles MA. Expression of hTERT immortalises normal human urothelial cells without inactivation of the p16/Rb pathway. Oncogene. 2006;25:5037–5045. doi: 10.1038/sj.onc.1209513. [DOI] [PubMed] [Google Scholar]
- Chen J, Williams IR, Lee BH, Duclos N, Huntly BJ, Donoghue DJ, et al. Constitutively activated FGFR3 mutants signal through PLCgamma-dependent and -independent pathways for hematopoietic transformation. Blood. 2005;106:328–337. doi: 10.1182/blood-2004-09-3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV, et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517–1525. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- d'Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. 1998;9:71–78. [PubMed] [Google Scholar]
- Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Garcia-Espana A, Salazar E, Sun TT, Wu XR, Pellicer A. Differential expression of cell cycle regulators in phenotypic variants of transgenically induced bladder tumors: implications for tumor behavior. Cancer Res. 2005;65:1150–1157. doi: 10.1158/0008-5472.CAN-04-2074. [DOI] [PubMed] [Google Scholar]
- Gibbs L, Legeai-Mallet L. FGFR3 intracellular mutations induce tyrosine phosphorylation in the Golgi and defective glycosylation. Biochim Biophys Acta. 2007;1773:502–512. doi: 10.1016/j.bbamcr.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Guy M, Moorghen M, Bond JA, Collard TJ, Paraskeva C, Williams AC. Transcriptional down-regulation of the retinoblastoma protein is associated with differentiation and apoptosis in human colorectal epithelial cells. Br J Cancer. 2001;84:520–528. doi: 10.1054/bjoc.2000.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner C, Vogt T, Hartmann A. FGFR3 mutations in benign skin tumors. Cell Cycle. 2006;5:2723–2728. doi: 10.4161/cc.5.23.3509. [DOI] [PubMed] [Google Scholar]
- Harada D, Yamanaka Y, Ueda K, Nishimura R, Morishima T, Seino Y, et al. Sustained phosphorylation of mutated FGFR3 is a crucial feature of genetic dwarfism and induces apoptosis in the ATDC5 chondrogenic cell line via PLCgamma-activated STAT1. Bone. 2007;41:273–281. doi: 10.1016/j.bone.2006.11.030. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2:E65–67. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Donoghue DJ. Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol Biol Cell. 2001;12:931–942. doi: 10.1091/mbc.12.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoui M, Hofmann-Radvanyi H, Zimmermann U, Couvelard A, Degott C, Faridoni-Laurens L, et al. No evidence of somatic FGFR3 mutation in various types of carcinoma. Oncogene. 2001;20:5059–5061. doi: 10.1038/sj.onc.1204651. [DOI] [PubMed] [Google Scholar]
- Korkolopoulou P, Lazaris A, Konstantinidou AE, Kavantzas N, Patsouris E, Christodoulou P, et al. Differential expression of bcl-2 family proteins in bladder carcinomas. Relationship with apoptotic rate and survival. Eur Urol. 2002;41:274–283. doi: 10.1016/s0302-2838(02)00003-9. [DOI] [PubMed] [Google Scholar]
- Legeai-Mallet L, Benoist-Lasselin C, Munnich A, Bonaventure J. Overexpression of FGFR3, Stat1, Stat5 and p21Cip1 correlates with phenotypic severity and defective chondrocyte differentiation in FGFR3-related chondrodysplasias. Bone. 2004;34:26–36. doi: 10.1016/j.bone.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35–44. doi: 10.1093/hmg/8.1.35. [DOI] [PubMed] [Google Scholar]
- Lievens PM, Liboi E. The thanatophoric dysplasia type II mutation hampers complete maturation of fibroblast growth factor receptor 3 (FGFR3), which activates signal transducer and activator of transcription 1 (STAT1) from the endoplasmic reticulum. J Biol Chem. 2003;278:17344–17349. doi: 10.1074/jbc.M212710200. [DOI] [PubMed] [Google Scholar]
- Lievens PM, Roncador A, Liboi E. K644E/M FGFR3 Mutants Activate Erk1/2 from the Endoplasmic Reticulum through FRS2alpha and PLCgamma-independent Pathways. J Mol Biol. 2006 doi: 10.1016/j.jmb.2006.01.058. [DOI] [PubMed] [Google Scholar]
