

Abstract
The presence of AβpE3 (N-terminal truncated Aβ starting with pyroglutamate) in Alzheimer’s disease (AD) has received considerable attention since the discovery that this peptide represents a dominant fraction of Aβ peptides in senile plaques of AD brains. This was later confirmed by other reports investigating AD and Down’s syndrome postmortem brain tissue. Importantly, AβpE3 has a higher aggregation propensity, and stability, and shows an increased toxicity compared to full-length Aβ. We have recently shown that intraneuronal accumulation of AβpE3 peptides induces a severe neuron loss and an associated neurological phenotype in the TBA2 mouse model for AD. Given the increasing interest in AβpE3, we have generated two novel monoclonal antibodies which were characterized as highly specific for AβpE3 peptides and herein used to analyze plaque deposition in APP/PS1KI mice, an AD model with severe neuron loss and learning deficits. This was compared with the plaque pattern present in brain tissue from sporadic and familial AD cases. Abundant plaques positive for AβpE3 were present in patients with sporadic AD and familial AD including those carrying mutations in APP (arctic and Swedish) and PS1. Interestingly, in APP/PS1KI mice we observed a continuous increase in AβpE3 plaque load with increasing age, while the density for Aβ1-x plaques declined with aging. We therefore assume that, in particular, the peptides starting with position 1 of Aβ are N-truncated as disease progresses, and that, AβpE3 positive plaques are resistant to age-dependent degradation likely due to their high stability and propensity to aggregate.
Keywords: Transgenic mice, Arctic mutation, Swedish mutation, Presenilin-1 mutation, Sporadic Alzheimer’s disease, Pyroglutamate Abeta, Biacore, Antibody specificity
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the presence of extracellular amyloid plaques composed of amyloid-β (Aβ) surrounded by dystrophic neurites and neurofibrillary tangles. The discovery that certain early-onset familial forms of AD may be caused by an enhanced production of Aβ peptides have led to the hypothesis that amyloidogenic Aβ is intimately involved in the AD pathogenic process (Selkoe 1998). Besides Aβ peptides starting with an aspartate at position 1, a variety of different N-truncated Aβ peptides have been identified in AD brains. Ragged peptides with a major species including those beginning with phenylalanine at position 4 of Aβ have been reported as early as 1985 by Masters et al. (1985a, b)). In contrast, no N-terminal sequence could be obtained from cores purified in a sodium dodecyl sulfate-containing buffer, which led to the assumption that the N-terminus could be blocked (Gorevic et al. 1986; Selkoe et al. 1986). The presence of AβpE3 (N-terminal truncated Aβ starting with pyroglutamate) in AD brain was subsequently shown using mass spectrometry of purified Aβ peptides, explaining at least partially initial difficulties in sequencing Aβ peptides purified from human brain tissue (Mori et al. 1992). The authors reported that only 10–15% of the total Aβ isolated by this method begins at position 3 with AβpE3. Saido et al. (1995) then showed that AβpE3 represents a dominant fraction of Aβ peptides in senile plaques of AD brains. This was later confirmed by other reports investigating AD and Down’s syndrome postmortem brain tissue (Guntert et al. 2006; Harigaya et al. 2000; Hosoda et al. 1998; Iwatsubo et al. 1996; Kuo et al. 1997, 2001; Miravalle et al. 2005; Piccini et al. 2005, 2007; Russo et al. 1997; Saido et al. 1996, Tekirian et al. 1998, Wegiel et al. 2007).
In general, N-terminal deletions in general enhance aggregation of β-amyloid peptides in vitro (Pike et al. 1995; Masters et al. 1985a, b). Importantly, AβpE3 has a higher aggregation propensity (He and Barrow 1999; Schilling et al. 2006), and stability (Kuo et al. 1998), and shows an increased toxicity compared to full-length Aβ (Russo et al. 2002). Ku et al. (2001) have used an integrated chemical and morphological comparison of the Aβ peptides and amyloid plaques present in the brains of APP23 transgenic mice and human AD patients. The lack of post-translational modifications such as N-terminal degradation, isomerization, racemization and pyroglutamyl formation of Aβ from APP23 mice provides an explanation for the differences in solubility of Aβ from human AD and transgenic mouse plaques. This group also demonstrated that in another transgenic mouse model (Tg2576), Aβ peptides lacked a pronounced N-terminal degradation, post-translational modifications and cross-linkages that were frequently observed in the compact Aβ peptide deposits found in AD brain. Thus, under in vivo conditions, truncated Aβ molecules appeared to be generated by hydrolysis at multiple sites rather than by postmortem N-terminal degradation (Kalback et al. 2002). Recently, it has been demonstrated that the N-terminal pE-formation can be catalyzed by glutaminyl cyclase (QC) which pharmacologically is interfered by a QC inhibitor, both in vitro (Cynis et al. 2008) and in vivo (Schilling et al. 2008). QC expression was found up-regulated in the cortex of patients with AD and correlated with the appearance of pE-modified Aβ. Oral application of a glutaminyl cyclase inhibitor resulted in reduced AβpE3-42 burden in two different transgenic mouse models of AD and in a new Drosophila model. Interestingly, treatment of these mice was accompanied by reductions in Aβx-40/42, diminished plaque formation and gliosis, as well as improved performance in context memory and spatial learning tests (Schilling et al. 2008).
