

Abstract
Cells that sustain double strand DNA breaks (DSBs) can develop genomic instability, which contributes to carcinogenesis, and agents that cause DSBs are considered potential carcinogens. We looked for evidence of acid-induced DNA damage, including DSBs, in benign Barrett’s epithelial (BAR-T) cell lines in vitro and in patients with Barrett’s esophagus in vivo. In BAR-T cells, we also explored the mechanisms underlying acid-induced DNA damage. We exposed BAR-T cells to acid in the presence of a fluorescent probe for reactive oxygen species (ROS), and in the presence or absence of DIDS (which prevents intracellular acidification) and NAC (a scavenger of ROS). DSBs were detected by Western blotting and immunofluorescence for histone H2AX phosphorylation, and by Comet assay. During endoscopy in patients with Barrett’s esophagus, we took biopsy specimens from the metaplastic mucosa before and after esophageal perfusion with 0.1N HCl for 3 minutes, and we sought DSBs by Western blotting for histone H2AX phosphorylation. In BAR-T cells, acid exposure resulted in ROS production and caused a time-dependent increase in levels of phospho-H2AX that continued for at least 48 hours. Pre-treatment with DIDS or NAC prevented the acid-induced increase in phospho-H2AX levels. DSBs also were detected in biopsy specimens of Barrett’s metaplasia following esophageal acid perfusion in all of 6 patients with Barrett’s esophagus. Acid exposure causes DSBs in Barrett’s epithelial cells through ROS produced as a consequence of intracellular acidification. These findings suggest that acid can be considered a carcinogen in Barrett’s esophagus.
Keywords: Barrett’s esophagus, gastroesophageal reflux, DNA damage, reactive oxygen species, double strand breaks
Introduction
In the last 3 decades, the incidence of esophageal adenocarcinoma has increased more than 6-fold in the United States (1). The major risk factors for this lethal cancer are gastroesophageal reflux disease (GERD) and Barrett’s esophagus, the condition in which a metaplastic columnar epithelium replaces esophageal squamous epithelium that has been damaged by GERD. Carcinogenesis in Barrett’s metaplasia involves the accumulation of genetic and epigenetic abnormalities that cause genomic instability (2–6). It has been proposed that acid reflux promotes carcinogenesis in Barrett’s esophagus, but it is not clear how, or even if, acid exposure contributes to genomic instability in Barrett’s epithelial cells (7).
A number of studies have found that cells exposed to acid can develop DNA damage that might result in genomic instability (8–13). Double-strand breaks (DSBs) are among the most pernicious forms of DNA damage (14; 15). Cells with persistent DSBs have been found to develop chromosomal abnormalities, like translocations and deletions, that can contribute to genomic instability and cancer formation (14; 15). Indeed, agents that cause DSBs, like ionizing radiation and ultraviolet light, can be considered carcinogens (15; 16). Clemons et al. have reported that protracted acid exposure results in DSBs in certain Barrett’s cell lines and in SEG-1 cancer cells, which originally were alleged to be a Barrett’s cancer cell line but which recently were discovered to be non-esophageal in origin (17). In the single non-dysplastic Barrett’s cell line (QhTERT) used in that study, however, it is not clear whether the DSBs were the direct result of acid injury to DNA, or merely a consequence of the marked (60%) increase in cell death induced by the protracted acid exposure (17). This distinction is critically important in determining whether acid is a potential carcinogen in Barrett’s esophagus. During apoptosis, the cell destroys itself by inflicting severe DNA damage, which includes DSBs. If the sequence of events induced by acid in Barrett’s cells is apoptosis followed by DSBs, then acid would not be considered a carcinogen because DSBs in a dying cell cannot contribute to carcinogenesis. If, on the other hand, acid causes DSBs directly, without causing cell death, then the DSBs could persist and acid could well be a carcinogen in Barrett’s esophagus.
Cellular DNA-damage-sensing molecules, like Chk2 and p53, become activated in response to DNA injury. That activation halts cell cycle progression to allow the cell to repair the damaged DNA before it is replicated (14; 18; 19). We have reported that, in response to acid exposure, non-neoplastic Barrett’s epithelial cell lines activate Chk2 and p53, and exhibit a delay in cell cycle progression (20; 21). We also showed that a 10-minute acid exposure did not affect the viability of BAR-T cells, and produced only a trivial (1.1%) increase in apoptosis (20). Those observations suggest that acid directly damages DNA in Barrett’s esophagus, in which case acid might be a carcinogen. To explore that possibility, we looked for evidence of acid-induced DNA damage, including DSBs, in non-neoplastic Barrett’s epithelial cell lines and in biopsy specimens of Barrett’s metaplasia that were taken from patients whose Barrett’s esophagus had been perfused with acid during an endoscopic examination. We also explored the molecular mechanisms underlying acid-induced DNA injury in our Barrett’s cell lines using agents that inhibit intracellular acidification and agents that scavenge reactive oxygen species (ROS).
Materials and Methods
Patients and Acid Exposures
This study was approved by the institutional review board of the Dallas VA Medical Center. Patients with long-segment Barrett’s esophagus (specialized intestinal metaplasia involving ≥ 3 cm of the distal esophagus) without dysplasia who were scheduled for elective endoscopic examinations were invited to participate in the study. All patients were male, their average age was 64.8 ± 2.4 SEM years, and all were on treatment with proton pump inhibitors. During endoscopy, six biopsy specimens of Barrett’s metaplasia were taken using a jumbo biopsy forceps (Olympus FB-50K-1) before and after perfusion of the distal esophagus with 10 cc of 0.1N HCl over 3 minutes as previously described by our laboratory (22).
