

Abstract
Genetic amplification, mutation, and translocation are known to play a causal role in the up-regulation of an oncogene in cancer cells. Here we report an emerging role of microRNA, the epigenetic deregulation of which may also lead to this oncogenic activation. SOX4, an oncogene belonging to the SRY-related high-mobility-group-box family, was found to be over-expressed (P<0.005) in endometrial tumors (n=74) compared with uninvolved controls (n=20). This gene is computationally predicted to be the target of a microRNA, miR-129-2. When compared to the matched endometria, the expression of miR-129-2 was lost in 27 of 31 primary endometrial tumors that also showed a concomitant gain of SOX4 expression (P<0.001). This inverse relationship is associated with hypermethylation of the miR-129-2 CpG island, which was observed in endometrial cancer cell lines (n=6) and 68% of 117 endometrioid endometrial tumors analyzed. Reactivation of miR-129-2 in cancer cells by pharmacological induction of histone acetylation and DNA demethylation resulted in decreased SOX4 expression. In addition, restoration of miR-129-2 by cell transfection led to decreased SOX4 expression and reduced proliferation of cancer cells. Further analysis found a significant correlation of hypermethylated miR-129-2 with microsatellite instability and MLH1 methylation status (P<0.001), and poor overall survival (P<0.039) in patients. Therefore, these results imply that the aberrant expression of SOX4 is, in part, caused by epigenetic repression of miR-129-2 in endometrial cancer. Unlike the notion that promoter hypomethylation may up-regulate an oncogene, we present a new paradigm in which hypermethylation-mediated silencing of a microRNA de-represses its oncogenic target in cancer cells.
Keywords: DNA methylation, epigenetics, miR-129-2, SOX4
Introduction
The SRY-related high-mobility group box 4 gene, or SOX4, is known to be over-expressed in prostate, hepatocellular, lung, bladder and medulloblastoma cancers with poor prognostic features and advanced disease status (1-6). Its oncogenic potentials have been demonstrated in knock-in cells leading to aberrant transformation while the proliferation and metastatic capability has been greatly reduced in knock-out cancer cells (1-4, 7). Functional analysis has shown that SOX4 belongs to the TCF/LEF family of transcription factors that mediate transcription responses to Wnt signaling (2, 8). More recently, a genome-wide chromatin immunoprecipitation study has further uncovered additional transcriptional targets of SOX4 that are associated with TGFβ, Hedgehog, and Notch pathways, microRNA processing, and tumor metastasis (7, 9).
Genetic mechanisms leading to aberrant expression of SOX4 have been explored in cancer cells. SOX4 is mapped to chromosome 6p22, a region frequently amplified in lung, bladder, endometrial cancers (2, 10-12). Somatic mutations have also been found in the exon region of this intronless gene in lung cancer (2). However, there is no experimental evidence to demonstrate positive correlation between these reported genetic alterations and the aberrantly increased SOX4. One emerging mechanism is microRNA (miRNA)-mediated oncogene expression.
miRNAs, a class of small non-coding RNAs (18-25 nt), are known to form imperfect paring at the 3′-end of untranslated regions (UTR) of a target locus, resulting in mRNA degradation or translational inhibition (13). Through this post-transcriptional regulatory mechanism, miRNAs control a variety of physiological processes in normal cells, and deregulation of miRNAs may promote tumorigenesis (14). Increasing evidence indicates that epigenetic perturbations may contribute to abnormal miRNA expression in cancer cells (15). One well-studied epigenetic phenomenon is DNA methylation frequently observed in the promoter CpG island regions of genes (16). While promoter hypermethylation is associated with transcriptional silencing of coding genes for tumor suppressor functions, promoter hypomethylation can be related to activation of oncogenes in cancer cells (16, 17). We therefore speculate whether methylation alteration may commonly occur in non-coding miRNAs, resulting in de-regulation of its target genes in cancer cells.
Here we report, for the first time, that the SOX4 oncogene is also over-expressed in endometrial cancer. A miRNA, miR-129-2, was computationally predicted and functionally validated to be an upstream regulator of SOX4. A CpG island encompassing the miR-129-2 locus was found to be hypermethylated in endometrial cancer cell lines and primary tumors. We further demonstrate that this methylation-mediated silencing has a causal role for SOX4 activation in endometrial cancer.
Materials and Methods
Endometrial specimens and cell lines
Tissue specimens (117 tumors and 8 uninvolved controls) were obtained as part of our ongoing work on characterizing molecular alterations in endometrioid endometrial carcinomas. All participants consented to both molecular analyses and follow-up studies, and the protocols were approved by the Human Studies Committee at the Washington University and the Ohio State University. Clinicopathological variables of tumors, including age, stage, grade, microsatellite instability (MSI) and MLH1 methylation, were summarized in Supplementary Table 1 and reported in our previous study (18). Human endometrial cancer cell lines, AN3CA, HEC1A, Ishikawa, KLE, RL95-2 and SK-UT-1B were routinely maintained in our laboratory (19), and ECC-1 cells were obtained from ATCC. For epigenetic studies, these cells were treated with 5-aza-2′-deoxycytidine (DAC, 0.5 μM, Sigma) for 48 h and/or trichostatin A (TSA, 5 μM, Sigma) for 24 h. DNA and RNA from treated and untreated cells were isolated using standard protocols (20).