A promising experimental approach to unravel the role of Aβ in AD pathology has been the generation of transgenic mice overexpressing the amyloid precursor protein (APP). All mouse models mimic the typical AD-like pathological deficits in synaptic transmission (Hsia et al. 1999), changes in behavior, differential glutamate responses and deficits in long-term potentiation. These characteristics are generally attributed to the overexpression of full-length APP (Moechars et al. 1999). Although learning deficits (Billings et al. 2005; Holcomb et al. 1998; Oakley et al. 2006; Puolivali et al. 2002) were evident in various APP models, the extent of Aβ-amyloid deposition did not correlate with the behavioral phenotype (Holcomb et al. 1999). In the past, extracellular Aβ has been regarded as the causative agent, whereas more recent evidence now points to toxic effects of Aβ in intracellular compartments (Tseng et al. 2004; Wirths et al. 2004). In addition, another concept proposes that the soluble oligomers and the β-sheet containing amyloid fibrils are the toxic forms of Aβ (Harmeier et al. 2009; Klein 2002; Selkoe 2001). It has further been demonstrated that soluble oligomeric Aβ42, but not plaque-associated Aβ, correlates best with cognitive dysfunction in AD (McLean et al. 1999; Naslund et al. 2000). Oligomers are formed preferentially intracellularly within neuronal processes and synapses rather than extracellularly (Takahashi et al. 2004; Walsh et al. 2000). Previously, we have reported that intraneuronal Aβ rather than extracellular plaque pathology correlates with neuron loss in the hippocampus (Casas et al. 2004), the frontal cortex (Christensen et al. 2008b) and the cholinergic system (Christensen et al. 2008a) of APP/PS1KI mice expressing transgenic human mutant APP751 including the Swedish and London mutations on a murine knock-in (KI) Presenilin 1 (PS1) background with two FAD-linked mutations (PS1M233T and PS1L235P). The APP/PS1KI mice exhibit robust learning deficits at the age of 6 months (Wirths et al. 2008), age-dependent axonopathy (Wirths et al. 2007), neuron loss in hippocampus CA1 together with synaptic deficits, and hippocampus atrophy coinciding with intraneuronal aggregation of N-terminal modified Aβ variants (Breyhan et al. 2009). Notably, the APP/PS1KI mouse model exhibits a large heterogeneity of N-truncated Aβx-42 variants (Casas et al. 2004). Recently, we generated a new mouse model expressing only N-truncated AβpE3 in neurons, and demonstrated for the first time that this peptide is neurotoxic in vivo inducing neuron loss and an associated neurological phenotype (Wirths et al. 2009).
Given the increasing interest in AβpE3, we have generated two novel monoclonal antibodies which we have characterized as highly specific for AβpE3 peptides and herein used to analyze plaque deposition in APP/PS1KI mice, an AD model with severe neuron loss and learning deficits, and compared this pattern with that present in brain tissue from sporadic and familial AD cases.
Materials and methods
Antibodies
Two novel antibodies 1-57 and 2-48 against N-terminal truncated Aβ starting with pyroglutamate (pGlu, pE) were generated by Synaptic Systems (Goettingen, Germany) by immunizing three Balb/c mice with a KLH-coupled pyroglutamate-Abeta-peptide with the peptide pEFRHD-C. After preparation of the lymph nodes, cells were fused with the myeloma cell line P3-×63-Ag8. The hybridoma supernatants of mixed clones were screened by ELISA and subcloned. The monoclonal antibodies 1-57 (IgG2b) and 2-48 (IgG1) were selected and further characterized for their specificity and affinity (see below). Other monoclonal anti-Aβ antibodies used for comparison are 4G8 (Aβ epitope 17-24; Covance), HJ5.1 (Aβ 13-28, courtesy of Hong Jiang and David Holtzman) and Aβ[N] (IBL, Hamburg, Germany).
Surface plasmon resonance analysis (BIAcore)
To obtain affinity data, a BIAcore3000 system (GE Healthcare, Uppsala, Sweden) with the BIAcontrol software (Version 4.1) was used. Aβ peptides (PSL, Heidelberg, Germany; Aβ1-38, Aβ3-38, AβpE3-38) were solubilized and monomerized in 50% formic acid, lyophilized, and re-solubilized in 0.12% ammonia to obtain stock solutions of 1 μg/μl [see also (Harmeier et al. 2009)]. The Aβ peptides were then immobilized by amine coupling (Amine Coupling Kit, GE Healthcare, Uppsala, Sweden) to CM5-chips at pH 4.5 after preparing dilutions (final peptide concentrations were 20 μg/ml) in coupling buffer (10 mM NaAcetate pH 4.5), as running buffer HBS-EP (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0,005% P20) was used. The monoclonal antibodies 1-57 and 2-48 were diluted in running buffer and tested in a concentration range from 1 to 35 μg/ml at flow rates of 15 or 20 μl/min, respectively. Injection times varied from 5 to 20 min. The antibodies Aβ[N] (IBL, Hamburg, Germany) and 4G8 (Covance, USA) were used at concentrations of 10 μg/ml. The chip surface was regenerated after each antibody injection by a 90 s pulse injection with buffer containing 8 M urea at a flow rate of 100 μl/min. Binding curves were evaluated with the BIA evaluation software (Version 4.1). All chemicals were obtained from Roth (Karlsruhe, Germany) except for HEPES (Sigma-Aldrich, München, Germany) and P20 (GE Healthcare, Uppsala, Sweden).
Transgenic mice
The generation of APP/PS1KI mice has been described previously (Casas et al. 2004). In brief, human mutant APP751 containing the Swedish and London mutations is overexpressed under the control of the murine Thy-1 promoter, whereas murine PS1 with the M233T and L235P FAD-linked mutations is expressed under the control of the endogenous mouse PS1 promoter. APP/PS1KI mice were a generous gift of Dr. Laurent Pradier, Sanofi-Aventis, France. The mice were backcrossed for more than 10 generations on a C57BL/6J genetic background. All animals were handled according to the German guidelines for animal care. All research involving animals have been conducted according to guidelines of the German animal protection law. According to that the local committee for animal welfare at the University Medicine of Goettingen approved the experiments before the mice were killed.