Cell Culture
We used 3 non-neoplastic, telomerase-immortalized Barrett’s epithelial cell lines (BAR-T, BAR-T9, and BAR-T10) that were created in our laboratory from endoscopic biopsy specimens of non-dysplastic Barrett’s specialized intestinal metaplasia taken from three patients with long segment Barrett’s esophagus (20; 23; 24). All of the BAR cell lines were co-cultured with a fibroblast feeder layer as previously described (25). Cells were maintained in monolayer culture at 37 °C in humidified air with 5% CO2 in growth media as previously described (23). For individual experiments, cells were equally seeded into collagen IV-coated wells (BD Biosciences, San Jose, CA) and maintained in growth media. We selected to use the BAR-T line for all experiments (unless otherwise indicated) because this line has been extensively characterized by our laboratory (20; 21; 23; 24; 26).
Acid Exposure and Inhibition of Intracellular Acidification
For individual experiments, the cells were cultured either in neutral full growth medium (pH 7.2) or in acidic full growth medium (brought to a pH of 4.0 with 1M HCl). Neutral or acidic medium was added for 10 minutes to equally-seeded wells of BAR cells, then removed and replaced with neutral pH medium for the remainder of the experiment unless otherwise indicated. The pH levels and durations of acid exposure were chosen to simulate typical episodes of gastroesophageal reflux in GERD patients (27). In experiments designed to prevent the intracellular acidification of cells exposed to acidic medium, cells were pretreated with 500 μM disodium 4,4′-diisothiocyanatostilbine-2,2′-disulfonate (DIDS; Sigma, St. Louis, MO). The DIDS was dissolved in 0.1M KHCO3 as a 100X stock solution, and cells were treated for 15 minutes prior to acid exposure.
Detection of Intracellular ROS
Equally seeded wells of cells were washed twice with Hank’s buffered saline solution (HBSS; Invitrogen, Carlsbad, CA) and incubated with 10 μM of a 5-(and -6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA; Molecular Probes, Carlsbad, CA) probe for 20 minutes at 37 C in the dark. Cells were exposed to neutral or acidic medium containing the probe for 10 minutes, after which the medium was removed and replaced with neutral pH medium containing the probe. After 30 minutes, cells were then washed twice with HBSS to remove any excess probe, placed in PBS, and fluorescent intensity was immediately detected using flow cytometry. To determine the specificity of the probe for ROS, cells were pretreated for 30 minutes with 10 mM N-Acety-L-cysteine [NAC (Sigma, St. Louis, MO)], which increases cellular pools of free radical scavengers, prior to incubation with carboxy-H2DCFDA.
Neutral and Alkaline Single-Cell Gel Electrophoresis (Comet Assay) for DNA Damage
We detected DNA damage using an alkaline and neutral CometAssay (Trevigen, Gaithersburg, MD) per the manufacturer’s instructions. The alkaline assay detects single-stranded DNA breaks, DSBs, apurinic and apyrimidinic sites, and alkali labile DNA adducts. If the assay is performed at neutral conditions (i.e. without the alkaline buffer) then it mainly detects DSBs (28). We mixed acid-exposed or control cells (100,000 cells/ml) with melted LMAgarose at 37°C in a ratio of 1:10 and then layered 75 μl of the mixture onto a CometSlide. The slide was maintained at 4°C for 10 minutes for gelling and then immersed in Lysis Solution at 4°C for 45 minutes. For the alkaline assay, the slide was placed in Alkali Unwinding Solution for 45 minutes at room temperature in the dark; this step was eliminated for the neutral comet assay. The slide was then placed in alkaline solution or TBE buffer (for the neutral comet assay) and electrophoresis was conducted for 80 minutes at 1 V/cm and 315 mA at 4°C for the alkaline comet assay or 20 minutes at 1V/cm and 6 mA at 4°C for the neutral comet assay. After the electrophoresis, slides were dipped in 70% ethanol for 5 minutes and allowed to air dry. Slides were then stained with silver staining or SYBR Green I solution and comet “tails” were visualized with a light microscope (Olympus, Melville, NY) or fluorescence microscope (Nikon, Melville, NY), respectively. All CometAssays were performed in duplicate. The comet extent tail moment values were quantitated from slides stained with SYBR Green using the CometScore software (TriTek Corp., Sumerduck, VA) from a minimum of 50 individual cells. This parameter measures the smallest detectable size of migrating DNA as indicated by the tail length, and the number of broken DNA pieces as indicated by the intensity of the DNA stain in the tail (28). BAR-T cells growing in culture served as negative controls; BAR-T cells treated with 200 μM hydrogen peroxide (H2O2) served as positive controls.
Western Blotting and Immunofluorescence
For the phospho-H2AX Western blots, cells were exposed to neutral media or acidic media for 1.5, 3, or 10 minutes, after which the acidic media were removed, the cells washed in cold PBS, and immediately lysed. For the phospho-H2AX time course Western blots, cells were exposed to neutral or acidic media for 10 minutes, after which the acidic media were removed and replaced with neutral media for 15 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 24 hours or 48 hours. Cells were then washed in cold PBS followed by immediate cell lysis. Cells were lysed in 1X cell lysis buffer (Cell Signaling Technology, Beverly, MA). The biopsy specimens were lysed in buffer containing 150 mM NaCl, 1% Nonidet P-40, 1% deoxycholate, 20 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 μg/ml leupeptin, 1 or 0.1 mM phenylmethylsulfonyl fluoride, and one Protease Inhibitor Cocktail Tablet per 50 ml of lysis buffer (Roche Applied Science, Indianapolis, IN). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis; protein concentrations were determined using the BCA-200 Protein Assay kit (Pierce, Rockford, IL). After separation and transfer to nitrocellulose membranes, the membranes were incubated with primary antibodies (1:1,000 dilutions) to phospho-H2AX (Cell Signaling Technology). Horseradish peroxidase secondary antibodies were used and chemiluminescence was determined using the Super Signal West Dura detection system (Pierce, Rockford, IL); β-actin (Sigma) was used to confirm equal loading. All Western blots were performed in duplicate.