Endometrial tissue microarray
Tissue microarray slides, each containing a total of 74 endometrial tumors and 20 normal specimens, were obtained from US Biomax. Patients' characteristics were summarized in Supplementary Table 2. These slides were pre-processed prior to immunohistostaining. Antigen retrieval was performed by heat-induced epitope retrieval, in which the slides were placed in Dako TRS solution (pH 6.1) for 25 min at 94°C. Slides were then placed on a Dako autostainer with primary antibody (SOX4, 1:50, Abcam) and incubated for 60 min at room temperature. Staining was visualized with DAB chromogen, and slides were then counterstained and dehydrated through graded ethanol solutions. Images were digitally scanned with iScan™ (BioImagene) and analyzed with the BioImagene TissueMine™ software for discriminating immunohistochemically stained cancer cells from the surrounding stromal tissue. Intensities of nuclear staining were measured as segmented images then quantified. The algorithm reported the number and percentage of positively stained and non-stained nuclei.
Cell transfection
ECC-1 and Ishikawa (3×106) cells were transfected with mature miR-129-2 mimics (miR-129-3p and -5p, 2.5 nM, Ambion), pre-miR negative control (#1, 2.5 nM, Ambion), SOX4 siGenome SMART pool siRNA (2.5 nM, Dharmacon), siGenome non-targeting siRNA pool (#1, 2.5 nM, Dharmacon) and plasmids using the Cell Line Nucleofector Kit (Lonza) according to manufacturer's instructions.
Reverse transcription and quantitative PCR (RT-qPCR)
Total RNA (1 μg) was reverse transcribed with the Superscript III reverse transcriptase (Invitrogen). PCR was performed as described previously (20). Specific primers for amplification were listed in the Supplementary Table 3. The relative expression of a coding gene in cells was determined by comparing the threshold cycle (Ct) of the gene against the Ct of GAPDH (20).
For detecting mature miRNA molecules (i.e., miR-129-3p and miR-129-5p), reverse transcription was performed following the Applied Biosystems Taq Man MicroRNA Assay protocol. This sensitive system has been designed to specifically detect mature miRNAs that are distinct from their precursors. In addition, the assay can often distinguish between miRNA targets that differ by only a single nucleotide (21). All reactions were performed in triplicate. The expression of miR-129-2-3p or -5p was normalized using RNU48 or U6. The expression relative to RNU48 or U6 was determined using the 2−ΔCT method.
Western blot analysis
Whole cell protein lysates were extracted with the M-PER Mammalian Protein Extraction Reagent (Pierce). Western blot analysis was conducted using antibodies against SOX4 (Abcam) and β-actin (Santa Cruz).
Cell proliferation assay
Cell proliferation was monitored using the CellTiter 96® Aqueous solution (Promega). Endometrial cancer cells (3,000/well) transfected with either miR-129-2, negative control miRNA, SOX4 siRNA or non-targeting siRNA pool were seeded in 96-well plates. Cell proliferation was documented every 24 h following the manufacturer's protocol. To measure cell proliferation, 20 μL of MTS labeling reagent was added to each well and incubated at 37°C for 1 h. Absorbance was measured at 490 nm in the ELISA reader (Molecular Devices).
3′-UTR reporter assay
The full-length 3′-UTRs of SOX4 and UBE2F, generous gifts from Dr. Joan Massague (Memorial Sloan-Kettering Cancer Center, New York), were cloned into the Psicheck 2 dual luciferase reporter vector (Promega). ECC-1 or Ishikawa cells were transfected with reporter constructs and a miR-129-2 and/or its antagomir targeting endogenous miR-129-2 (Ambion). Cells were lysed at 24 h after transfection and the ratio of Renilla to firefly luciferase was measured using the dual luciferase assay (Promega). Normalized Renilla to firefly ratios were determined in the presence or absence of miR-129-2 inhibition on SOX4 UTR luciferase activities.
Combined bisulfite restriction analysis (COBRA)
Genomic DNA (500 ng) was treated sodium bisulfite using the EZ DNA Methylation kit (Zymo Reasearch) following manufacturers' recommended protocols. COBRA analysis was used to evaluate methylation of miR-129-2. Target sequences were amplified by PCR, and the products were digested with AciI, (New England Biolabs) to identify methylated sequences. Primer sequences and PCR conditions are presented in Supplementary Table 3. Digested and non-digested PCR products were resolved on 2% agarose gels stained with ethidium bromide. DNA fragments digested by AciI were scored as “methylated” in a given sample.
MassARRAY analysis
To quantify methylation levels of the miR-129-2 CpG islands in clinical samples, the high-throughput MassARRAY platform (Sequenom) was carried out as described previously (22). Briefly, bisulfite-treated DNA was amplified with primers for the miR-129-2 CpG island. The PCR products were spotted on a 384-pad SpectroCHIP (Sequenom) and followed by spectral acquisition on a MassARRAY Analyzer. Methylation data of individual units (1-3 CpG sites per unit) were generated by the EpiTyper software (Sequenom).