Immunohistochemistry on paraffin sections
Human brain samples from frontal cortex were obtained from (1) the Netherlands Brain Bank (NBB), Amsterdam, The Netherlands; (2) the APP Swedish and APP arctic FAD brain samples from Uppsala University, and the Presenilin-1 FAD brain samples from the Hopital del la Salpetrière, a generous gift of Prof. Dr. Charles Duyckaerts and Dr. Veronique Sazdovitch. We did not receive information of the cause of death and postmortem delay in all cases, therefore the information was not added to Table 1. Tissue was post-fixed in 4% buffered formalin at 4°C for several weeks. Mice were transcardially perfused with 4% PFA in PBS and brains were carefully dissected. Post-fixation was carried out in 4% buffered formalin at 4°C before the tissue was embedded in paraffin. Immunohistochemistry was performed on 4 μm sagittal paraffin sections, as described previously (Wirths et al. 2002). In brief, sections were deparaffinized in xylene and rehydrated in a series of ethanol. After treatment with 0.3% H2O2 in PBS to block endogenous peroxidases, antigen retrieval was achieved by boiling sections in 0.01 M citrate buffer pH 6.0, followed by 3 min incubation in 88% formic acid. Non-specific binding sites were blocked by treatment with skim milk and fetal calf serum in PBS, prior to the addition of the primary antibodies. Primary antibodies 4G8 (1 μg/ml), Aβ[N] (2 μg/ml); 2-48 (10 μg/ml) and 1-57 (10 μg/ml), were incubated overnight in a humid chamber at room temperature, followed by incubation with biotinylated secondary antibodies (DAKO, Glostrup, DK) before staining was visualized using the ABC method with Vectastain kit (Vector Laboratories, Burlingame, USA) and diaminobenzidine as chromogen providing a reddish brown color. Counterstaining was carried out with hematoxylin.
Table 1.
Demographic data of the human specimen, semi-quantitative plaque load assessment, vessel pathology (congophilic angiopathy, CAA) and Braak staging
| Sex | Age | Plaque load | CAA | Braak stage | |||
|---|---|---|---|---|---|---|---|
| 2-48 | 4G8 | Aβ[N] | |||||
| Control 1 | M | 74 | + | + | (+) | ✓ | III |
| Control 2 | F | 82 | − | − | − | − | I |
| Control 3 | F | 73 | − | − | − | ✓ | I |
| Control 4 | F | 79 | ++ | ++ | + | ✓ | II |
| Control 5 | M | 70 | ++ | +++ | + | − | 0 |
| Control 6 | F | 73 | ++ | ++ | (+) | − | 0 |
| AD 1 | M | 72 | + | ++ | (+) | − | VI |
| AD 2 | M | 70 | +++ | +++ | +++ | ✓ | V |
| AD 3 | M | 78 | + | ++ | (+) | ✓ | IV |
| AD 4 | M | 60 | +++ | +++ | + | − | VI |
| AD 5 | M | 93 | + | + | + | ✓ | IV |
| AD 6 | M | 64 | +/++ | ++ | + | ✓ | VI |
| AD 7 | F | 77 | ++ | +++ | + | − | VI |
| AD 8 | F | 81 | ++ | +++ | + | − | IV |
| AD 9 | F | 78 | +++ | +++ | + | − | IV |
| AD 10 | F | 64 | (+) | ++ | − | − | III |
| AD 11 | F | 88 | +++ | +++ | − | ✓ | IV |
| AD 12 | M | 81 | +++ | +++ | +++ | ✓ | IV |
| AD 13 | F | 84 | +++ | +++ | − | − | IV |
| AD 14 | F | 84 | +++ | +++ | (+) | ✓ | IV |
| FAD APP arc | M | 64 | +++ | +++ | +/++ | ✓ | n.a. |
| FAD APP swe | F | 61 | +++ | +++ | +++ | ✓ | n.a. |
| FAD PS1 P264L | M | 54 | +++ | +++ | (+) | ✓ | VI |
The three familial AD (FAD) patients harbored an APP Swedish mutation (APP swe), an APP arctic mutation (APP arc), and a Presenilin-1 mutation (FAD P264L). Most but not all cases also showed CAA. Antibodies: 4G8 (Aβ 17-24), 2-48 (pGlu Aβ) and Aβ[N] (specific for Aβ at position 1)
Quantification of Aβ load
For each tissue block, five paraffin-embedded sections, at least 25 μm apart from each other, were stained simultaneously with DAB as chromogen. The relative Aβ load was evaluated using an Olympus BX-51 microscope equipped with an Olympus DP-50 camera and the ImageJ software (NIH, USA). Representative pictures of 10× magnification were systematically captured from the cortex. The pictures were binarized to 8-bit black using ImageJ and white pictures and a fixed intensity threshold was applied defining the DAB staining. Measurements were performed for a percentage of the area covered by the Aβ DAB staining.
Western blot
Peptides including Aβ1-42, Aβ3-42, AβpE11-42 and AβpE3-42 were purchased from Anaspec (San Jose, CA) and reconstituted in conditions specific to each peptide. For each Western blot, 10 ng of peptide was loaded per lane of a 12% Bis–Tris gel in MES buffer and run at 150 V for 1 h. The peptides were then transferred to 0.45 μm nitrocellulose for 1 h at 100 mA per membrane using wet transfer in Towbin buffer. Post-transfer, membranes were incubated in PBS, pH 9.0 at 95°C for 5 min, which allows for improved access to the antigen. Membranes were then blocked in 2%NFDM/PBS, pH 9.0 for 1 h at room temperature while gently mixing, before overnight incubation at 4°C using the primary antibody (1-57, 2-48 or HJ5.1) at 1 μg/ml in blocking buffer, again while gently mixing. Membranes were rinsed in PBS/0.05%Tween-20 (pH 9.0) three times for 5 min. Secondary anti-mouse IgG-HRP was applied in blocking conditions at a 1:10,000 dilution for 1 h at room temperature. Membranes were rinsed in PBS/0.05%Tween-20 (pH 9.0) three times for 10 min. The blots were developed using ECL Advance (GE Healthcare) by applying the chemiluminescent reagents for 1 min in reduced light conditions and then imaged on standard film emulsion.
