

Abstract
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is carcinogenic in multiple organs and numerous species. Bioactivation of PhIP is initiated by PhIP N2- hydroxylation catalyzed by cytochrome P450s. Following N-hydroxylation, O-acetylation catalyzed by N-acetyltransferase 2 (NAT2) is considered a further possible activation pathway. Genetic polymorphisms in NAT2 may modify cancer risk following exposure.
Nucleotide excision repair-deficient Chinese hamster ovary (CHO) cells stably transfected with human cytochrome P4501A1 (CYP1A1) and a single copy of either NAT2*4 (rapid acetylator) or NAT2*5B (slow acetylator) alleles were used to test the effect of CYP1A1 and NAT2 polymorphism on PhIP genotoxicity.
Cells transfected with NAT2*4 had significantly higher levels of N-hydroxy-PhIP O-acetyltransferase (P = 0.0150) activity than cells transfected with NAT2*5B. Following PhIP treatment, CHO cell lines transfected with CYP1A1, CYP1A1/NAT2*4 and CYP1A1/NAT2*5B each showed concentration-dependent cytotoxicity and hypoxanthine phosphoribosyl transferase (hprt) mutagenesis not observed in untransfected CHO cells.
dG-C8-PhIP was the primary DNA adduct formed and levels were dose-dependent in transfected CHO cells in the order: CYP1A1 < CYP1A1 & NAT2*5B < CYP1A1 & NAT2*4 although levels did not differ significantly (P>0.05) following one-way analysis of variance.
These results strongly support activation of PhIP by CYP1A1 with little effect of human NAT2 genetic polymorphism on mutagenesis and DNA damage.
Keywords: PhIP; 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine; CYP1A1; cytochrome P4501A1; NAT2; N-acetyltransferase 2
Introduction
2-Amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine (PhIP) is the most abundant heterocyclic amine mutagen formed during high temperature cooking of meats, accounting for ∼70% mean dietary intake of heterocyclic amines in the United States (Keating and Bogan, 2004). PhIP has been identified in smoke condensates from the frying of meat (Thiebaud et al., 1995) and has been detected in airborne particles, diesel-exhaust particulates and incinceration ash from garbage-burning plants (Manabe et al., 1993). PhIP is found in cigarette smoke (Manabe et al., 1991) and is a urinary mutagen in smokers of black tobacco (Peluso et al., 1991). PhIP causes mammary, prostate, and colon tumors in the rat and is classified as a probable human carcinogen (Sinha et al., 2000; National Toxicology Program, 2005).
PhIP-induced DNA adduct formation and mutagenesis require metabolic activation, and one important pathway includes N-hydroxylation catalyzed by cytochrome P450s (Turesky 2004; 2007; Kim and Guengerich, 2005). Although many arylamine carcinogens undergo N-hydroxylation by cytochrome P4501A2, this isoform is primarily restricted to the liver and PhIP carcinogenesis is independent of this P450 in mice (Kimura et al., 2003). Arylamine carcinogens also are activated by cytochrome P4501A1 (CYP1A1) (Murray et al., 1993; Edwards et al., 1994; Hammons et al., 1997; Yamazaki et al., 2004; Kim and Guengerich, 2005; Turesky, 2007) and cDNA-expressed human CYP1A1 exhibited over 15-fold higher affinity for PhIP than human CYP1A2 (Crofts et al., 1998). Since PhIP induces tumors in extrahepatic organs and has relatively high affinity for CYP1A1, it is important to evaluate the role of CYP1A1 in PhIP-induced genotoxicity. N-hydroxy-PhIP can be further O-acetylated by N-acetyltransferase 2 (NAT2) to the acetoxy-derivatives that are highly unstable, leading to electrophilic intermediates that are mutagenic and form DNA adducts (Schut and Snyderwine, 1999).
PhIP-induced DNA adducts primarily form at the C8 position of deoxyguanosine (dG-C8) and can be removed by nucleotide excision repair (NER) (Schut and Snyderwine, 1999). Because DNA adduct levels are a function of environmental exposure and polymorphism in genes involved in carcinogen metabolism, DNA adducts are an informative biomarker for investigations of genetic variation in carcinogen metabolism.
Since heterocyclic amines require metabolic activation to exert their carcinogenic effects, genetic polymorphisms in the enzymes catalyzing the activation and/or detoxification pathways of carcinogen metabolism may account for differences in susceptibility to carcinogens between individuals (Turesky, 2004). Humans exhibit genetic polymorphism in NAT2 resulting in rapid and slow acetylator phenotypes. Relative to the NAT2*4 reference allele, NAT2*5B possesses three polymorphisms in the NAT2 open reading frame: T341C (I114T), C481T (synonymous) and A803G (K268R) and is associated with slow acetylator phenotype (Zang et al., 2004). Epidemiologic studies suggest a role for NAT2 genetic polymorphism in susceptibility to various cancers, but laboratory-based experiments are valuable for investigating metabolism of carcinogens that may contribute to these findings.