Statistical and survival analyses
Student's t or Wilcox test was used to compare the immunohistostaining, cell proliferation, invasion, apoptosis, and RT-qPCR results in different treatment groups. A significance was assigned as * if P < 0.05. The relationship between methylation levels of miR-129-2 and relevant categorical clinicopathological covariates was performed using the Wilcox rank sum test for binary variables. The Kruskal Wallis test was used followed by a pairwise Wilcoxon rank sum test. Overall survival (OS) was defined as the time interval from the date of diagnosis to the date of death or latest follow-up if alive. Recurrence-free survival (RFS) was defined as the time interval from surgery to recurrence, disease progression or latest follow-up. The Kaplan-Meier product limit method was used to estimate the empirical survival functions for categorical covariates accompanied with P value from the Log-rank test. Univariate Cox proportional hazard models were used to access effect of a continuous covariate on survival outcomes. Multivariate Cox proportional hazard models were fitted to examine the potential predictive effect of covariates of interest on survival outcomes after adjustment for confounding factors. All tests were two-sided and all analyses were performed using R.
Results
SOX4 is over-expressed in endometrioid endometrial carcinomas
To determine whether SOX4 is aberrantly expressed in endometrial tumors, we conducted tissue microarray analysis in a panel of 74 endometrioid endometrial carcinomas and 20 uninvolved controls (see representative immunohistostaining images in Fig. 1A, left). The nuclear staining intensities of SOX4 were significantly higher in tumor sections than those of normal tissue sections (P < 0.005, Fig. 1A, right). This finding is consistent with the RT-qPCR results in which the levels of SOX4 mRNA were higher in tumors (n=31) compared to the adjacent normal counterparts (P < 0.001, Fig. 2D, left).
Figure 1.
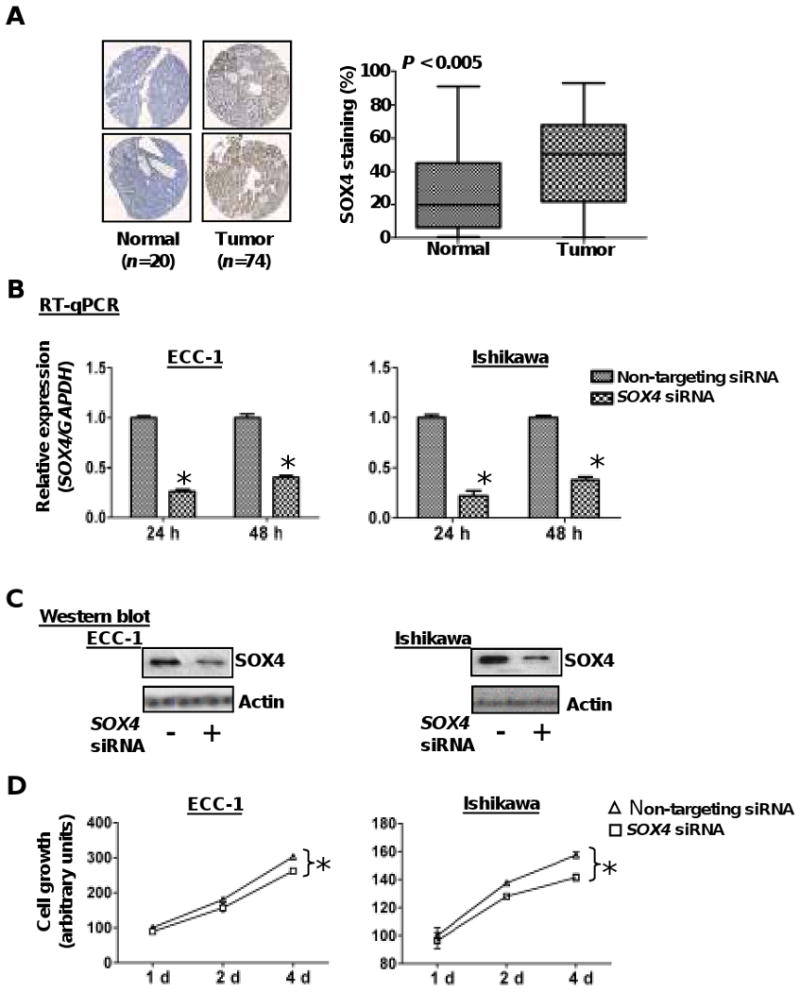
SOX4 is overexpressed in endometrial tumors. A, representative photographs of endometrial tissue microarrays (1.5mm core diameter) which underwent immunohisochemical staining for SOX4 and were scored for nuclear staining by the TissueMine software. Boxplots of SOX4 nuclear staining for normal tissue (n=20) and endometrial tumors (n=74) P < 0.005. B & C, relative expression levels of SOX4 mRNA and protein in ECC-1 and Ishikawa cells after transient transfection with SOX4 siRNA or a pool of non-targeting siRNA oligonucleotides for 24 and/or 48 h. GADPH and β-actin served as internal controls for RT-qPCR and western blotting respectively. Error bars represent SD among triplicates; *, P<0.05. D, cellular proliferation was measured by MTS assay in endometrial cancer cells transfected with SOX4 siRNA or non-targeting siRNA. Transfectants (3,000/well) were placed in 96-well plates and proliferation was measured every 24 h. Each point represents the mean value of at least triplicates. *, P < 0.05.
Figure 2.