Statistical analysis
Stereological quantifications were analyzed using two-way ANOVA followed by t tests. Data are presented as mean ± SEM. Significance levels were given as follows: ***P < 0.001, **P < 0.01 and *P < 0.05. All statistical analysis was performed using GraphPad Prism version 4.03 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Antibody staining against AβpE3-x
In order to characterize the staining pattern of the two novel AβpE3 antibodies, formalin-fixed and paraffin-embedded cortical tissue sections from 14 patients with AD were used (Table 1). Figure 1 shows plaque staining in frontal cortex of sporadic AD with the Aβ antibody 4G8. There were no significant differences between the AβpE3 antibodies 1-57 and 2-48.
Fig. 1.
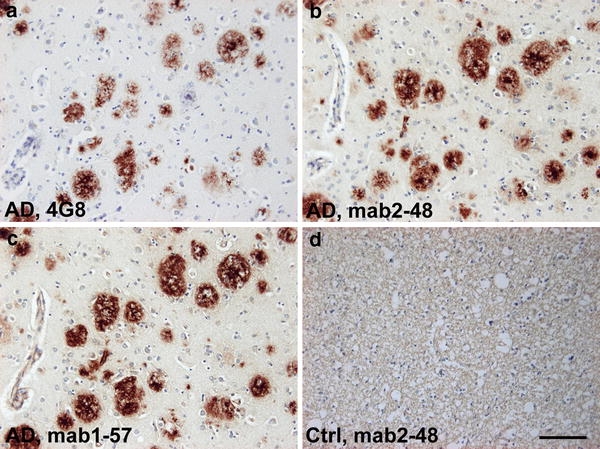
Plaque staining with pGlu Aβ (AβpE3) antibodies in frontal cortex in sporadic AD brain. Plaques staining with antibody 4G8 (a), pGlu Aβ antibodies 2-48 (b) and 1-57(c); but no staining in plaque-free healthy control tissue (d, 2-48 staining). Scale bar 100 μm
Surface plasmon resonance analysis of antibodies
The specificity and affinity of the antibodies used for immunohistochemical staining was determined with the help of surface plasmon resonance analyses (BIAcore). Equal amounts of the Aβ peptides pE3-38, 3-38, and 1-38 were immobilized to a sensor chip surface and various dilutions of the individual antibody were applied to quantify binding. The resulting sensorgrams clearly demonstrate an unequivocal specificity of 1-57 and 2-48 for AβpE3-38 (Fig. 2a, b), while we could hardly detect any binding to Aβ3-38 or 1-38. In contrast to the pGlu-specific antibodies, the commercially available antibody Aβ[N] exhibited specific binding to Aβ1-38 when subjected to the respective sensor surface (Fig. 2c). Hence, this result implies that this antibody is highly suitable to reveal the presence and quantity also in brain tissue of full-length Aβ species compared to the shorter species and AβpE3. To further characterize 1-57 and 2-48, the avidities of both antibodies were calculated from the steady state of binding activity at various tested concentrations. The resulting K D-values of 17.7 nM for 1-57 and 9.1 nM for 2-48 confirmed a very high affinity for both antibodies specifically recognizing AβpE3-38.
Fig. 2.
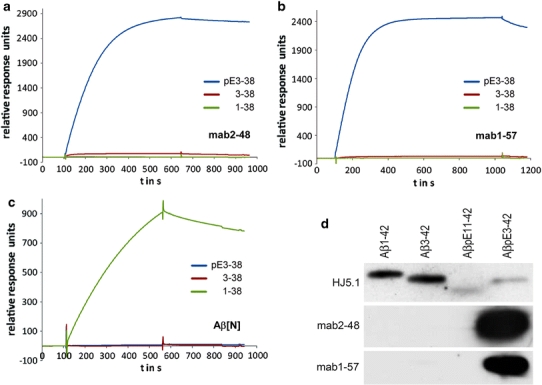
Binding specificity of pGlu Aβ (AβpE3) antibodies. (a) Biacore analysis revealed that pGlu antibodies 2-48 (b) and 1-57 specifically bind to AβpE3-38, not to Aβ3-38 and Aβ1-38. (c) The commercial antibody Aβ[N] recognizes only Aβ1-38, not Aβ3-38 or AβpE3-38. (d) 2-48 and 1-57 recognize Aβ3p(E)-42, but not Aβ1-42, Aβ3-42, or Aβ11p(E)-42 under these conditions. Lane 1 Aβ1-42, Lane 2 Aβ3-42, Lane 3 Aβ11p(E)-42, Lane 4 Aβ3p(E)-42. 10 ng per well of each peptide was loaded on 3 separate 12% Bis–Tris gels, transferred to nitrocellulose and blotted with 1 μg/ml of each primary antibody. 2-48 and 1-57 recognize Aβ3p(E)-42, Aβ1-42, Aβ3-42, or Aβ11p(E)-42 under these conditions
Western blot showing specificity of AβpE3-x antibodies 1-57 and 2-48
In the presence of SDS and under denaturing conditions the two antibodies against pGlu Aβ specifically detected AβpE3-42 without any visible cross-reactivity against other Aβ epitopes or pGlu epitope starting with Aβ11. The antibody HJ5.1 recognizes a central epitope of Aβ and therefore stains all peptides loaded (Fig. 2d). Staining specificity of transgenic mouse brain lysates using 2-48 has been shown previously (Wirths et al. 2009).