For the phospho-H2AX immunofluorescence assays, cells were exposed to neutral or acidic media for 10 minutes after which the media were removed and the cells were immediately fixed with cold methanol (−20°C) for 10 minutes at −20°C followed by incubation in 3% BSA in PBS for 45 minutes to block the non-specific binding sites. Primary antibody to phospho-H2AX (1:500 dilution) was added for 1 hour at room temperature. Secondary antibodies were conjugated with Alexa Fluor 488 and used at 1:1000 dilutions of goat anti-rabbit for phospho-H2AX. Specificity was determined by omitting the primary antibodyfrom the incubation. The slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for analysis. BAR-T cells treated with 200 μM H2O2 for 10 minutes served as a positive control for phospho-H2AX
Statistical Analyses
The data were collected from at least three independent experiments. Quantitative data are expressed as the mean ± the standard error of the mean (SEM). Statistical analysis was performed using ANOVA and the Student-Newman-Keuls multiple-comparison test with the Instat for Windows statistical software package (GraphPad Software, San Diego, CA). P values <0.05 were considered significant for all analyses.
Results
Acid Exposure Generates ROS in Barrett’s Epithelial Cells
In earlier experiments, we found that a single, 3-minute acid exposure (pH 4.0) induces ROS production in BAR-T cells (29). To determine the effect of a single 10-minute acid exposure (pH 4.0) on ROS production, we incubated BAR-T cells with carboxy-H2DCFDA, and determined fluorescence after acid exposure using flow cytometry. We found that a 10-minute acid exposure significantly increased ROS production in BAR-T cells (Figure 1). To confirm this finding, BAR-T cells incubated with carboxy-H2DCFDA were treated with a single, 10 minute-exposure to acidic medium in the presence of 10 mM NAC, a general scavenger of ROS. NAC treatment significantly decreased ROS levels (Figure 1).
Figure 1.
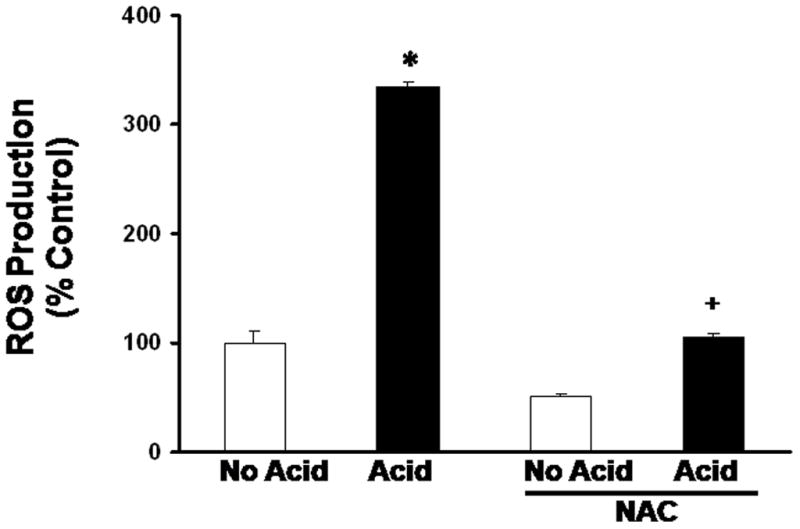
Results of ROS production in BAR-T cells after a single, 10-minute acid exposure. The bar graphs depict the mean + SEM of at least 3 individual experiments. (*, p<0.001 compared with non-acid treated controls; +, p<0.001 compared with acid treated cells)
Acid Exposure Causes DNA Damage in Barrett’s Epithelial Cells
Having found that acid exposure generates ROS in BAR-T cells, we next determined whether acid induces DNA damage. BAR-T cells were treated with a single, 10-minute exposure to acidic medium, and DNA damage was assessed using the CometAssay at alkaline conditions, which detects single- and double-stranded DNA breaks, apurinic and apyrimidinic sites, and alkali labile DNA adducts by the presence of comet “tails” (28). We found that acid exposure induced a significant increase in damaged DNA fragments detected in the comet “tails” in BAR-T cells (Figure 2A & B).
Figure 2.
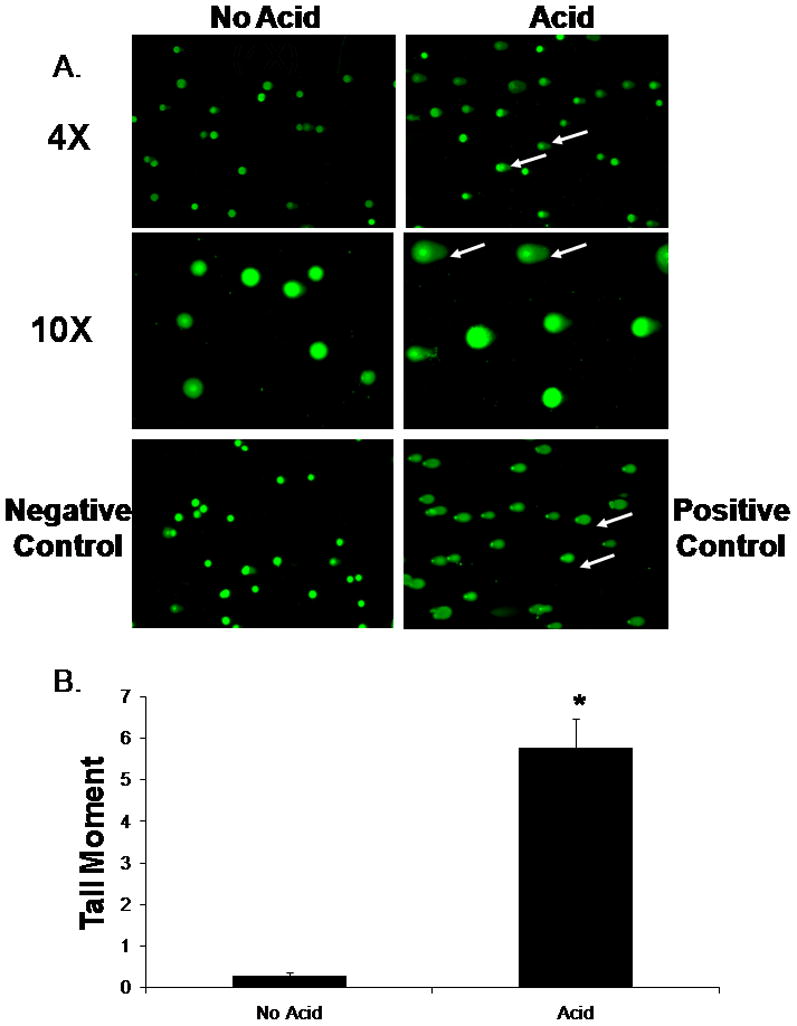
(A.) Representative experiment showing the results of the CometAssay performed at alkaline conditions in BAR-T cells following a 10-minute acid exposure at low magnification (4X) and high magnification (10X). Note the presence of comet “tails” indicating DNA damage in the acid-exposed cells (arrows). BAR-T cells treated with H2O2 served as a positive control. (B.) Bar graphs depict the comet tail moment values + SEM from a minimum of 50 individual cells. (*, p<0.0001 compared with non-acid treated controls)
Acid Exposure Causes Double-Stranded DNA Breaks in Barrett’s Epithelial Cells
BAR-T cells were treated with a single, 10-minute acid exposure, and DNA damage was assessed using the CometAssay at neutral conditions, which detects primarily DSBs. We found that acid exposure induced a significant increase in damaged DNA fragments detected in the comet “tails,” suggesting that acid causes DSBs in BAR-T cells (Figure 3A & B).