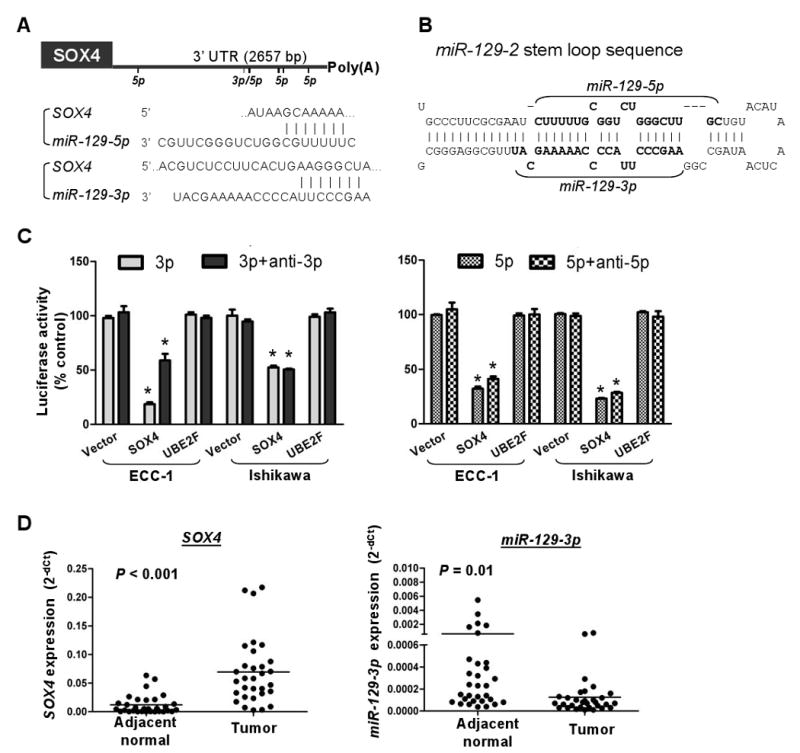
miR-129-2 directly targets SOX4. A, bioinformatic analysis of miR-129-3 and -5p (the mature forms of miR-129-2) predicted binding sites in the SOX4 3′-UTR (bars under line). The predicted pairing of mRNA target region (top) and miRNA (bottom) is as indicted, where a line (“|”) indicates hydrogen bonding. B, predicted secondary structure of miR-129-2. The stem loop structure of miR-129-2 is a precursor to two mature miRNAs: miR-129-3p, and miR-129-5p (bold text). C, miR-129-3p and -5p suppressed the expression of a luciferase vector with the SOX4 3′-UTR. A luciferase expression vector with the 3′-UTR of SOX4 and UBE2F or an empty vector was transfected into ECC-1 and Ishikawa cells. At the same time, anti-miR-129-3p or -5p (anti-3p or anti-5p) and/or miR-129-3p or 5p (3p or 5p) were introduced. Twenty-four hours after the transfection, the cells were harvested and assayed for luciferase activity. Renilla luciferase was used for normalization to empty vector for the transfection efficiency. Error bars, SD; *, P < 0.05 compared with empty vector. D, dot plots showing an inverse relationship between SOX4 (left) and miR-129-3p (right) mRNA expression in 31 pairs of endometrial tumors and adjacent normal tissues. Horizontal bars represent mean expression levels. Significant differences were determined using Student's t tests.
In a knock-out study, we assessed the role of SOX4 in endometrial cancer cells. ECC-1 and Ishikawa cells were transiently transfected with SOX4 siRNA or a non-targeting control. Both protein and mRNA levels of SOX4 were found to be reduced to ≤50% in transfectants (Fig. 1B & C), resulting in attenuation of cell growth (Fig. 1D). This SOX4 knock-out, however, had no effects on cell invasion or migration (data not shown).
miR-129-2 is a negative regulator of SOX4
Because miRNA may have a potential role in mediating oncogene repression (14), we searched potential target sequences at the 3′-UTR of SOX4 using three software programs PicTar, TargetScan, and miRanda (23-25). Putative binding sites were found in a microRNA locus, miR-129-2, which is the precursor of two mature forms, miR-129-3p and -5p (Fig. 2A & B). To further validate this computational finding that miR-129-2 may negatively regulate SOX4, we assessed the expression of 3′-UTR of SOX4 in luciferase reporter assays (Fig. 2C). The expression of the SOX4 reporter was significantly reduced to ≤55% in miR-129-3p- or -5p-transfected ECC-1 and Ishikawa cells, but not in control cells. Transfection with either miRNA did not affect the reporter activity of a negative control gene, UBE2F, which has no known miR-129-2 binding sites on its UTR. Moreover, inhibition of miR-129-3p or -5p by antagomirs slightly enhanced the expression of SOX4, suggesting that this gene is a direct target of miR-129-2. We additionally confirmed this inverse relationship between miR-129-3p and SOX4 mRNA and expression using the aforementioned 31-paired samples (Fig. 2D).