Dynamic changes of plaque-associated Aβ in the APP/PS1KI transgenic mouse brain
APP/PS1KI mice at 2, 6 and 10 months of age (Fig. 3) were stained with all four Aβ antibodies and plaque load was quantified as described earlier (Christensen et al. 2008b). One-way analysis of variance demonstrated significant differences between the groups using Aβ[N] (P < 0.0005; F = 19.39), 4G8 (P < 0.01; F = 14.77), 2-48 (P < 0.001; F = 20.67) and 1-57 (P < 0.01; F = 9.196). After Bonferroni correction (statistical significances between the groups are presented in Fig. 3) the mean values were as follows: Aβ[N] (mean ± SEM at 2 months 1.7 ± 0.2, 6 months 5.9 ± 0.7 and 10 months 3 ± 0.3); 4G8 (mean ± SEM at 2 months 3.2 ± 0.3, 6 months 9.6 ± 0.6 and 10 months 9.1 ± 1.2); 2-48 (mean ± SEM at 2 months 0.2 ± 0.1, 6 months 2.0 ± 0.4 and 10 months 4.7 ± 0.8); 1-57 (mean ± SEM at 2 months 0.1 ± 0.1, 6 months 0.8 ± 0.2 and 10 months 2.9 ± 0.9). While there was a peak of Aβ1-x at 6 months of age, total Aβ (4G8) reached a plateau at that time point, and AβpE steadily increased from 2 to 10 months of age. These data indicate that (1) total Aβ plaque burden reaches a certain level of saturation, (2) Aβ starting at position 1 is degraded and/or truncated at later time points and (3) N-truncated pGlu Aβ is constantly increasing.
Fig. 3.
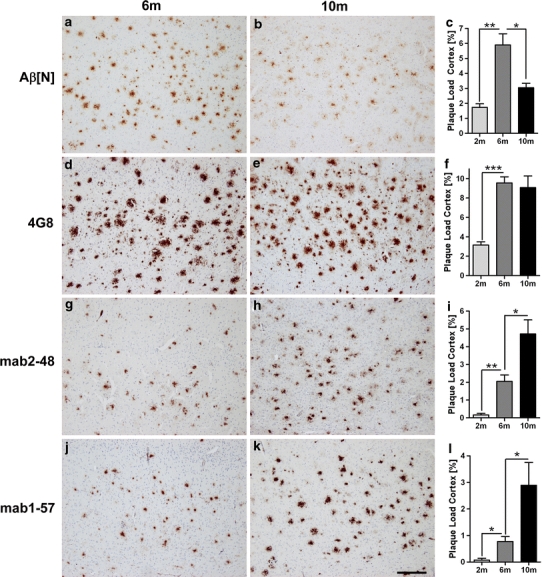
Plaque load analysis in APP/PS1KI mice. Plaque load was quantified at 2, 6 and 10 months of age (n = 5, female mice only). a–c Aβ[N] an antibodies specific for position 1 of Aβ showed a peak at six and a decline at 10 months of age. d–f 4G8 recognizing Aβ 17-24 showed an increase between 2 and 6 month of age, while at 10 months the plaque load did not increase any more. Analysis with the Aβ pyroglutamate (AβpE3) antibodies 2-48 (g–i) and 1-57 (j–l) demonstrated a continuous increase. Scale bar 200 μm
Comparison of Aβ plaque load in sporadic and familial AD brain
The plaque staining pattern (Fig. 1), BIAcore and Western blot analysis (Fig. 2) together with the staining pattern in the APP/PS1KI mice (Fig. 3) revealed that the specificity and affinity of the antibodies 1-57 and 2-48 was very similar under native and denaturing conditions. Therefore, only 2-48 was used as an additional antibody for confirmation of findings obtained with the following experiments. Human postmortem brain tissue (frontal cortex from sporadic AD, FAD and non-demented individuals) was used to compare staining patterns obtained with this set of Aβ antibodies (Fig. 4). Most of the sporadic and all of the familial AD (FAD) cases demonstrated high abundance and similar amount of plaques stained with 4G8 and 2-48. Plaques stained with Aβ[N] were less frequently observed. We also identified cases with similar staining using 4G8 and 2-48 (Table 1) indicating that N-terminal truncation has occurred. While the plaque load per se did not differ between non-demented controls and AD cases, it is interesting to note that the pattern in human cases resembled the pattern in APP/PS1KI mice. For example, case AD 5 demonstrated low plaque load according to all three antibodies resembling the pattern seen in 2-month-old mice. The case AD 9 showed a pattern as seen in 10-month-old mice. Interestingly, we never observed high Aβ1-x levels in combination with low AβpE3 reaction as in 6-month-old mice. All three antibodies stained Aβ in blood vessels, a typical feature of congophilic angiopathy (CAA). We did not observe any correlation between Braak staging and plaque pathology. Interestingly, all FAD cases showed abundant AβpE3 plaques. The FAD cases having an APP Swedish (APP swe) or APP arctic (APP arc) mutation demonstrated a similar extent of Aβ plaque load. The Presenilin-1 case with P264L mutation had only a minor amount of Aβ[N] positive plaques.
Fig. 4.