O-acetylation of N-hydroxy-PhIP has been shown to generate DNA adducts (Snyderwine et al., 2002) suggesting that PhIP- induced DNA damage and subsequent mutagenesis would be greater in rapid than slow NAT2 acetylators. On the other hand, several previous studies have shown that metabolic activation of N-hydroxy-PhIP did not require O-acetylation (Buonarati and Felton, 1990; Wild et al., 1995; Wu et al., 1997). Thus, we investigated the effects of human CYP1A1 and NAT2 genetic polymorphism on PhIP-induced DNA adducts and mutagenesis in nucleotide excision repair-deficient Chinese hamster ovary (CHO) cells transfected with human CYP1A1, human NAT2*4 (rapid acetylator allele) or human NAT2*5B (slow acetylator allele).
Materials and Methods
Cell culture
The UV5-CHO cell line, a nucleotide excision repair-deficient derivative of the AA8 line, was obtained from the ATCC (Catalog number: CRL-1865). UV5-CHO is hypersensitive to bulky adduct mutagens and belongs to the excision repair cross complementation group 2. All cells were grown in alpha-modified minimal essential medium (Cambrex) without L-glutamine, ribosides, and deoxyribosides supplemented with 10% fetal bovine serum (Hyclone), 100 units/ml penicillin, 100 units/ml streptomycin (Cambrex), and 2 mM L-glutamine (Cambrex) at 37 °C in 5% CO2. Media were supplemented with appropriate selective agents to maintain stable transfectants. Construction of UV5/CHO cells expressing human CYP1A1 and NAT2*4 or NAT2*5B was recently reported and characterized (Bendaly et al., 2007). Briefly, the pFRT/lacZeo plasmid (Invitrogen) was transfected into nucleotide excision repair-deficient UV5 cell lines to generate a UV5 cell line containing a single integrated FRT site (UV5FRT). Purified human NADPH cytochrome P450 reductase (POR) and CYP1A1 polymerase chain reaction (PCR) products were digested and ligated into similarly treated pIRES vector and transformed into DH5α competent cells. The pIRES plasmid containing cDNAs of human CYP1A1 and POR was transfected into the newly established UV5FRT cell line. Those UV5 cells expressing a single FRT site, CYP1A1, and POR were expanded, and intact geneticin-resistant cells assayed for CYP1A1 activity by measuring 7-ethoxyresorufin O-deethylase activity as described previously (Metry et al., 2007). Cell lines with similar levels of 7-ethoxyresorufin O-deethylase activity were selected for additional transfection with either NAT2*4 or NAT2*5B.
The open reading frames of NAT2*4 and NAT2*5B were amplified by PCR, digested with NheI and XhoI (New England Biolabs), and inserted into the similarly prepared pcDNA5/FRT vector (Invitrogen). The pcDNA5/FRT plasmid containing human NAT2*4 or NAT2*5B was co-transfected with pOG44, a Flp recombinase expression plasmid, into UV5FRT cells. Integration of the pcDNA5/FRT construct into the FRT site was confirmed by PCR (Metry et al., 2007). The NAT2*4- and NAT2*5B-transfected cells were characterized for N-acetylation of sulfamethazine, a NAT2-selective substrate (Bendaly et al., 2007).
O-Acetyltransferase assays
N-hydroxy-PhIP O-acetyltransferase assays were determined by high performance liquid chromatography (HPLC) as previously described (Metry et al., 2007). Briefly, reaction mixtures containing equal amounts of cell lysate protein, 1 mg/ml deoxyguanosine, 100 μM N-hydroxy-PhIP (Toronto Research Chemicals), and 1 mM acetyl-coenzyme A were incubated at 37°C for 10 min and stopped by the addition of water-saturated ethyl acetate. The reactions were centrifuged for 10 min and the organic phase was transferred, evaporated to dryness, and resuspended in 100 μl of 10% acetonitrile. HPLC separation was achieved using a gradient of 85:15 sodium perchlorate (pH 2.5)/acetonitrile to 0:100 sodium perchlorate (pH 2.5)/acetonitrile over 10 min. Baseline measurements using extracts of UV5 and UV5/CYP1A1 cells were subtracted from measurements in the NAT2*4- and NAT2*5B-transfected CHO cell lines.
Cytotoxicity and mutagenesis
Assays for cytotoxicity and mutagenesis were carried out as described previously (Metry et al., 2007). Briefly, cells were grown for 12 doublings, with selective agents in complete hypoxanthine-aminopterin-thymidine medium (30 μM hypoxanthine, 0.1 μM aminopterin, and 30 μM thymidine). Cells were plated at a density of 5 × 105 cells/T-25 flask and incubated for 24 h, after which media were changed and the cells were treated separately for 48 h with various concentrations of PhIP (Toronto Research Chemicals) or vehicle control (0.5% dimethyl sulfoxide). Survival was determined by colony-forming assay and expressed as percent of vehicle control. The remaining cells were replated and subcultured. After 7 days of growth, cultures were plated for cloning efficiency in complete media and for mutations in complete media containing 40 μM 6-thioguanine (Sigma). Dishes were seeded with 1 × 105 cells/100 mm dish (10 replicates) and incubated for 7 days; cloning efficiency dishes were seeded with 100 cells/well/six-well plate in triplicate and incubated for 6 days.