Figure 3.
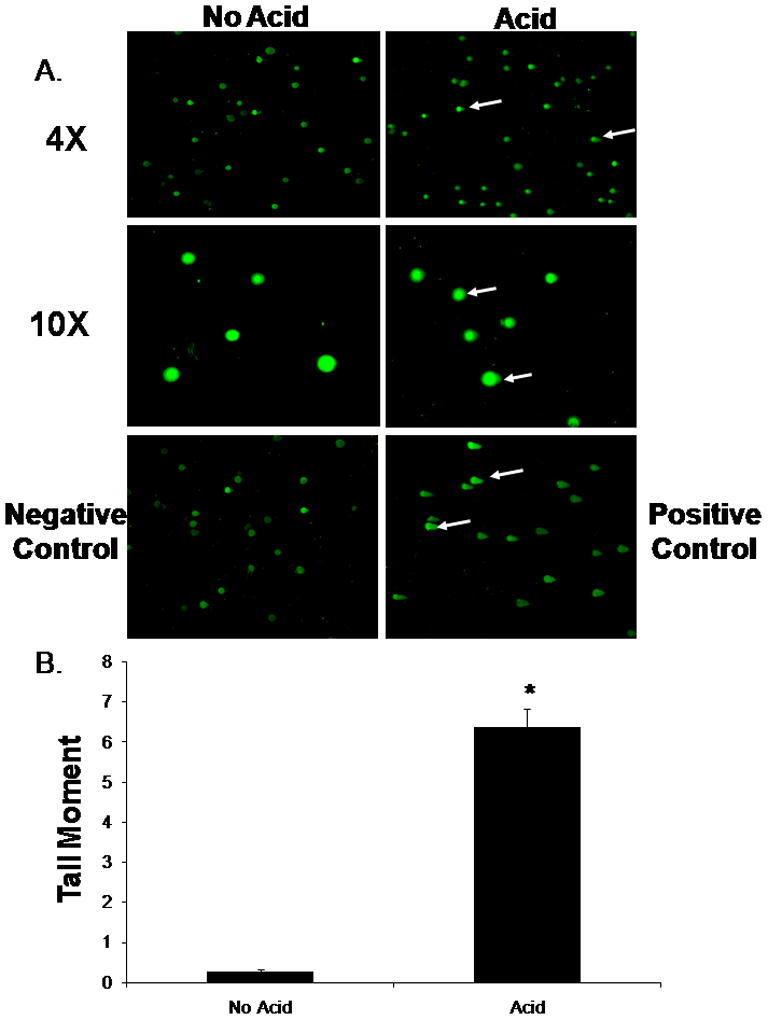
(A.) Representative experiment showing the results of the CometAssay performed at neutral conditions in BAR-T cells following a single, 10-minute acid exposure at low (4X) and high magnification (10X). Note the presence of comet “tails” indicating DSBs in the acid-exposed cells (arrows). BAR-T cells treated with H2O2 served as a positive control. (B.) Bar graphs depict the comet tail moment values + SEM from a minimum of 50 individual cells. (*, p<0.0001 compared with non-acid treated controls)
Recently, the detection of histone H2AX phosphorylation has been found to be more sensitive assay than the CometAssay for detecting DSBs in live cells (16; 30; 31). To confirm our findings from the CometAssay, we treated the BAR-T cells with a single, 1.5-, 3-, or 10-minute acid exposure, and performed Western blotting for phospho-H2AX expression. We found that acid exposure induced a time-dependent increase in phospho-H2AX expression (Figure 4A). We then determined the effect of a single, 10-minute acid exposure on phospho-H2AX expression in two additional telomerase-immortalized metaplastic Barrett’s cell lines (BAR-T9 and BAR-T10). As in the BAR-T cells, a single 10-minute acid exposure increased the expression of phospho-H2AX in the BAR-T9 and BAR-T10 cell lines (Figure 4B).
Figure 4.