Methylation-mediated silencing of miR-129-2 derepresses SOX4 expression
Since the expression of miR-129-2 is frequently lost in endometrial tumors (see Fig. 2D) and the 5′-end of this locus has a canonical CpG island (Fig. 3A), we determined whether this down-regulation is mediated by epigenetic mechanisms. Hypermethylation of this promoter CpG island was detected in six endometrial cancer cell lines, ECC-1, HE1A, Ishikawa, KLE, RL95-2, and SK-UT-1B, based on a COBRA assay (Fig. 3B). When these cells were treated with a demethylating agent, DAC (0.5 μM), a histone deacetylase inhibitor, TSA (5 μM), or in combination, reactivation of miR-129-3p was observed in 4 (Ishikawa, KLE, RL95-2, and SK-UT-1B) of the 6 cell lines analyzed treated with DAC (Fig. 3C). More profound effects of this re-expression were found in all these cell lines treated with TSA and the combination (Fig. 3C & Supplementary Fig. S1). These results suggest that the loss of miR-129-2 expression is associated with promoter hypermethylation in endometrial cancer cells. Interestingly, these epigenetic treatments might lead to the suppression of SOX4 in endometrial cancer cells. The effect occurred at the mRNA level in ECC-1, HEC1A, KLE, RL95-2 and SK-UT-1B cells (Fig. 3D, left). This suppression, however, was more prominent at the post-translational level for Ishikawa and RL95-2 cells (Fig. 3D, right). In addition, we showed that the expression of four other miR-129-2 targets, EIF2C3, PLCG1, RAB21 and STAT5B, was repressed in cancer cells treated with DAC and/or TSA (Supplementary Fig. S2). Taken together, the observation indirectly indicates that these epigenetic treatments may lead to reactivation of miR-129-2, which in turn represses the expression of SOX4.
Figure 3.

Reactivation of miR-129-2 in cancer cells by pharmacological induction of hyperacetylation and DNA demethylation lead to reduced SOX4 expression at both the mRNA and protein levels. A, genomic map of miR-129-2 CpG island and amplicon. Bar under line, CpG site; ↓, AciI cutting sites. B, COBRA analysis in endometrial cancer cell lines. u, unmethylated band; m, methylated bands; SssI, 100% methylated control; Blood, a mix of 4 normal peripheral blood samples as negative control; +, AciI restriction enzyme added; -, without AciI. C, relative expression levels of miR-129-3p in endometrial cancer cell lines treated with DAC and/or TSA in relation to untreated controls was determined by RT-qPCR analysis. RNU48 was used as internal control gene. Error bar, SD; *, P < 0.05 compared with untreated control of the same cell type. D, relative expression levels of SOX4 mRNA (right) and protein (right) indicating a down-regulation of SOX4 in endometrial cancer cells after treatment with DAC and/or TSA. GAPDH or β-actin was used as internal or loading control, respectively.
To further prove this inverse relationship, we conducted a functional knock-in study in two endometrial cell lines, ECC-1 and Ishikawa, harboring the epigenetically silenced miR-129-2. Transient transfection of these cells with either miR-129-3p or miR-129-5p resulted in reduction of both SOX4 mRNA and protein (Fig. 4A & B) (Note: Transfection efficiency was examined by measuring each mature microRNA, see Supplementary Fig. S3). Moreover, the knock-in of one of these mimics, miR-129-5p, greatly reduced the proliferation of these cancer cells (P < 0.05, Fig. 4C). In addition, the expression of three SOX4-regulated genes (DHX9, FZD5 and SEMA3C) (3, 9) was partially reduced in ECC-1 cells treated with DAC and/or TSA or ectopically transfected with miR-129-3p or -5p (Supplementary Fig. S4). Taken together, these in vitro studies suggest that miR-129-2 negatively regulates SOX4 and that promoter hypermethylation of this miRNA derepresses its expression.
Figure 4.
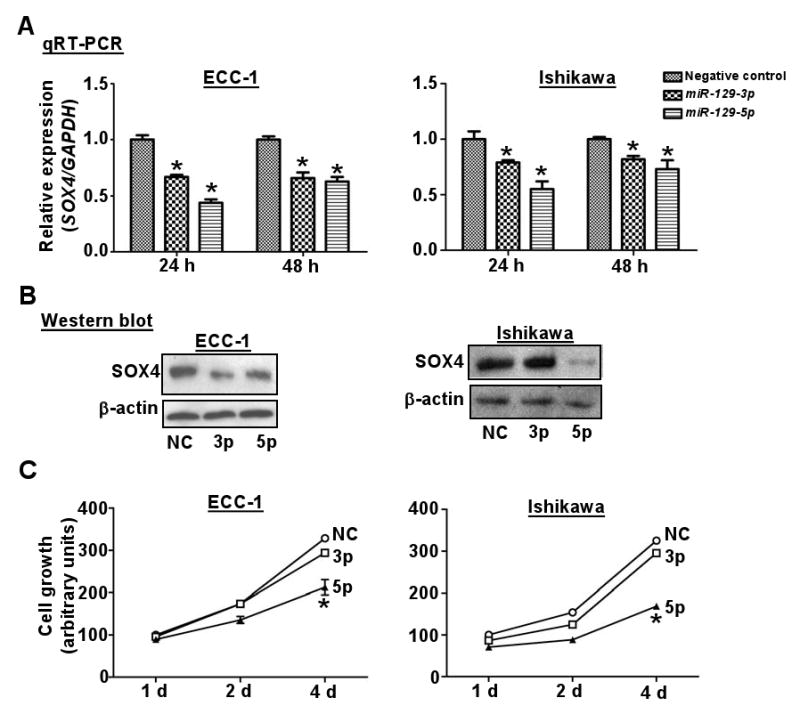
Functional analysis of miR-129-2 in endometrial cancer cell lines. A & B, relative levels of SOX4 mRNA (A) or protein (B) expression in ECC-1 and Ishikawa cells after transient transfection with miRNAs or negative control (NC) RNA oligonucleotides for 24 or 48 h. GADPH and β-actin served as an internal control of mRNA or protein, respectively. Error bars, SD from triplicates; *, P < 0.05 compared with NC at the same time point. C, cellular proliferation by MTS assay in the endometrial cancer cell lines ECC-1 and Ishikawa transfected with miR-129-3p (3p), miR-129-5p (5p), or negative control (NC). Proliferation measured as described in Fig. 1C.