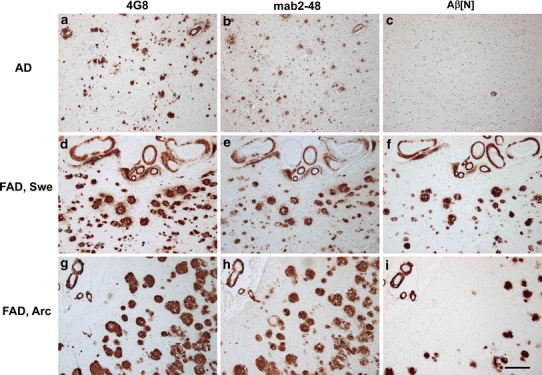
Staining pattern in frontal cortex of sporadic and familial AD brain. Upper panel showing the staining pattern in a sporadic AD case using 4G8 (Aβ 17-24), 2-48 (pGlu Aβ) and Aβ[N] (specific for Aβ at position 1) (a–c). While the staining pattern of 4G8 and 2-48 is comparable, there is no staining with Aβ[N]. Middle panel comparable levels of plaque load with all three antibodies in a FAD case with the Swedish mutation. Note abundant vessel staining with all three antibodies (d–f). Lower panel the same is true for the staining pattern in a case with the arctic mutation; however, Aβ[N] staining is less abundant (g–i). Scale bar 200 μm
Discussion
In the present work, we show that large numbers of plaques positive for AβpE3 are present in patients with sporadic AD as has been shown earlier using two novel antibodies. In addition, we demonstrate that also familial AD including those carrying mutations in APP (arctic and Swedish) and PS1 harbor abundant AβpE3plaques. In contrast to a recent publication (Schilling et al. 2008), we observed that control cases also harbored AβpE3 positive plaques. The amount of plaques positive for Aβ1-x is less abundant in the control cases. Interestingly, in APP/PS1KI mice, we observed a continuous increase in AβpE3 plaque load with increasing age, while the density for Aβ1-x plaques declined with aging. We therefore assume that, in particular, the peptides starting with position 1 of Aβ are N-truncated as disease progresses. On the other side, we have demonstrated that intraneuronal AβpE3-42 expression is highly toxic in vivo, and that the induced neuron loss is associated with a lethal neurological phenotype in TBA2 transgenic mice (Wirths et al. 2009). Saido et al. suggested that the removal of N-terminal amino acids 1 and 2 of Aβ could be carried out by a hypothetical peptidase (Saido et al. 1995), and very recently it has been shown that aminopeptidase A may be responsible for the N-terminal truncation of full-length Aβ peptides (Sevalle et al. 2009). If a subsequent glutamate cyclization by a glutamate cyclase occurs leading to pGlu formation, this could protect the peptide from degradation and even make it more prone to accumulate. The increasing levels of AβpE3 plaques in APP/PS1KI mice with age could be due to this effect. To further verify this in vivo, we have recently generated transgenic mice expressing Aβ3E starting at position 3 with glutamine and ending at position 42 (TBA2 mouse line (Wirths et al. 2009)). The severity of the neurological phenotype observed in TBA2 mice, accompanied by Purkinje cell loss and premature mortality reflects the in vivo toxicity of AβpE3-42. Interestingly, 85% of Aβ peptides in the APP/PS1KI mice terminated at position 42, whereas the N-terminus shows a large heterogeneity including AβpE3. The time point of high levels of AβpE3-42 coincided with the onset of behavioral deficits in both mouse models. We have previously shown that at 6 months, the APP/PS1KI mice exhibit a neuron loss in CA1 of the hippocampus (Breyhan et al. 2009; Casas et al. 2004), the frontal cortex (Christensen et al. 2008b), and in distinct cholinergic nuclei (Christensen et al. 2008a). Overall, the pathological events seen in the APP/PS1KI mouse model might be at least partly triggered by AβpE3-42 accumulation.
N-terminal truncation of Aβ seems to be a common phenomenon in brain material that has been stored for a long time. In brain material that has been stored only a few years, much more N-terminally intact Aβ is found. The conclusion from this preliminary study is that the process of N-terminal truncation might continue while the brain tissue is kept at −80°C (Lars Lannfelt and Martin Ingelsson, unpublished data). This phenomenon is not likely to occur in paraffin-embedded tissue. However, it cannot be completely ruled out.
N-truncated AβpE3 peptides have been identified by several groups from AD brains (Guntert et al. 2006; Harigaya et al. 2000; Hosoda et al. 1998; Iwatsubo et al. 1996; Kuo et al. 1997, 2001; Miravalle et al. 2005; Mori et al. 1992; Piccini et al. 2005, 2007; Russo et al. 1997; Saido et al. 1995, 1996; Tekirian et al. 1998). N-terminal deletions, in general, enhance aggregation of β-amyloid peptides in vitro (Pike et al. 1995). AβpE3 has a higher aggregation propensity (He and Barrow 1999; Schilling et al. 2006), and stability (Kuo et al. 1998), and shows an increased toxicity compared to full-length Aβ (Russo et al. 2002). It has been suggested that N-truncated Aβ peptides are formed directly by BACE and not through a progressive proteolysis of full-length Aβ1-40/42 (Russo et al. 2001). In our view this concept is not likely to be the explanation of the present data, because the increase in pyroglutamate formation appeared to happen on the expense of full-length N-terminal Abeta. Our data are in good agreement with a previously published paper by Guntert et al. (2006). It was found that in cored plaques there was an increase of N-terminal truncations of approximately 20% between Braak stages IV–VI. In contrast, diffuse plaques of AD and control cases showed consistently only low levels of amino-terminal truncations. Whether or not AβpE3 is acting as a seed for plaque formation is a matter of current scientific debate. Up to now there is no sufficient proof (or disproof) that this is actually the case. AβpE3 formation (in plaques) appears to be a late process involved in the maturation of plaques rather than in the seeding of plaques. On the other side, it is obvious that intraneuronal AβpE3-42 accumulation initiates an early lethal phenotype in TBA2 mice associated with massive neuron loss, atrophy and neurological behavioral deficits (Wirths et al. 2009). In in vitro experiments Schilling et al. have shown that cyclization of glutamate at position 3 of Aβ can be driven enzymatically by glutaminyl cyclase (QC) (Schilling et al. 2004). In addition, it has been demonstrated that QC inhibition significantly reduced AβpE3 formation, emphasizing the importance of QC activity during cellular maturation of pyroglutamate-containing peptides. The pharmacological inhibition of QC activity by the QC inhibitor P150, which significantly reduced the level of AβpE3 in vitro (Cynis et al. 2006) and in vivo (Schilling et al. 2008), suggests that QC inhibition might serve as a new therapeutic approach. In addition, we have investigated the level of IgM autoantibodies directed against different Aβ epitopes as potential diagnostic biomarker for AD (Marcello et al. 2009). Anti-Aβ autoantibody levels were measured in 75 plasma samples from patients with AD, individuals with mild cognitive impairment (MCI), and healthy age- and sex-matched controls. The mean level of AβpE3-IgM was significantly decreased in AD patients as compared to healthy controls. In the group of MCI patients there was a significant positive correlation between AβpE3-IgM and cognitive decline analyzed by MMSE (rho = 0.58, df = 13, P = 0.022). These observations indicate that the level of IgM autoantibodies against AβpE3 is a promising plasma biomarker for AD and correlates with the cognitive status of individuals at risk to develop AD. It is, however, tempting to speculate that the correlation of the cognitive status in MCI patients with lower levels of AβpE3-IgM in plasma might be due to early changes in AβpE3-induced neurodegeneration prior to clinical diagnosis of AD. Overall, the importance of AβpE3 in AD pathology is an issue of increasing interest. We believe that the present manuscript adds further evidence that N-terminal pE-modified Aβ in plaques is continuously increasing on the expense of Aβ1-x. The consequences of continuous rearrangement of plaque Aβ by pyroglutamate formation are presently unclear.