Identification and quantitation of PhIP- DNA adducts
Cells grown in 15- cm plates were treated separately with PhIP as described above for the cytotoxicity and mutagenesis assays. Cells were harvested after 48 h of treatment and DNA was extracted and quantified as previously described (Metry et al., 2007). dG-C8-PhIP and dG-C8-PhIP-D3 adduct standards (>95% purity) were obtained from Toronto Research Chemicals. One-tenth volumes each of proteinase K solution (20 mg/mL) and 10% SDS were added to the cell lysate, and the mixture was incubated at 37°C for 60 min. One volume of phenol, equilibrated with 10 mM Tris HCl (pH 8.0), was added to the mixture, which was then vortexed and centrifuged at 3,600 x g for 15 min. The aqueous layer was removed and added to 1 volume of phenol/chloroform/isoamyl alcohol (25:24:1) saturated with 10 mM Tris HCl (pH 8.0), which was vortexed and centrifuged. The aqueous layer was removed and added to 1 volume of cold (-20°C) isopropanol, and the mixture was vortexed and centrifuged. The DNA pellet was washed with 70% ethanol and redissolved in 5 mM Tris HCl (pH 7.4) containing 1 mM CaCl2, 1 mM ZnCl2, and 10 mM MgCl2. DNA was quantified by UV spectroscopy using A260 nm. DNA quality was monitored by UV spectroscopy using A260/280 nm and this ratio was consistently above 1.9. DNA samples (200 μg) added to 1 ng (3.3 adducts per 106 DNA bases) deuterated internal standard (dG-C8-PhIP-D3) were digested at 37°C with 10 units DNAse I (US Biological) for 1 h followed by 5 units micrococcal nuclease (Sigma), 5 units nuclease P1 (US Biological), 0.01 units spleen phosphodiesterase (Sigma), and 0.01 units snake venom phosphodiesterase (Sigma) for 6 h followed by 5 units alkaline phosphatase (Sigma) overnight. Two volumes of acetonitrile were added to the digest, which was then filtered and concentrated to 100 μl in a speed vacuum.
Samples were subjected to binary gradient HPLC and introduced into a Micromass Quattro LC triple quadrupole mass spectrometer using a custom-built nanospray as described previously (Metry et al., 2007; Neale et al., 2008). Samples were loaded onto a Inertsil C18 precolumn (5 mm × 300 μm i.d., 5 μm; LC Packings) using Perkin Elmer ABI 140D syringe pumps and a Hewlett Packard 1100 Series autosampler. Multiple reaction monitoring (dwell time, 0.5 s; span, 0.4 Da) was used to measure the [M+H]+ to [(M-116) + H]+ (loss of deoxyribose) mass transition. A Quattro LC micromass triple quadrupole mass spectrometer equipped with a nanoelectrospray ion source was used for PhIP-DNA adduct quantitation. Multiple reaction monitoring in the electrospray ionization-positive ion mode was carried out using argon as the collision gas. Capillary and cone voltages and collision energies were optimized for cleavage of the glycosidic bond. The dG-C8-PhIP adduct was monitored using the transition from m/z 490 to m/z 374 and the deuterated internal standard (dG-C8-PhIP-D3) was monitored using the transition from m/z 493 to m/z 377.
UvrABC in vitro assay
UvrABC endonuclease was cloned from UvrABca, UvrBBca, and UvrCTma, and those individual recombinant protein subunits were overexpressed in and purified from E.coli (Jiang et al., 2003; 2004). Plasmid pTHQB04 (5.45 kbp, which contains a 1.5 kbp fragment of the human β-globin gene) was propagated in XL1-Blue (Quan and States, 1996). Form I plasmid DNA was purified by CsCl-ethidium bromide sedimentation equilibrium ultracentrifugation. DNA concentrations were determined by measuring the A260. DNA quality was checked by agarose gel electrophoresis as well as measurement of the A260/A280 ratios (which were 1.8-2.0).
N-hydroxy-PhIP-damaged DNA was prepared with 2 μg of pTHQB04 plasmid DNA, 2.5 mM acetyl coenzyme A and varying concentrations of N-hydroxy-PhIP. Water was substituted for acetyl coenzyme A in controls. The total reaction was incubated at 37°C for 5 min, and stopped with an equal volume of ethyl acetate. The aqueous layer was extracted from the ethyl acetate mixture 4 times. DNA was precipitated by adding 1/10 volume of 3 M sodium acetate (pH = 5.6), and 2.2 volume of cold ethanol. DNA concentrations were determined by measuring the A260. DNA quality was checked by agarose gel electrophoresis as well as measurement of the A260/A280 ratios (which were 1.8-2.0).