Hypermethylation of miR-129-2 is associated with shorter patient survival, MSI, and MLH1 methylation in endometrial tumors
To confirm our in vitro findings in primary tumors, we first studied methylation patterns of the miR-129-2 CpG island (see Fig. 3A) in eight tumors with matched normal endometria by COBRA and quantitative MassARRAY. We showed that tumors were hypermentylated compared with the corresponding adjacent normal tissues (Supplementary Fig. S5). We then extended this methylation study to117 (34 recurrent and 83 non-recurrent) primary endometrioid endometrial tumors and 8 uninvolved controls. Quantitative analysis indicated that 80 of these primary tumors exhibited extensive methylation in 14 units (1-3 CpG sites per unit) of the miR-129-2 CpG island relative to those of uninvolved controls (P < 0.0005) (Fig. 5A). This methylation survey in the patient cohort also uncovered a pattern in which methylation accumulation may begin at the flanking regions and progressively invade to the core of this CpG island, consistent with the so-called methylation spread theory (26).
Figure 5.
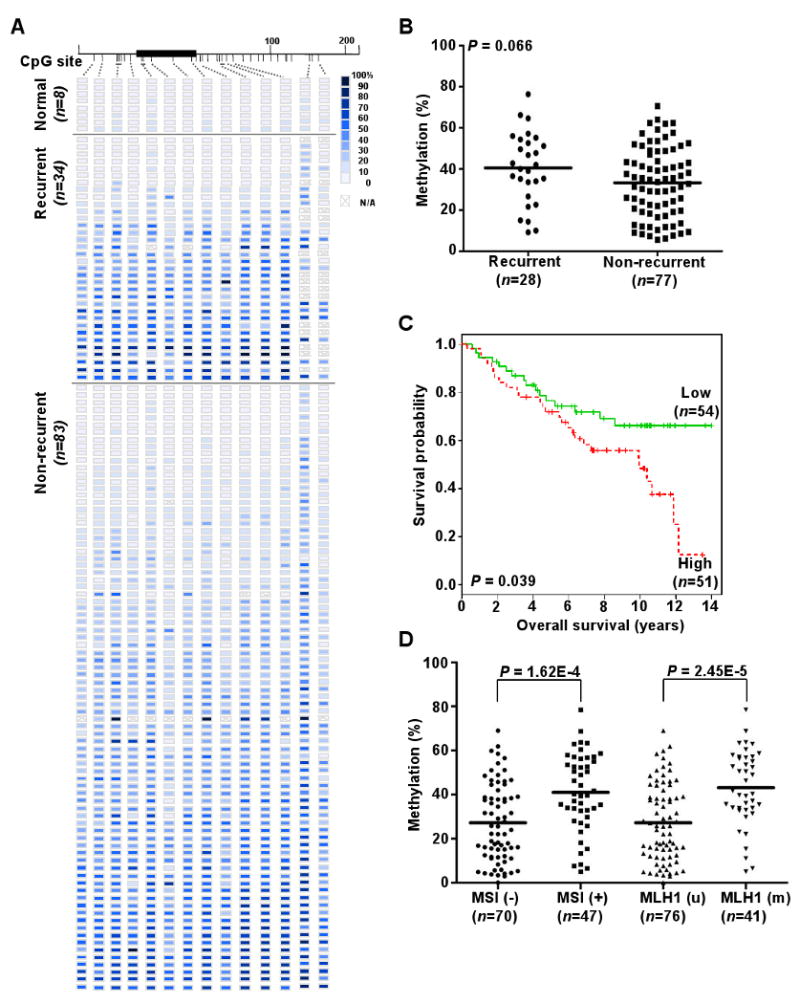
Methylation of miR-129-2 CpG island and clinic-pathological covariates analyses in primary endometrioid endometrial tumors. A, methylation profiles of 8 normal endometrial tissues, 34 recurrent, and 83 non-recurrent primary tumors created following MassARRAY analysis. Each row represents a sample and each column represents a CpG unit. Color-coding depicts the degree of methyltion with dark blue being 100% and white being 0%; N/A, not analyzable. B, dot plots point that miR-129-2 hypermethylation is moderately correlated with recurrent diseases. The mean of normal specimens in Fig. 5A was set as a threshold for analysis. Each dot represents the mean of each specimen on the first five CpG sites in Fig. 5A. Horizontal lines, mean values. P value was calculated by Wilcox test. C, Kaplan-Meier curves for overall survival. Samples were grouped according to the mean level of methylation for the first five CpG units of miR-129-2, when the mean exceeded the mean of normal specimens. Vertical bars represent excluded cases. P value estimated from Log-rank test. D, dot plots indicating the level of miR-129-2 promoter methylation is positively correlated with MSI and MLH1 methylation status.