Animal models aim to replicate the symptoms, the lesions or the cause(s) of Alzheimer disease. Mutated human APP expression results in the deposition of Aβ peptide, similar but not identical to the Aβ peptide of human senile plaque (reviewed in Duyckaerts et al. 2008). APP transgenic mouse models have been reported to show no (Kuo et al. 2001) or low AβpE3 levels (Guntert et al. 2006). Maeda et al. have demonstrated that the localization and abundance of [11C]PIB autoradiographic signals were closely associated with those of amino-terminally truncated and modified Aβ3pE deposition in AD and different APP transgenic mouse brains, implying that the detectability of amyloid by [11C]PIB-PET is dependent on the accumulation of specific Aβ subtypes (Maeda et al. 2007). An emerging role of intracellular Aβ accumulation has been previously shown in human AD (D’Andrea et al. 2002; Gouras et al. 2000) while it has been observed that Aβ localizes predominantly to abnormal endosomes (Cataldo et al. 2004), multivesicular bodies and within pre- and postsynaptic compartments (Langui et al. 2004; Takahashi et al. 2002). Takahashi et al. demonstrated that Aβ42 aggregates into oligomers within endosomal vesicles and along microtubules of neuronal processes, both in cultured neurons of Tg2576 mice, as well as in Tg2576 and human AD brain (Takahashi et al. 2004). In in vitro experiments, Schilling et al. have shown that cyclization of glutamate at position 3 of Aβ can be driven enzymatically by glutaminyl cyclase (QC) (Schilling et al. 2004). In addition, it has been shown that QC inhibition significantly reduced AβpE3 formation, emphasizing the importance of QC activity during cellular maturation of pyroglutamate-containing peptides (Cynis et al. 2006). The pharmacological inhibition of QC activity by the QC inhibitor P150, which significantly reduced the level of AβpE3 in vitro (Cynis et al. 2006) and in vivo (Schilling et al. 2008) suggests that QC inhibition might serve as a new therapeutic approach to rescue AβpE3 triggered neurodegeneration in AD.
In summary, we have shown that AβpE3 plaques are abundant in sporadic and familial AD cases, and that at least in the APP/PS1KI mouse model, the AβpE3 plaque load increases in an age-dependent manner, which is not the case for total Aβ and Aβ1-x. Most recently, it has been shown that plaque load in non-demented individuals constantly increases with aging, while it decreases in AD patients older than 95 years (Savva et al. 2009). At present there is a lack of information on AβpE3 plaque load in such a cohort of individuals. It would be interesting to study the heterogeneity of N-terminal variant Aβ peptides, since we observed that only Aβ1-x plaques declined with aging in the APP/PS1KI mice and were less frequent in sporadic and familial AD cases. It is tempting to speculate that the continuous increase in AβpE3 between 6 and 10 months in APP/PS1KI mice is due to a corresponding truncation of full-length Aβ-peptides, as the overall plaque load, as shown by 4G8 staining, is stable in that time period. Future studies will provide a better understanding of the consequence of N-truncated Aβ for AD progression.
Acknowledgments
This work was supported by the European Commission, Marie Curie Early Stage Training, MEST-CT-2005-020013 (NEURAD), Alzheimer PhD Graduate School.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Billings LM, Oddo S, Green KN, McGaugh JL, et al. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Breyhan H, Wirths O, Duan K, Marcello A, et al. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. 2009;117(6):677–685. doi: 10.1007/s00401-009-0539-7. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated A{beta}42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165(4):1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25(10):1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Christensen DZ, Bayer TA, Wirths O (2008a) Intracellular Abeta triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2008.07.022 [DOI] [PubMed]
- Christensen DZ, Kraus SL, Flohr A, Cotel MC, et al. Transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008;116(6):647–655. doi: 10.1007/s00401-008-0451-6. [DOI] [PubMed] [Google Scholar]
- Cynis H, Schilling S, Bodnar M, Hoffmann T, et al. Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim Biophys Acta. 2006;1764(10):1618–1625. doi: 10.1016/j.bbapap.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Cynis H, Scheel E, Saido TC, Schilling S, et al. Amyloidogenic processing of amyloid precursor protein: evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-beta. Biochemistry. 2008;47(28):7405–7413. doi: 10.1021/bi800250p. [DOI] [PubMed] [Google Scholar]
- D’Andrea MR, Nagele RG, Wang HY, Lee DH. Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer’s disease entorhinal cortex. Neurosci Lett. 2002;333(3):163–166. doi: 10.1016/S0304-3940(02)00875-3. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorevic PD, Goni F, Pons-Estel B, Alvarez F, et al. Isolation and partial characterization of neurofibrillary tangles and amyloid plaque core in Alzheimer’s disease: immunohistological studies. J Neuropathol Exp Neurol. 1986;45(6):647–664. doi: 10.1097/00005072-198611000-00004. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156(1):15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntert A, Dobeli H, Bohrmann B. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience. 2006;143(2):461–475. doi: 10.1016/j.neuroscience.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Saido TC, Eckman CB, Prada CM, et al. Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer’s disease brain. Biochem Biophys Res Commun. 2000;276(2):422–427. doi: 10.1006/bbrc.2000.3490. [DOI] [PubMed] [Google Scholar]
- Harmeier A, Wozny C, Rost BR, Munter LM, et al. Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. J Neurosci. 2009;29(23):7582–7590. doi: 10.1523/JNEUROSCI.1336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Barrow CJ. The A beta 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater beta-sheet forming and aggregation propensities in vitro than full-length A beta. Biochemistry. 1999;38(33):10871–10877. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4(1):97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, et al. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29(3):177–185. doi: 10.1023/A:1021691918517. [DOI] [PubMed] [Google Scholar]