The plasmid relaxation assay was adapted from that described previously (Jiang et al., 2004). N-hydroxy-PhIP-treated plasmid (20 fmol, equivalent to 71 ng pTHQB04, 1 nM of final DNA substrate) was preincubated with UvrA and UvrB in 20 μl of UvrABC buffer (50 mM Tris-HCl, pH 7.5, 50 mM KCl, 10 mM MgCl2, 5 mM dithiothreitol, and 1 mM ATP) at 55°C for 30 min, UvrCTma was added and incubated at 55°C for additional 30 min. Reactions without UvrABC were performed as a no incision control. Reactions were terminated by adding 2 μl of stop buffer (1% SDS and 200 mM EDTA-Na2). Cleavage of damaged plasmid-DNA was monitored by following the conversion of Form I plasmid to Form II plasmid. A 20 ng sample of the UvrABCTma incised DNA was resolved by 1% agarose gel electrophoresis and visualized by staining with SYBR-Gold (Molecular Probes, Eugene Oregon, USA). Fluorescence of resolved bands was quantitated using fluorescence detection mode (Blue excitation, 537 nm) of a Molecular Dynamics Storm 860 Phosphorimager (Amersham Pharmacia Biotech, Piscataway, NJ).
Results
N-hydroxy-PhIP O-acetyltransferase activities
Cell lysates from UV5 and each of the transfected CHO cell lines were tested for their capacity to activate N-hydroxy-PhIP to form dG-C8-PhIP adducts. N-hydroxy-PhIP O-acetyltransferase activity in CYP1A1/NAT2*4 -transfected cell line was 2.5-fold higher (P=0.0150) than in the CYP1A1/NAT2*5B-transfected cell line (Fig. 1). Low but detectable levels of N-hydroxy-PhIP activation were detected in the UV5 and the CYP1A1-transfected cell lines that were subtracted from the experimental measurements in the CYP1A1/NAT2-transfected cells.
Figure 1.

N-hydroxy-PhIP O-acetyltransferase activities in cell lysates of CYP1A1/NAT2*4- and CYP1A1/NAT2*5B-transfected CHO cells. Low but detectable levels of N-hydroxy-PhIP activation detected in the UV5 and the CYP1A1-transfected cell lines were subtracted from the experimental measurements in the CYP1A1/NAT2-transfected cells. Each bar represents Mean ± S.E.M. for three experiments. *N-hydroxy-PhIP O-acetyltransferase activities were significantly (p=0.0150) higher in CYP1A1/NAT2*4-than CYP1A1/NAT2*5B-transfected CHO cells.
PhIP-induced cytotoxicity and mutagenesis
The CYP1A1-, CYP1A1/NAT2*4- and CYP1A1/NAT2*5B-transfected CHO cell lines each showed concentration-dependent cytotoxicity (Fig. 2) and hypoxanthine phosphoribosyl transferase mutagenesis (Fig. 3) following PhIP treatment, whereas they were not observed in untransfected CHO cells.
Figure 2.

PhIP-induced cytotoxicity in UV5 CHO cell lines. Percent survival on the ordinate is plotted versus PhIP treatment concentration on the abscissa. Each data point represents Mean ± S.E.M. for three experiments.
Figure 3.

PhIP-induced hprt mutants in UV5 CHO cell lines. PhIP-induced hprt mutants are plotted on the ordinate versus treatment concentration on the abscissa. Each data point represents Mean ± S.E.M. for three experiments. *PhIP-induced hprt mutants were significantly higher (p<0.05) in the CYP1A1/NAT2*4 cells at the two highest doses.
PhIP-induced DNA adduct formation
The dG-C8-PhIP standards were characterized by HPLC-tandem mass spectrometry and used to verify the identity of DNA adducts formed in vitro. They were quantitated relative to dG-C8-PhIP-D3 internal standard. One principal PhIP adduct (dG-C8-PhIP), corresponding to m/z 490, was identified in CHO cells incubated with PhIP (Fig. 4). Following PhIP treatment, dG-C8-PhIP DNA adduct levels were dose dependent in cells transfected with CYP1A1 while the further addition of NAT2*4 or NAT2*5B did not have an appreciable effect (Fig 5). Adduct levels were undetectable in untransfected cells. The observation that cells transfected only with CYP1A1 were able to bioactivate PhIP raised the question of whether N-hydroxy-PhIP required further metabolism by phase II enzymes to generate the DNA reactive electrophile. In order to investigate whether N-hydroxy-PhIP could be activated by acetyl coenzyme A to form DNA adducts without catalysis by NAT2, we incubated N-hydroxy-PhIP and acetyl coenzyme A but without NAT2 in the presence of supercoiled plasmid DNA. We assayed for the formation of DNA adducts by UvrABC incision in a plasmid relaxation assay as outlined in Methods. UvrABC is a prokaryotic endonuclease that recognizes and excises bulky DNA adducts such as dG-C8-PhIP. DNA adduct formation, detected by UvrABC incision, was observed in the presence of acetyl coenzyme A despite the absence of catalysis by NAT2 (Fig 6).
Figure 4.

Top panel: mass spectrometric data of dG-C8-PhIP (490 m/z) and dG-C8-PhIP-D3 (493 m/z). Middle panel: main daughter fragment of dG-C8-PhIP (top; 374 m/z) and dG-C8-PhIP-D3 (bottom; 377 m/z). Bottom panel: HPLC chromatograms of dG-C8-PhIP (top; 11.67 min) and dG-C8-PhIP-D3 (bottom; 11.69 min).
Figure 5.