The mean methylation level of each CpG unit was used to compare between the recurrent and non-recurrent group. More accumulation of miR-129-2 methylation was found in the former group, but the sample size of this cohort could be too small to reach statistical significance (P = 0.066, Fig. 4B). Detailed analysis of individual CpG units is shown in Supplementary Fig. S6. We also evaluated the association between miR-129-2 hypermethylation and patient survival. On univariate analysis, miR-129-2 hypermethylation was correlated with shorter overall survival (Cox Hazard Ratios, 1.02; P = 0.018), but not with recurrence-free survival (Cox Hazard Ratios, 1.02; P = 0.067) when the mean methylation level of normal controls was used to set the threshold. The Kaplan-Meier survival analysis indicated that patients with this hypermethylation had poor overall survival (Fig. 5C, P = 0.039, Log-rank test). Statistical analysis further revealed that miR-129-2 hypermethylation was significantly associated with MSI and MLH1 methylation (P < 0.0001, Fig. 5D). Endometrial tumors exhibiting the MSI phenotype usually have high replication error rates and genomic instability (18, 27). This defect is attributed, in part, to epigenetic repression of MLH1 that is responsible for DNA mismatch repair in normal cells (27).
Discussion
In addition to genetic alterations, promoter hypomethylation has been recognized as a epigenetic mechanism that contributes to oncogene activation in cancer cells (16, 17). In this case, a demethylation event is supposed to occur in an inactive, methylated promoter, leading to transcriptional reactivation of an oncogene. However, experimental proof for genuine promoter hypomethylation is frequently difficult and inconclusive because the outgrowth of a subpopulation of cancer cells may confound this epigenetic observation. For example, the oncogene of interest may have never been silent in a minor population of cancer-initiating cells while the majority of other cells display promoter hypermethylation of the gene. The increased expression of this oncogene may simply result from rapid expansion of these few cells that eventually take over the whole population during tumor progression. If this scenario indeed occurs, it cannot be a bona fide epigenetic event for oncogene activation.
In this study, we demonstrate that promoter hypermethylation can be directly associated with the activation of an oncogene. Specifically, this epigenetic event occurs in an upstream regulator, miR-129-2, that was shown to negatively regulate SOX4 oncogene in both knock-in and -out assays. miR-129-2 is embedded in a canonical CpG island on chromosome 11, which was found to be frequently hypermethylated in endometrial cancer. This epigenetic event results in miR-129-2 silencing, which in turn derepresses SOX4 expression. While we still cannot rule out hypomethylation of the SOX4 promoter CpG island as one of the causes, our present observation conclusively suggest that promoter hypermethylation of miR-129-2 is a common mechanism leading to the SOX4 over-expression in endometrial cancer.
It should be noted that a second CpG island is found 1.2-kb upstream from the first one analyzed in this study. In addition, the 5′-ends of two transcripts, BG120451 and BI964058, are located in this upstream region. It is possible that these transcripts are the primary RNAs for miR-129-2 (28). Future mapping of putative transcription start sites located in these 5′-end areas will provide insight into the transcriptional control of this microRNA locus. Additional methylation analysis may further determine the role of this second CpG island in the silencing of miR-129-2 during endometrial tumorigenesis.
We have additionally searched the miRBase database and found that the expression of SOX4 may be regulated by at least 16 putative miRNAs, including miR-129-1 located on chromosome 7. Similar to miR-129-2, the miR129-1 precursor produces mature miR-129-5p, which negatively regulates SOX4 as described in this study. Since there is no known CpG island located near or within the miR-129-1 locus, it remains to be determined whether this miRNA is transcriptionally silenced by other epigenetic mechanisms (e.g., EZH2-mediated histone modifications, 29) in endometrial cancer.
Five (miR-203, miR-335, miR-219-2, miR-596, and miR-618) of the other 15 miRNAs are located close to CpG islands based on our computational analysis (data not shown). Among these loci, miR-335 is the only one currently reported to be lost in primary breast tumors of recurrent patients (7). However, it remains to be determined whether promoter hypermethylation plays a role in the silencing of this locus. Future studies will therefore be important to study the coordinate regulation of these miRNAs on SOX4 repression. It is also possible that concurrent hypermethylation of these loci contributes to a CpG island methylator phenotype (30) and may improve the statistical power for predicting disease recurrence in our endometrial patient cohort (see Fig. 5B).
Extending from our present observation, epigenetically mediated silencing of other miRNAs that lead to tumor progression has recently been reported in the literature (15, 31-33). For example, ABL1 was showed to be a direct target of miR-203 (32). This miRNA was genetically and epigenetically down-regulated in leukemia cells expressing ABL1 or BCR-ABL1 fusion protein (32). Restoration of miR-203 in vitro led to reduced ABL1 and BCR-ABL1 expression and decreased proliferation of malignant cells (32). Taken together, these and our studies clearly indicate that epigenetic silencing of tumor-suppressive miRNAs can be important constituents in cancer epigenomes and that the event is as significant as hypomethylation of oncogenes and hypermethylation of tumor suppressor genes.
In conclusion, our findings support a comprehensive screening of miRNA regulators at the 3′-UTR regions of all known oncogenes. High-throughout functional studies can be developed to establish the inverse relationship between these tumor-suppressive miRNA loci and their target oncogenes. This type of omics study may find that epigenetically mediated silencing of these miRNAs can be as common as genetic alterations that contribute to oncogene activation in cancer cells. As such, the combined epigenetic and miRNA-based therapies can be feasible options for future treatments in cancer patients.
Supplementary Material
Acknowledgments
Grant support: NIH grants R01 CA069065, U01 ES015986 and U54 CA113001 (T. Huang), and R01 CA071754 (P. Goodfellow) and funds from the Ohio state University Comprehensive Cancer Center (T. Huang).