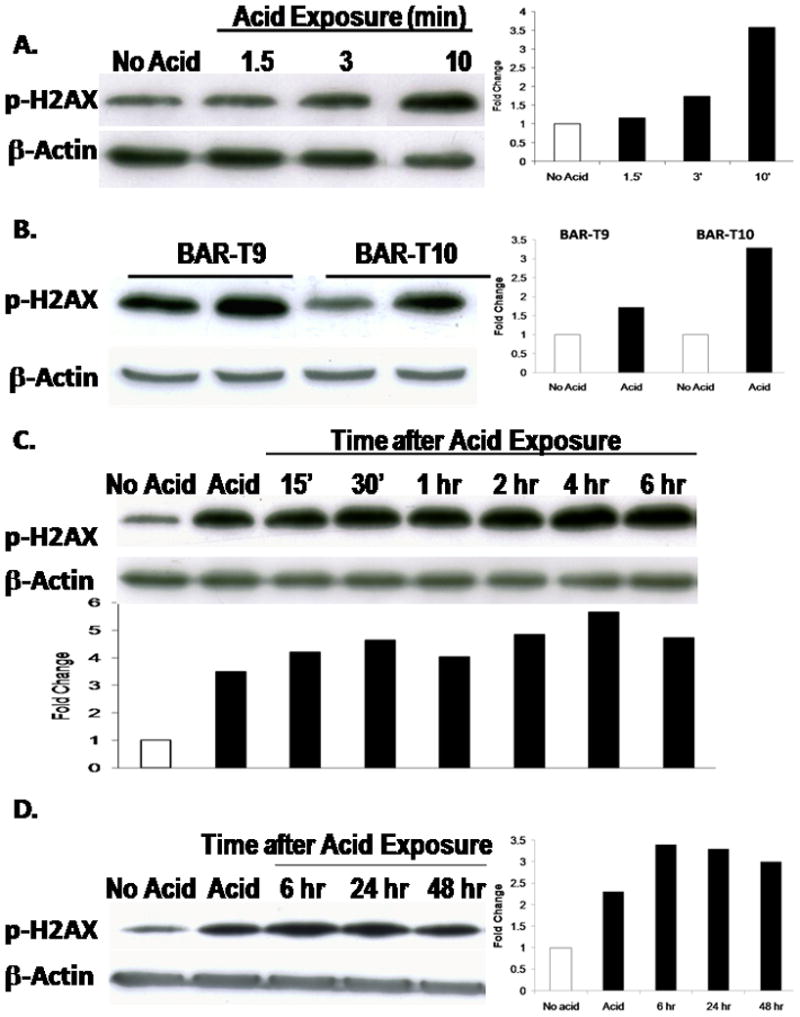
(A) Representative Western blots demonstrating expression of phospho-H2AX in BAR-T cells following exposure to acid for 1.5, 3, or 10 minutes. (B) Representative Western blots demonstrating expression of phospho-H2AX in BAR-T9 and BAR-T10 cells following a single, 10-minute acid exposure. (C & D) Representative Western blots demonstrating the time course for phospho-H2AX expression in BAR-T cells following a single, 10-minute acid exposure. β-actin served as a loading control.
We next sought to determine the duration for which H2AX remains phosphorylated in response to acid exposure. We found that phospho-H2AX expression persisted for up to 48 hours after a single, 10-minute acid exposure in BAR-T cells (Figure 4C & D). In addition to an increase in expression, phospho-H2AX also localizes to the sites of damaged DNA and forms nuclear foci. Using immunofluoresence, we observed phospho-H2AX-containing nuclear foci in acid treated, but not in untreated, BAR-T cells (Figure 5). These findings demonstrate that acid exposure causes DSBs in Barrett’s epithelial cells.
Figure 5.
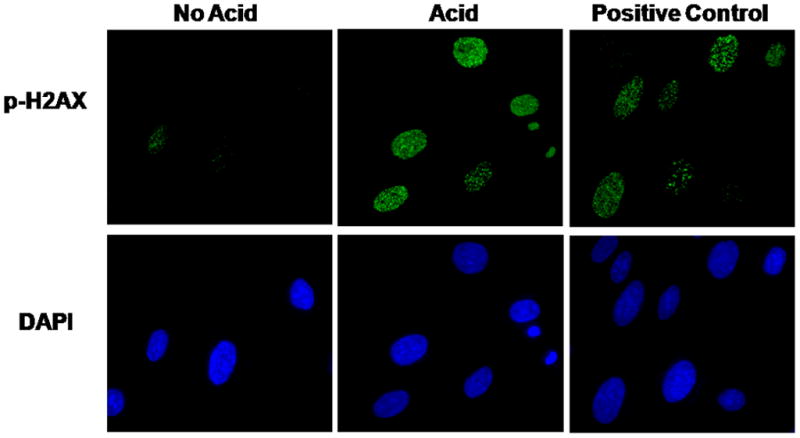
A representative experiment showing the results of p-H2AX nuclear foci formation in BAR-T cells after a single, 10-minute acid exposure. Note the increase in p-H2AX-containing nuclear foci in the acid-treated cells, but not in the untreated control cells. DAPI staining demonstrates the total number of cell nuclei in the same field. BAR-T cells treated with H2O2 served as a positive control. (Magnification: 60X)
DIDS Prevents Double-Stranded DNA Breaks in Barrett’s Epithelial Cells Exposed to Acid
Acid exposure has been shown to cause intracellular acidification via a DIDS-inhibitable mechanism in esophageal epithelial cells (including BAR-T cells) (R.C. Orlando, personal communication) (32; 33). To explore whether intracellular acidification is responsible for causing DSBs in Barrett’s epithelial cells, we studied the effects of DIDS on DSBs in acid-treated BAR-T cells. We treated BAR-T cells with a single, 10-minute exposure to acidic medium in the presence of 500 μM DIDS, and assessed DSBs by phospho-H2AX expression. We found that DIDS prevented the acid-induced increase in phospho-H2AX, suggesting that intracellular acidification is the mechanism whereby acid exposure causes DSBs in Barrett’s epithelial cells (Figure 6A).
Figure 6.