PhIP -induced DNA adduct levels in UV5 CHO cell lines. Adduct levels are plotted on the ordinate versus PhIP treatment concentration on the abscissa. Each data point represents Mean ± S.E.M. for three experiments (the S.E.M. sometimes falls within the symbol).
Figure 6.
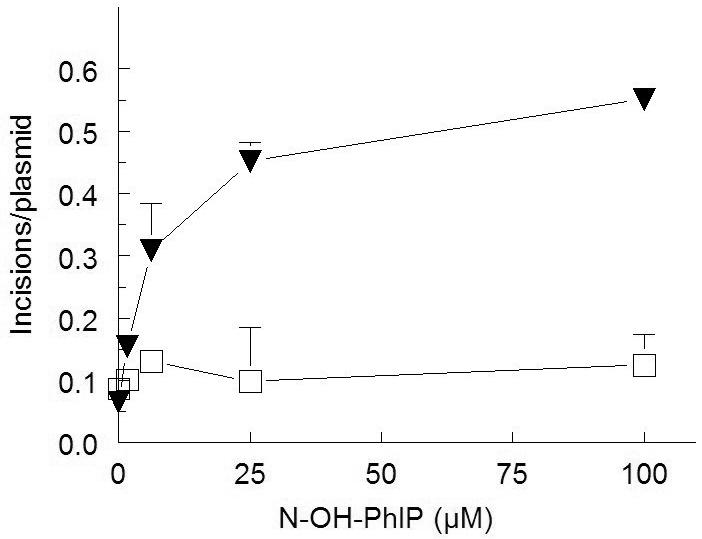
UvrABC incision of damaged plasmid DNA as a function of N-hydroxy-PhIP concentration in the absence (open squares) and presence (closed inverted triangles) of 2.5 mM AcCoA. Data points represent Means ± S.E.M. from triplicate samples (the S.E.M. sometimes falls within the symbol).
Discussion
CYP1A1 and NAT2 genotypes may increase risk of colorectal cancer from heterocyclic amines (Murtaugh et al., 2005). Recent studies have shown that lung PhIP-DNA adducts were significantly lower in lung tissue of CYP1A1 null mice but not cytochrome P4501A2 null mice (Ma et al., 2007). Our study tested the effect of human CYP1A1 in combination with rapid or slow acetylator NAT2 on mutagenesis and DNA adducts following PhIP exposure. Our study showed that PhIP induced cytotoxicity and mutagenesis in all CYP1A1-transfected cell lines but not in the untransfected UV5 cell line. Our plasmid relaxation studies suggest that enzymatic catalysis of N-hydroxy-PhIP is not required to generate a DNA reactive electrophile. We observed induction of UvrABC sensitive sites in plasmid DNA incubated with N-hydroxy-PhIP in the presence of AcCoA concentrations as low as 20 μM.
Heterocyclic amines can also be detoxified through glucuronidation and glutathione-S-transferase or via aromatic ring hydroxylation. Once the N-hydroxy-metabolite is formed, it can be reduced to the initial carcinogen, deactivated by glutathione-S-transferases or glucuronosyltransferases, or further activated by N-acetyltransferases and sulfotransferases. Although we observed differences in N-hydroxy-PhIP O-acetyltransferase catalytic activity between the CYP1A1/NAT2*4- and CYP1A1/NAT2*5B- transfected CHO cell lines, NAT2 did not affect PhIP-induced mutagenesis significantly, consistent with results previously reported in cytochrome P4501A2/NAT2-transfected CHO cell lines (Metry et al., 2007). Although the NAT2- effect was slight and not significant (p >0.05), N-hydroxy-PhIP-induced mutagenesis and DNA adduct levels were slightly higher in the CYP1A1/NAT2*4- than the CYP1A1/NAT2*5B-transfected CHO cell line. Previous studies have reported that PhIP DNA adduct levels in human breast tissue are higher in rapid than slow NAT2 acetylators (Zhu et al., 2003) and have suggested that rapid acetylator NAT2 phenotype increases colorectal (Lang et al., 1994; Chen et al., 1998; Le Marchand et al., 2001; Lilla et al., 2006; Ognjanovic et al., 2006; Cotterchio et al., 2008), breast (Deitz et al., 2000; Gallicchio et al., 2006), and lung (Chiou et al., 2005) cancer risk in individuals exposed to heterocyclic amine carcinogens. In contrast, other studies carried out with colorectal (Barrett et al., 2003; Barlak et al., 2006), breast (van der Hel et al., 2004; Ochs-Balcome et al., 2007; Mignone et al., 2009), and lung (Barlak et al., 2006) cancer do not support the association with rapid acetylator NAT2 phenotype. A much more robust effect of NAT2 phenotype has been noted for mutagenicity and DNA adduct formation from 2-amino-3,8-dimethylimidazo-[4,5-f]quinoxaline (MeIQx) (Bendaly et al., 2007). Thus, the effects of NAT2 phenotype in the gene-environmental studies may reflect heterocyclic amines such as MeIQx more than they do PhIP as noted for colorectal cancer (Ishibe et al., 2002). In summary, these results strongly support activation of PhIP by CYP1A1 with little effect of human NAT2 genetic polymorphism on mutagenesis and DNA damage.