We thank Chieh Ti Kuo, Xiao-Hong Gu, Geoffrey Tsoi, Mary Ann Mallon and Drs. Kurtis H. Yearsley and Yu-I Weng for their technical assistance, and Drs. Joan Massague and Sohail F. Tavazoie (Memorial Sloan-Kettering Cancer Center, New York) for providing SOX4 and UBE2F 3′-UTR plasmids.
Footnotes
Supplementary data: The supplementary data include 6 figures and 3 tables.
References
- 1.Pramoonjago P, Baras AS, Moskaluk CA. Knockdown of Sox4 expression by RNAi induces apoptosis in ACC3 cells. Oncogene. 2006;25:5626–39. doi: 10.1038/sj.onc.1209566. [DOI] [PubMed] [Google Scholar]
- 2.Medina PP, Castillo SD, Blanco S, et al. The Sry-HMG box gene, SOX4, is a target of gene amplification at chromosome 6p in lung cancer. Hum Mol Genet. 2009;18:1343–52. doi: 10.1093/hmg/ddp034. [DOI] [PubMed] [Google Scholar]
- 3.Liao YL, Sun YM, Chau GY, et al. Identification of SOX4 target genes using phylogenetic footprinting-based prediction from expression microarrays suggests that overexpression of SOX4 potentiates metastasis in hepatocellular carcinoma. Oncogene. 2008;27:5578–89. doi: 10.1038/onc.2008.168. [DOI] [PubMed] [Google Scholar]
- 4.Liu P, Ramachandran S, Ali Seyed M, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;66:4011–9. doi: 10.1158/0008-5472.CAN-05-3055. [DOI] [PubMed] [Google Scholar]
- 5.Aaboe M, Birkenkamp-Demtroder K, Wiuf C, et al. SOX4 expression in bladder carcinoma: clinical aspects and in vitro functional characterization. Cancer Res. 2006;66:3434–42. doi: 10.1158/0008-5472.CAN-05-3456. [DOI] [PubMed] [Google Scholar]
- 6.Neben K, Korshunov A, Benner A, et al. Microarray-based screening for molecular markers in medulloblastoma revealed STK15 as independent predictor for survival. Cancer Res. 2004;64:3103–11. doi: 10.1158/0008-5472.can-03-3968. [DOI] [PubMed] [Google Scholar]
- 7.Tavazoie SF, Alarcon C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinner D, Kordich JJ, Spence JR, et al. Sox17 and Sox4 differentially regulate β-Catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol Cell Biol. 2007;27:7802–15. doi: 10.1128/MCB.02179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scharer CD, McCabe CD, Ali-Seyed M, Berger MF, Bulyk ML, Moreno CS. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res. 2009;69:709–17. doi: 10.1158/0008-5472.CAN-08-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurst CD, Fiegler H, Carr P, Williams S, Carter NP, Knowles MA. High-resolution analysis of genomic copy number alterations in bladder cancer by microarray-based comparative genomic hybridization. Oncogene. 2004;23:2250–63. doi: 10.1038/sj.onc.1207260. [DOI] [PubMed] [Google Scholar]
- 11.Levan K, Partheen K, Osterberg L, Helou K, Horvath G. Chromosomal alterations in 98 endometrioid adenocarcinomas analyzed with comparative genomic hybridization. Cytogenet Genome Res. 2006;115:16–22. doi: 10.1159/000094796. [DOI] [PubMed] [Google Scholar]
- 12.Heidenblad M, Lindgren D, Jonson T, et al. Tiling resolution array CGH and high density expression profiling of urothelial carcinomas delineate genomic amplicons and candidate target genes specific for advanced tumors. BMC Med Genomics. 2008;1:3. doi: 10.1186/1755-8794-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 14.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 15.Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 18.Zighelboim I, Goodfellow PJ, Gao F, et al. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J Clin Oncol. 2007;25:2042–8. doi: 10.1200/JCO.2006.08.2107. [DOI] [PubMed] [Google Scholar]
- 19.Byron SA, Gartside MG, Wellens CL, et al. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res. 2008;68:6902–7. doi: 10.1158/0008-5472.CAN-08-0770. [DOI] [PubMed] [Google Scholar]
- 20.Cheng ASL, Jin VX, Fan M, et al. Combinatorial analysis of transcription factor partners reveals recruitment of c-Myc to estrogen receptor-α responsive promoters. Mol Cell. 2006;21:393–404. doi: 10.1016/j.molcel.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin HJL, Zuo T, Lin CH, et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of cystatin M in epithelial cells. Cancer Res. 2008;68:10257–66. doi: 10.1158/0008-5472.CAN-08-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis BP, Shih Ih, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 25.Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 26.Stirzaker C, Song JZ, Davidson B, Clark SJ. Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res. 2004;64:3871–7. doi: 10.1158/0008-5472.CAN-03-3690. [DOI] [PubMed] [Google Scholar]
- 27.Laghi L, Bianchi P, Malesci A. Differences and evolution of the methods for the assessment of microsatellite instability. Oncogene. 2008;27:6313–21. doi: 10.1038/onc.2008.217. [DOI] [PubMed] [Google Scholar]
- 28.Marson A, Levine SS, Cole MF, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–33. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741–50. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 30.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JPJ. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 32.Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 33.Kozaki Ki, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.