- Martinez-Torrecuadrada J, Cifuentes G, Lopez-Serra P, Saenz P, Martinez A, Casal JI. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clin Cancer Res. 2005;11:6280–6290. doi: 10.1158/1078-0432.CCR-05-0282. [DOI] [PubMed] [Google Scholar]
- Monsonego-Ornan E, Adar R, Rom E, Yayon A. FGF receptors ubiquitylation: dependence on tyrosine kinase activity and role in downregulation. FEBS Lett. 2002;528:83–89. doi: 10.1016/s0014-5793(02)03255-6. [DOI] [PubMed] [Google Scholar]
- Murakami M, Elfenbein A, Simons M. Non-canonical fibroblast growth factor signalling in angiogenesis. Cardiovasc Res. 2008;78:223–231. doi: 10.1093/cvr/cvm086. [DOI] [PubMed] [Google Scholar]
- Naski MC, Wang Q, Xu J, Ornitz DM. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchetti D, Greco A, Compasso S, Colombo G, Dell'Era P, Otsuki T, et al. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene. 2001;20:3553–3562. doi: 10.1038/sj.onc.1204465. [DOI] [PubMed] [Google Scholar]
- Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D, Basilico C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1361–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- Sibley K, Cuthbert-Heavens D, Knowles MA. Loss of heterozygosity at 4p16.3 and mutation of FGFR3 in transitional cell carcinoma. Oncogene. 2001a;20:686–691. doi: 10.1038/sj.onc.1204110. [DOI] [PubMed] [Google Scholar]
- Sibley K, Stern P, Knowles MA. Frequency of fibroblast growth factor receptor 3 mutations in sporadic tumours. Oncogene. 2001b;20:4416–4418. doi: 10.1038/sj.onc.1204543. [DOI] [PubMed] [Google Scholar]
- Su WC, Kitagawa M, Xue N, Xie B, Garofalo S, Cho J, et al. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature. 1997;386:288–292. doi: 10.1038/386288a0. [DOI] [PubMed] [Google Scholar]
- Tomlinson DC, Baldo O, Harnden P, Knowles MA. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol. 2007a;213:91–98. doi: 10.1002/path.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson DC, Hurst CD, Knowles MA. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene. 2007b doi: 10.1038/sj.onc.1210399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson DC, L'Hote CG, Kennedy W, Pitt E, Knowles MA. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Res. 2005;65:10441–10449. doi: 10.1158/0008-5472.CAN-05-1718. [DOI] [PubMed] [Google Scholar]
- van Oers JM, Adam C, Denzinger S, Stoehr R, Bertz S, Zaak D, et al. Chromosome 9 deletions are more frequent than FGFR3 mutations in flat urothelial hyperplasias of the bladder. Int J Cancer. 2006;119:1212–1215. doi: 10.1002/ijc.21958. [DOI] [PubMed] [Google Scholar]
- van Rhijn BW, van Tilborg AA, Lurkin I, Bonaventure J, de Vries A, Thiery JP, et al. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur J Hum Genet. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- Wallerand H, Bakkar AA, de Medina SG, Pairon JC, Yang YC, Vordos D, et al. Mutations in TP53, but not FGFR3, in urothelial cell carcinoma of the bladder are influenced by smoking: contribution of exogenous versus endogenous carcinogens. Carcinogenesis. 2005;26:177–184. doi: 10.1093/carcin/bgh275. [DOI] [PubMed] [Google Scholar]
- Webster MK, D'Avis PY, Robertson SC, Donoghue DJ. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol Cell Biol. 1996;16:4081–4087. doi: 10.1128/mcb.16.8.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. Enhanced signaling and morphological transformation by a membrane-localized derivative of the fibroblast growth factor receptor 3 kinase domain. Mol Cell Biol. 1997;17:5739–5747. doi: 10.1128/mcb.17.10.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells A, Grandis JR. Phospholipase C-gamma1 in tumor progression. Clin Exp Metastasis. 2003;20:285–290. doi: 10.1023/a:1024088922957. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Monden T, Ikeda K, Izawa H, Fukuda K, Fukunaga M, et al. Coexpression of cdk2/cdc2 and retinoblastoma gene products in colorectal cancer. Br J Cancer. 1995;71:1231–1236. doi: 10.1038/bjc.1995.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.