Acknowledgments
Declaration of interest
This study was supported by United States Public Health Service grants R01-CA034627 and P30-ES014443.
References
- Barrett JH, Smith G, Waxman R, Gooderham N, Lightfoot T, Garner RC, Augustsson K, Wolf CR, Bishop DT, Forman D. Investigation of interaction between N-acetyltransferase 2 and heterocyclic amines as potential risk factors for colorectal cancer. Carcinogenesis. 2003;24:275–282. doi: 10.1093/carcin/24.2.275. [DOI] [PubMed] [Google Scholar]
- Bendaly J, Zhao S, Neale JR, Metry KJ, Doll MA, States JC, Pierce WM, Jr., Hein DW. 2-Amino-3,8-dimethylimidazo-[4,5-f]quinoxaline-induced DNA adduct formation and mutagenesis in DNA repair-deficient Chinese hamster ovary cells expressing human cytochrome P4501A1 and rapid or slow acetylator N-acetyltransferase 2. Cancer Epidemiology Biomarkers and Prevention. 2007;16:1503–1509. doi: 10.1158/1055-9965.EPI-07-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlak J, Reamon-Buettner SM. N-acetyltransferase 2 (NAT2) gene polymorphisms in colon and lung cancer patients. BMC Medical Genetics. 2006;7:58. doi: 10.1186/1471-2350-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonarati MH, Felton JS. Activation of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) to mutagenic metabolites. Carcinogenesis. 1990;11:1133–1138. doi: 10.1093/carcin/11.7.1133. [DOI] [PubMed] [Google Scholar]
- Chen J, Stampfer MJ, Hough HL, Garcia-Closas M, Willett WC, Hennekens CH, Kelsey KT, Hunter DJ. A prospective study of N-acetyltransferase genotype, red meat intake, and risk of colorectal cancer. Cancer Research. 1998;58:3307–3311. [PubMed] [Google Scholar]
- Chiou HL, Wu MF, Chien WP, Cheng YW, Wong RH, Chen CY, Lin TS, Lee H. NAT2 fast acetylator genotype is associated with an increased risk of lung cancer among never-smoking women in Taiwan. Cancer Letters. 2005;223:93–101. doi: 10.1016/j.canlet.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Cotterchio M, Boucher BA, Manno M, Gallinger S, Okey AB, Harper PA. Red Meat Intake, Doneness, Polymorphisms in Genes that Encode Carcinogen-Metabolizing Enzymes, and Colorectal Cancer Risk. Cancer Epidemiology Biomarkers and Prevention. 2008;17:3098–3107. doi: 10.1158/1055-9965.EPI-08-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts FG, Sutter TR, Strickland PT. Metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by human cytochrome P4501A1, P4501A2 and P4501B1. Carcinogenesis. 1998;19:1969–1973. doi: 10.1093/carcin/19.11.1969. [DOI] [PubMed] [Google Scholar]
- Deitz AC, Zheng W, Leff MA, Gross M, Wen WQ, Doll MA, Xiao GH, Folsom AR, Hein DW. N-Acetyltransferase-2 genetic polymorphism, well-done meat intake, and breast cancer risk among postmenopausal women. Cancer Epidemiology Biomarkers and Prevention. 2000;9:905–910. [PubMed] [Google Scholar]
- Edwards RJ, Murray BP, Murray S, Schulz T, Neubert D, Gant TW, Thorgeirsson SS, Boobis AR, Davies DS. Contribution of CYP1A1 and CYP1A2 to the activation of heterocyclic amines in monkeys and human. Carcinogenesis. 1994;15:829–836. doi: 10.1093/carcin/15.5.829. [DOI] [PubMed] [Google Scholar]
- Gallicchio L, McSorley MA, Newschaffer CJ, Thuita LW, Argani P, Hoffman SC, Helzlsouer KJ. Flame-broiled food, NAT2 acetylator phenotype, and breast cancer risk among women with benign breast disease. Breast Cancer Research and Treatment. 2006;99:229–233. doi: 10.1007/s10549-006-9203-2. [DOI] [PubMed] [Google Scholar]
- Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, Kadlubar FF. Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis. 1997;18:851–854. doi: 10.1093/carcin/18.4.851. [DOI] [PubMed] [Google Scholar]
- Ishibe N, Sinha R, Hein DW, Kulldorff M, Strickland P, Fretland AJ, Chow WH, Kadlubar FF, Lang NP, Rothman N. Genetic polymorphisms in heterocyclic amine metabolism and risk of colorectal adenomas. Pharmacogenetics. 2002;12:145–150. doi: 10.1097/00008571-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Jiang G, Jankowiak R, Grubor N, Banasiewicz M, Small GJ, Skorvaga M, Van Houten B, States JC. Supercoiled DNA promotes formation of intercalated cis-N2-deoxyguanine adducts and base-stacked trans-N2-deoxyguanine adducts by (+)-7R,8S-dihydrodiol-9S,10R-epoxy-7,8,9,10-tetra- hydrobenzo[a]pyrene. Chemical Research in Toxicology. 2004;17:330–339. doi: 10.1021/tx034184h. [DOI] [PubMed] [Google Scholar]