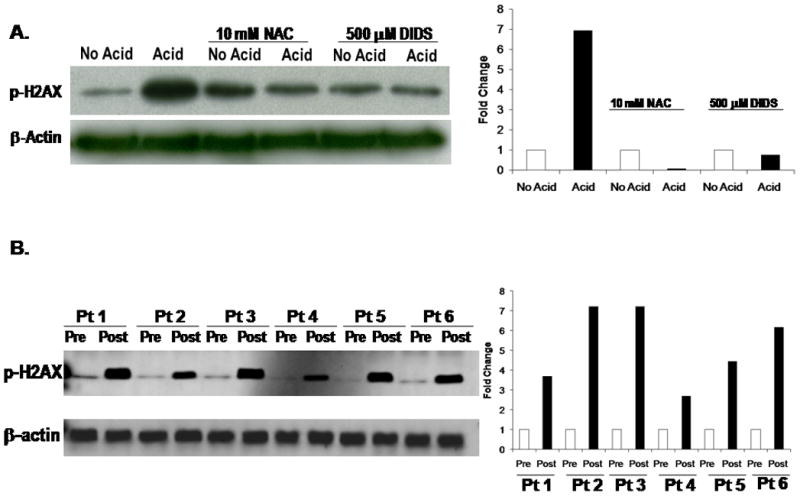
(A) Representative Western blots demonstrating the results of NAC or DIDS treatment on acid-induced phospho-H2AX expression in BAR-T cells. Note that both NAC and DIDS prevent the acid-induced increase in phospho-H2AX expression. (B) Representative Western blot demonstrating phospho-H2AX expression before and after esophageal acid perfusion in biopsy specimens of Barrett’s metaplasia taken from 6 patients with Barrett’s esophagus during endoscopic examinations. Note that acid increased phospho-H2AX expression in the metaplastic Barrett’s mucosa in all 6 patients. β-actin served as a loading control.
NAC Prevents Double-Stranded DNA Breaks in Barrett’s Epithelial Cells Exposed to Acid
To determine whether the acid-induced ROS caused the DSBs, we treated BAR-T cells with a single, 10 minute-exposure to acidic medium in the presence of 10 mM NAC, and assessed for DSBs by determining phospho-H2AX expression. NAC prevented the acid-induced increase in phospho-H2AX expression, suggesting that the ROS produced in response to acid- exposure are responsible for inducing DSBs in BAR-T cells (Figure 6A).
Esophageal Acid Exposure Causes Double-Stranded DNA Breaks in Patients with Barrett’s Esophagus
Having found that acid exposure increases phosphorylation of H2AX in three Barrett’s epithelial cell lines, we sought to confirm that these same effects occur in vivo by obtaining endoscopic biopsy specimens of Barrett’s epithelium before and after esophageal acid perfusion in 6 patients with Barrett’s esophagus. In agreement with our in vitro data, we found that acid increased phospho-H2AX expression in biopsy specimens of Barrett’s metaplasia from all 6 patients (Figure 6B).
Discussion
We have shown that acid exposure induces DNA damage, including DSBs, in Barrett’s epithelial cells both in vitro and in vivo. In BAR-T cells, we have shown evidence of acid-induced DNA damage by Comet assays performed under neutral and alkaline conditions, by H2AX phosphorylation, and by the demonstration of nuclear foci containing phospho-H2AX. We have found that H2AX phosphorylation increases immediately after acid exposure and continues even up to 48 hours, and that phosphorylation increases with the duration of acid exposure.
To explore whether the induction of DSBs by acid is a general feature of Barrett’s epithelial cells or a unique feature of BAR-T cells, we studied 2 additional telomerase-immortalized Barrett’s epithelial cell lines that had been established in our laboratory (24). We found that acid exposure caused an increase in the phosphorylation of H2AX in all 3 Barrett’s cell lines, suggesting that DNA vulnerability to acid is a common property of non-neoplastic BAR-T cells. In confirmation of these in vitro experiments, we found evidence of DSBs by H2AX phosphorylation in biopsy specimens of Barrett’s metaplasia taken from all of six patients who had the esophagus perfused with acid during endoscopic examinations. In cultures of esophageal cells (including rabbit esophageal squamous cells, primary cultures of Barrett’s epithelial cells and BAR-T cells), extracellular acid has been found to cause intracellular acidification through activation of the Na+-independent Cl−/HCO3− exchanger, which can be inhibited by DIDS (R.C. Orlando, personal communication)(32; 33). Our finding that DIDS prevents phospho-H2AX production in BAR-T cells exposed to acid suggests that it is intracellular acidification that mediates the generation of DSBs.
Several lines of evidence suggested to us that ROS would be involved in the DSBs induced by acid exposure in Barrett’s epithelial cells. For example, a number of studies had shown that esophageal epithelial cells exposed to acid and bile salts increased their production of ROS (13; 34–37). In an earlier study, we also found that a 3-minute acid exposure significantly increased ROS production in BAR-T cells (29). ROS are well known to cause oxidative damage to DNA (38; 39), and biopsy specimens of Barrett’s metaplasia have been found to exhibit evidence of oxidative DNA injury (11–13). Moreover, ROS have been shown to cause DSBs. Cultures of fibroblasts from SOD1 transgenic mice, which have abnormally high levels of endogenous ROS production, develop more DSBs than fibroblasts from wild-type control mice (40).
In this study, we have found that a 10-minute acid exposure increases fluorescence in BAR-T cells that were treated with carboxy-H2DCFDA, indicating that acid induces ROS production. Using another benign Barrett’s epithelial cell line (QhTERT), Clemons also found that a 10-minute acid exposure increased the generation of intracellular ROS (17). That study did not determine whether the ROS caused the DSBs in the non-neoplastic cell line, however. Our finding that treatment with an ROS scavenger (NAC) prevents the development of DSBs after acid exposure indicates that acid-induced DNA damage is mediated by ROS production. Taken together, our results suggest that exposure of Barrett’s epithelial cells to acid results in intracellular acidification that induces the production of ROS, which cause DSBs.