- Jiang G, Skorvaga M, Van Houten B, States JC. Reduced sulfhydryls maintain specific incision of BPDE-DNA adducts by recombinant thermoresistant Bacillus caldotenax UvrABC endonuclease. Protein Expression and Purification. 2003;31:88–98. doi: 10.1016/s1046-5928(03)00137-2. [DOI] [PubMed] [Google Scholar]
- Keating GA, Bogen KT. Estimates of heterocyclic amine intake in the US population. J Chromatogr B Analytical Technology Biomedical Life Sciences. 2004;802:127–133. doi: 10.1016/j.jchromb.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Kim D, Guengerich FP. Cytochrome P450 activation of arylamines and heterocyclic amines. Annual Review of Pharmacology and Toxicology. 2005;45:27–49. doi: 10.1146/annurev.pharmtox.45.120403.100010. [DOI] [PubMed] [Google Scholar]
- Kimura S, Kawabe M, Yu A, Morishima H, Fernandez-Salguero P, Hammons GJ, Ward JM, Kadlubar FF, Gonzalez FJ. Carcinogenesis of the food mutagen PhIP in mice is independent of CYP1A2. Carcinogenesis. 2003;24:583–587. doi: 10.1093/carcin/24.3.583. [DOI] [PubMed] [Google Scholar]
- Lang NP, Butler MA, Massengill J, Lawson M, Stotts RC, Hauer-Jensen M, Kadlubar FF. Rapid metabolic phenotypes for acetyltransferase and cytochrome P4501A2 and putative exposure to food-borne heterocyclic amines increase the risk for colorectal cancer or polyps. Cancer Epidemiology Biomarkers and Prevention. 1994;3:675–682. [PubMed] [Google Scholar]
- Le Marchand L, Hankin JH, Wilkens LR, Pierce LM, Franke A, Kolonel LN, Seifried A, Custer LJ, Chang W, Lum-Jones A, Donlon T. Combined effects of well-done red meat, smoking, and rapid N-acetyltransferase 2 and CYP1A2 phenotypes in increasing colorectal cancer risk. Cancer Epidemiology Biomarkers and Prevention. 2001;10:1259–1266. [PubMed] [Google Scholar]
- Lilla C, Verla-Tebit E, Risch A, Jager B, Hoffmeister M, Brenner H, Chang-Claude J. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiology Biomarkers and Prevention. 2006;15:99–107. doi: 10.1158/1055-9965.EPI-05-0618. [DOI] [PubMed] [Google Scholar]
- Ma X, Idle JR, Malfatti MA, Krausz KW, Nebert DW, Chen CS, Felton JS, Waxman DJ, Gonzalez FJ. Mouse lung CYP1A1 catalyzes the metabolic activation of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Carcinogenesis. 2007;28:732–737. doi: 10.1093/carcin/bgl184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe S, Kurihara N, Wada O, Izumikawa S, Asakuno K, Morita M. Detection of a carcinogen, 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine, in airborne particles and diesel-exhaust particles. Environmental Pollution. 1993;80:281–286. doi: 10.1016/0269-7491(93)90049-t. [DOI] [PubMed] [Google Scholar]
- Manabe S, Tohyama K, Wada O, Aramaki T. Detection of a carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), in cigarette smoke condensate. Carcinogenesis. 1991;12:1945–1947. doi: 10.1093/carcin/12.10.1945. [DOI] [PubMed] [Google Scholar]
- Metry KJ, Zhao S, Neale JR, Doll MA, States JC, McGregor WG, Pierce WM, Jr., Hein DW. 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine-induced DNA adducts and genotoxicity in chinese hamster ovary (CHO) cells expressing human CYP1A2 and rapid or slow acetylator N-acetyltransferase 2. Molecular Carcinogenesis. 2007;46:553–563. doi: 10.1002/mc.20302. [DOI] [PubMed] [Google Scholar]
- Mignone LI, Giovannucci E, Newcomb PA, Titus-Ernstoff L, Trentham-Dietz A, Hampton JM, Orav EJ, Willett WC, Egan KM. Meat consumption, heterocyclic amines, NAT2, and the risk of breast cancer. Nutrition and Cancer. 2009;61:36–46. doi: 10.1080/01635580802348658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray BP, Edwards RJ, Murray S, Singleton AM, Davies DS, Boobis AR. Human hepatic CYP1A1 and CYP1A2 content, determined with specific anti-peptide antibodies, correlates with the mutagenic activation of PhIP. Carcinogenesis. 1993;14:585–592. doi: 10.1093/carcin/14.4.585. [DOI] [PubMed] [Google Scholar]
- Murtaugh MA, Sweeney C, Ma KN, Caan BJ, Slattery ML. The CYP1A1 genotype may alter the association of meat consumption patterns and preparation with the risk of colorectal cancer in men and women. Journal of Nutrition. 2005;135:179–186. doi: 10.1093/jn/135.2.179. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program . Report on Carcinogenesis. Eleventh Edition U.S. Department of Health and Human Services, Public Health Service; Research Triangle Park, NC: 2005. [Google Scholar]