DSBs are especially dangerous mutations because, if they persist, they can cause genomic instability and contribute to carcinogenesis (14; 15). In earlier studies, we showed that acid activates the Chk2 and p53 DNA-damage-response pathways and delays cell cycle progression in non-neoplastic Barrett’s epithelial cells (20; 21). DSBs are the primary stimuli that activate the Chk2 DNA-damage-response pathway, and p53 is one of its major downstream effectors (41; 42). In our non-neoplastic Barrett’s cells, we found an increase in H2AX phosphorylation (a sensitive marker of DSBs) immediately after acid exposure, suggesting that DNA damage is the likely trigger for the early, acid-induced activation of the Chk2 pathway that we observed in our previous studies (20). After a 10-minute acid exposure, moreover, the increase in H2AX phosphorylation persisted up to 48 hours, which is more than enough time to activate the p53 DNA damage-response pathway that we also observed in our earlier studies. In non-neoplastic Barrett’s epithelial cells, therefore, we have shown that acid is a genotoxin which causes DSBs that persist for at least 48 hours. These observations suggest that acid can be considered a carcinogen in Barrett’s esophagus.
Our study has potential clinical implications. Our finding that acid causes DSBs in non-neoplastic Barrett’s epithelial cells and, therefore, can be considered a carcinogen in Barrett’s esophagus, supports the practice of aggressive acid suppression for patients with this condition. In an earlier study using BAR-T cells, we demonstrated that acid exposure had potentially beneficial anti-proliferative effects that were mediated by activation of the p53 and Chk2 pathways. In that study, however, it was not clear whether those pathways were activated directly by acid (which would be beneficial) or indirectly through DNA damage (which would be detrimental). Our present study showing that acid causes severe DNA damage suggests that the apparent anti-proliferative effects of acid are merely a response to genetic injury. The molecular mechanisms elucidated by our study support the prescription of aggressive acid suppression as a chemotherapeutic strategy for patients with Barrett’s esophagus. This issue requires further investigation in prospective, controlled clinical trials.
In conclusion, we have shown that benign Barrett’s epithelial cells exposed to acid in vitro produce ROS and develop severe DNA damage, including DSBs, which persist for at least 48 hours. We have also documented the development of DSBs in Barrett’s metaplasia following esophageal acid perfusion in vivo in patients with Barrett’s esophagus. Our findings that acid-induced DSBs can be prevented by treating Barrett’s epithelial cells with NAC or DIDS suggests that the DNA damage is caused by ROS produced as a consequence of intracellular acidification. Taken together, these observations suggest that acid should be considered a carcinogen in Barrett’s esophagus.
Acknowledgments
This work was supported by the Office of Medical Research, Department of Veterans Affairs, Dallas, TX (R.F.S.), the Harris Methodist Health Foundation, Dr. Clark R. Gregg Fund (R.F.S., K.H-C.), and the National Institutes of Health (R01-DK63621 to R.F.S)
Reference List
- 1.Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:142–146. doi: 10.1093/jnci/dji024. [DOI] [PubMed] [Google Scholar]
- 2.Klump B, Hsieh CJ, Holzmann K, Gregor M, Porschen R. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology. 1998;115:1381–1386. doi: 10.1016/s0016-5085(98)70016-2. [DOI] [PubMed] [Google Scholar]
- 3.Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res. 1997;57:2619–2622. [PubMed] [Google Scholar]
- 4.Maley CC, Galipeau PC, Li X, et al. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004;64:7629–7633. doi: 10.1158/0008-5472.CAN-04-1738. [DOI] [PubMed] [Google Scholar]
- 5.Casson AG, Mukhopadhyay T, Cleary KR, Ro JY, Levin B, Roth JA. p53 gene mutations in Barrett’s epithelium and esophageal cancer. Cancer Res. 1991;51:4495–4499. [PubMed] [Google Scholar]
- 6.Blount PL, Meltzer SJ, Yin J, Huang Y, Krasna MJ, Reid BJ. Clonal ordering of 17p and 5q allelic losses in Barrett dysplasia and adenocarcinoma. Proc Natl Acad Sci U S A. 1993;90:3221–3225. doi: 10.1073/pnas.90.8.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzgerald RC, Lascar R, Triadafilopoulos G. Review article: Barrett’s oesophagus, dysplasia and pharmacologic acid suppression. Aliment Pharmacol Ther. 2001;15:269–276. doi: 10.1046/j.1365-2036.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 8.Xiao H, Li TK, Yang JM, Liu LF. Acidic pH induces topoisomerase II-mediated DNA damage. Proc Natl Acad Sci U S A. 2003;100:5205–5210. doi: 10.1073/pnas.0935978100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita T. Low pH leads to sister-chromatid exchanges and chromosomal aberrations, and its clastogenicity is S-dependent. Mutat Res. 1995;334:301–308. doi: 10.1016/0165-1161(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 10.Morita T, Nagaki T, Fukuda I, Okumura K. Clastogenicity of low pH to various cultured mammalian cells. Mutat Res. 1992;268:297–305. doi: 10.1016/0027-5107(92)90235-t. [DOI] [PubMed] [Google Scholar]
- 11.Olliver JR, Hardie LJ, Dexter S, Chalmers D, Wild CP. DNA damage levels are raised in Barrett’s oesophageal mucosa relative to the squamous epithelium of the oesophagus. Biomarkers. 2003;8:509–521. doi: 10.1080/13547500310001644961. [DOI] [PubMed] [Google Scholar]
- 12.Olliver JR, Hardie LJ, Gong Y, et al. Risk factors, DNA damage, and disease progression in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2005;14:620–625. doi: 10.1158/1055-9965.EPI-04-0509. [DOI] [PubMed] [Google Scholar]