- Neale JR, Smith NB, Pierce WM, Hein DW. Methods for aromatic and heterocyclic amine carcinogen-DNA adduct analysis by liquid chromatography-tandem mass spectrometry. Polycyclic Aromatic Compounds. 2008;28:402–417. doi: 10.1080/10406630802377773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs-Balcom HM, Wiesner G, Elston RC. A meta-analysis of the association of N-acetyltransferase 2 gene (NAT2) variants with breast cancer. American Journal of Epidemiology. 2007;166:246–254. doi: 10.1093/aje/kwm066. [DOI] [PubMed] [Google Scholar]
- Ognjanovic S, Yamamoto J, Maskarinec G, Le Marchand L. NAT2, meat consumption and colorectal cancer incidence: an ecological study among 27 countries. Cancer Causes and Control. 2006;17:1175–1182. doi: 10.1007/s10552-006-0061-3. [DOI] [PubMed] [Google Scholar]
- Peluso M, Castegnaro M, Malaveille C, Friesen M, Garren L, Hautefeuille A, Vineis P, Kadlubar F, Bartsch H. 32P Postlabelling analysis of urinary mutagens from smokers of black tobacco implicates 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) as a major DNA-damaging agent. Carcinogenesis. 1991;12:713–717. doi: 10.1093/carcin/12.4.713. [DOI] [PubMed] [Google Scholar]
- Quan T, States JC. Preferential DNA damage in the p53 gene by benzo[a]pyrene metabolites in cytochrome P4501A1-expressing xeroderma pigmentosum group A cells. Molecular Carcinogenesis. 1996;16:32–43. doi: 10.1002/(SICI)1098-2744(199605)16:1<32::AID-MC5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–368. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- Sinha R, Gustafson DR, Kulldorff M, Wen WQ, Cerhan JR, Zheng W. 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, a carcinogen in high-temperature-cooked meat, and breast cancer risk. Journal of the National Cancer Institute. 2000;92:1352–1354. doi: 10.1093/jnci/92.16.1352. [DOI] [PubMed] [Google Scholar]
- Snyderwine EG, Venugopal M, Yu M. Mammary gland carcinogenesis by food-derived heterocyclic amines and studies on the mechanisms of carcinogenesis of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Mutation Research. 2002;506-507:145–152. doi: 10.1016/s0027-5107(02)00161-6. [DOI] [PubMed] [Google Scholar]
- Thiebaud HP, Knize MG, Kuzmicky PA, Hsieh DP, Felton JS. Airborne mutagens produced by frying beef, pork and a soy-based food. Food and Chemical Toxicology. 1995;33:821–828. doi: 10.1016/0278-6915(95)00057-9. [DOI] [PubMed] [Google Scholar]
- Turesky RJ. The role of genetic polymorphisms in metabolism of carcinogenic heterocyclic aromatic amines. Current Drug Metabolism. 2004;5:169–180. doi: 10.2174/1389200043489036. [DOI] [PubMed] [Google Scholar]
- Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicology Letters. 2007;168:219–227. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- van der Hel OL, Peeters PH, Hein DW, Doll MA, Grobbee DE, Ocke M, Bueno de Mesquita HB. GSTM1 null genotype, red meat consumption and breast cancer risk (The Netherlands) Cancer Causes and Control. 2004;15:295–303. doi: 10.1023/B:CACO.0000024255.16305.f4. [DOI] [PubMed] [Google Scholar]
- Wild D, Feser W, Michel S, Lord HL, Josephy PD. Metabolic activation of heterocyclic aromatic amines catalyzed by human arylamine N-acetyltransferase isozymes (NAT1 and NAT2) expressed in Salmonella typhimurium. Carcinogenesis. 1995;16:643–648. doi: 10.1093/carcin/16.3.643. [DOI] [PubMed] [Google Scholar]
- Wu RW, Tucker JD, Sorensen KJ, Thompson LH, Felton JS. Differential effect of acetyltransferase expression on the genotoxicity of heterocyclic amines in CHO cells. Mutation Research. 1997;390:93–103. doi: 10.1016/s0165-1218(97)00005-0. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Fujita K, Nakayama K, Suzuki A, Nakamura K, Yamazaki H, Kamataki T. Establishment of ten strains of genetically engineered Salmonella typhimurium TA1538 each co-expressing a form of human cytochrome P450 with NADPH-cytochrome P450 reductase sensitive to various promutagens. Mutation Research. 2004;562:151–162. doi: 10.1016/j.mrgentox.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Zang Y, Zhao S, Doll MA, States JC, Hein DW. The T341C (Ile114Thr) polymorphism of N-acetyltransferase 2 yields slow acetylator phenotype by enhanced protein degradation. Pharmacogenetics. 2004;14:717–723. doi: 10.1097/00008571-200411000-00002. [DOI] [PubMed] [Google Scholar]
- Zhu J, Chang P, Bondy ML, Sahin AA, Singletary SE, Takahashi S, Shirai T, Li D. Detection of 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine-DNA adducts in normal breast tissues and risk of breast cancer. Cancer Epidemiology Biomarkers and Prevention. 2003;12:830–837. [PubMed] [Google Scholar]