- 13.Dvorak K, Payne CM, Chavarria M, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett’s Oesophagus. Gut. 2007;56:763–71. doi: 10.1136/gut.2006.103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev. 2003;194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 16.Albino AP, Huang X, Jorgensen E, et al. Induction of H2AX phosphorylation in pulmonary cells by tobacco smoke: a new assay for carcinogens. Cell Cycle. 2004;3:1062–1068. [PubMed] [Google Scholar]
- 17.Clemons NJ, McColl KE, Fitzgerald RC. Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology. 2007;133:1198–1209. doi: 10.1053/j.gastro.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 18.Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27:589–605. doi: 10.1038/emboj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartek J, Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001;490:117–122. doi: 10.1016/s0014-5793(01)02114-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhang HY, Zhang X, Hormi-Carver K, Feagins LA, Spechler SJ, Souza RF. In Non-neoplastic Barrett’s Epithelial Cells, Acid Exerts Early Antiproliferative Effects through Activation of the Chk2 Pathway. Cancer Res. 2007;67:8580–8587. doi: 10.1158/0008-5472.CAN-07-2023. [DOI] [PubMed] [Google Scholar]
- 21.Feagins LA, Zhang HY, Hormi-Carver K, et al. Acid has antiproliferative effects in nonneoplastic Barrett’s epithelial cells. Am J Gastroenterol. 2007;102:10–20. doi: 10.1111/j.1572-0241.2006.01005.x. [DOI] [PubMed] [Google Scholar]
- 22.Souza RF, Shewmake KL, Shen Y, et al. Differences in ERK Activation in Squamous Mucosa in Patients Who Have Gastroesophageal Reflux Disease with and without Barrett’s Esophagus. Am J Gastroenterol. 2005;100:551–559. doi: 10.1111/j.1572-0241.2005.41122.x. [DOI] [PubMed] [Google Scholar]
- 23.Jaiswal KR, Morales CP, Feagins LA, et al. Characterization of telomerase-immortalized, non-neoplastic, human Barrett’s cell line (BAR-T) Dis Esophagus. 2007;20:256–264. doi: 10.1111/j.1442-2050.2007.00683.x. [DOI] [PubMed] [Google Scholar]
- 24.Hormi-Carver K, Zhang X, Zhang HY, et al. Unlike esophageal squamous cells, Barrett’s epithelial cells resist apoptosis by activating the nuclear factor-kappaB pathway. Cancer Res. 2009;69:672–677. doi: 10.1158/0008-5472.CAN-08-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez RD, Morales CP, Herbert BS, et al. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001;15:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hormi-Carver K, Feagins LA, Spechler SJ, Souza RF. All trans-retinoic acid induces apoptosis via p38 and caspase pathways in metaplastic Barrett’s cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G18–G27. doi: 10.1152/ajpgi.00237.2006. [DOI] [PubMed] [Google Scholar]
- 27.Richter JE, Bradley LA, DeMeester TR, Wu WC. Normal 24-hr ambulatory esophageal pH values. Influence of study center, pH electrode, age, and gender. Dig Dis Sci. 1992;37:849–856. doi: 10.1007/BF01300382. [DOI] [PubMed] [Google Scholar]
- 28.Liao W, McNutt MA, Zhu WG. The Comet assay: A sensitive method for detecting DNA damage in individual cells. Methods. 2009 doi: 10.1016/j.ymeth.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 29.Feagins LA, Zhang HY, Zhang X, et al. Mechanisms of oxidant production in esophageal squamous cell and Barrett’s cell lines. Am J Physiol Gastrointest Liver Physiol. 2008;294:G411–G417. doi: 10.1152/ajpgi.00373.2007. [DOI] [PubMed] [Google Scholar]
- 30.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 31.Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Tobey NA, Reddy SP, Keku TO, Cragoe EJ, Jr, Orlando RC. Mechanisms of HCl-induced lowering of intracellular pH in rabbit esophageal epithelial cells. Gastroenterology. 1993;105:1035–1044. doi: 10.1016/0016-5085(93)90946-a. [DOI] [PubMed] [Google Scholar]
- 33.Lao-Sirieix P, Corovic A, Jankowski J, Lowe A, Triadafilopoulos G, Fitzgerald RC. Physiological and molecular analysis of acid loading mechanisms in squamous and columnar-lined esophagus. Dis Esophagus. 2008;21:529–538. doi: 10.1111/j.1442-2050.2007.00807.x. [DOI] [PubMed] [Google Scholar]
- 34.Farhadi A, Fields J, Banan A, Keshavarzian A. Reactive oxygen species: are they involved in the pathogenesis of GERD, Barrett’s esophagus, and the latter’s progression toward esophageal cancer? Am J Gastroenterol. 2002;97:22–26. doi: 10.1111/j.1572-0241.2002.05444.x. [DOI] [PubMed] [Google Scholar]
- 35.Iijima K, Henry E, Moriya A, Wirz A, Kelman AW, McColl KE. Dietary nitrate generates potentially mutagenic concentrations of nitric oxide at the gastroesophageal junction. Gastroenterology. 2002;122:1248–1257. doi: 10.1053/gast.2002.32963. [DOI] [PubMed] [Google Scholar]
- 36.Jenkins GJ, D’Souza FR, Suzen SH, et al. Deoxycholic acid at neutral and acid pH, is genotoxic to oesophageal cells through the induction of ROS: The potential role of anti-oxidants in Barrett’s oesophagus. Carcinogenesis. 2007;28:136–142. doi: 10.1093/carcin/bgl147. [DOI] [PubMed] [Google Scholar]
- 37.Song S, Guha S, Liu K, Buttar N, Bresalier RS. COX-2 Induction by Unconjugated Bile Acids Involves Reactive Oxygen Species-Mediated Signaling Pathways in Barrett’s Oesophagus and Oesophageal Adenocarcinoma. Gut. 2007 doi: 10.1136/gut.2007.121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 39.Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. 1997;272:19095–19098. doi: 10.1074/jbc.272.31.19095. [DOI] [PubMed] [Google Scholar]
- 40.Karanjawala ZE, Murphy N, Hinton DR, Hsieh CL, Lieber MR. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr Biol. 2002;12:397–402. doi: 10.1016/s0960-9822(02)00684-x. [DOI] [PubMed] [Google Scholar]
- 41.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 42.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]