- Hosoda R, Saido TC, Otvos L, Jr, Arai T, et al. Quantification of modified amyloid beta peptides in Alzheimer disease and Down syndrome brains. J Neuropathol Exp Neurol. 1998;57(11):1089–1095. doi: 10.1097/00005072-199811000-00012. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96(6):3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo T, Saido TC, Mann DM, Lee VM, et al. Full-length amyloid-beta (1-42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am J Pathol. 1996;149(6):1823–1830. [PMC free article] [PubMed] [Google Scholar]
- Kalback W, Watson MD, Kokjohn TA, Kuo YM, et al. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry. 2002;41(3):922–928. doi: 10.1021/bi015685+. [DOI] [PubMed] [Google Scholar]
- Klein WL. Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41(5):345–352. doi: 10.1016/S0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Emmerling MR, Woods AS, Cotter RJ, et al. Isolation, chemical characterization, and quantitation of A beta 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem Biophys Res Commun. 1997;237(1):188–191. doi: 10.1006/bbrc.1997.7083. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Webster S, Emmerling MR, De Lima N, et al. Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of Abeta peptides of Alzheimer’s disease. Biochim Biophys Acta. 1998;1406(3):291–298. doi: 10.1016/s0925-4439(98)00014-3. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Kokjohn TA, Beach TG, Sue LI, et al. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem. 2001;276(16):12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, et al. Subcellular topography of neuronal A{beta} peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165(5):1465–1477. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Ji B, Tomiyama T, Maruyama M, et al. Longitudinal, quantitative assesment of amyloid, neuroinflammation and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by PET. J Neurosci. 2007;27(41):10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello A, Wirths O, Schneider-Axmann T, Degerman-Gunnarsson M et al. (2009) Reduced levels of IgM autoantibodies against N-truncated pyroglutamate Aβ in plasma of patients with Alzheimer’s disease. Neurobiol Aging 10.1016/j.neurobiolaging.2009.08.011 [DOI] [PubMed]
- Masters CL, Multhaup G, Simms G, Pottgiesser J, et al. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. Embo J. 1985;4(11):2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Miravalle L, Calero M, Takao M, Roher AE, et al. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry. 2005;44(32):10810–10821. doi: 10.1021/bi0508237. [DOI] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reverse D, et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274(10):6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem. 1992;267(24):17082–17086. [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283(12):1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccini A, Russo C, Gliozzi A, Relini A, et al. {beta}-Amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280(40):34186–34192. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- Piccini A, Zanusso G, Borghi R, Noviello C, et al. Association of a presenilin 1 S170F mutation with a novel Alzheimer disease molecular phenotype. Arch Neurol. 2007;64(5):738–745. doi: 10.1001/archneur.64.5.738. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Overman MJ, Cotman CW. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J Biol Chem. 1995;270(41):23895–23898. doi: 10.1074/jbc.270.41.23895. [DOI] [PubMed] [Google Scholar]
- Puolivali J, Wang J, Heikkinen T, Heikkila M, et al. Hippocampal A beta 42 levels correlate with spatial memory deficit in APP and PS1 double transgenic mice. Neurobiol Dis. 2002;9(3):339–347. doi: 10.1006/nbdi.2002.0481. [DOI] [PubMed] [Google Scholar]
- Russo C, Saido TC, DeBusk LM, Tabaton M, et al. Heterogeneity of water-soluble amyloid beta-peptide in Alzheimer’s disease and Down’s syndrome brains. FEBS Lett. 1997;409(3):411–416. doi: 10.1016/S0014-5793(97)00564-4. [DOI] [PubMed] [Google Scholar]
- Russo C, Salis S, Dolcini V, Venezia V, et al. Amino-terminal modification and tyrosine phosphorylation of [corrected] carboxy-terminal fragments of the amyloid precursor protein in Alzheimer’s disease and Down’s syndrome brain. Neurobiol Dis. 2001;8(1):173–180. doi: 10.1006/nbdi.2000.0357. [DOI] [PubMed] [Google Scholar]
- Russo C, Violani E, Salis S, Venezia V, et al. Pyroglutamate-modified amyloid–peptides–AbetaN3(pE)—strongly affect cultured neuron and astrocyte survival. J Neurochem. 2002;82(6):1480–1489. doi: 10.1046/j.1471-4159.2002.01107.x. [DOI] [PubMed] [Google Scholar]
- Saido TC, Iwatsubo T, Mann DM, Shimada H, et al. Dominant and differential deposition of distinct beta-amyloid peptide species, Abeta N3(pE), in senile plaques. Neuron. 1995;14(2):457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett. 1996;215(3):173–176. doi: 10.1016/0304-3940(96)12970-0. [DOI] [PubMed] [Google Scholar]
- Savva GM, Wharton SB, Ince PG, Forster G, et al. Age, neuropathology, and dementia. N Engl J Med. 2009;360(22):2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- Schilling S, Hoffmann T, Manhart S, Hoffmann M, et al. Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS Lett. 2004;563(1–3):191–196. doi: 10.1016/S0014-5793(04)00300-X. [DOI] [PubMed] [Google Scholar]
- Schilling S, Lauber T, Schaupp M, Manhart S, et al. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro) Biochemistry. 2006;45(41):12393–12399. doi: 10.1021/bi0612667. [DOI] [PubMed] [Google Scholar]
- Schilling S, Zeitschel U, Hoffmann T, Heiser U, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat Med. 2008;14(10):1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8(11):447–453. doi: 10.1016/S0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Abraham CR, Podlisny MB, Duffy LK. Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer’s disease. J Neurochem. 1986;46(6):1820–1834. doi: 10.1111/j.1471-4159.1986.tb08501.x. [DOI] [PubMed] [Google Scholar]
- Sevalle J, Amoyel A, Robert P, Fournie-Zaluski MC, et al. Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J Neurochem. 2009;109(1):248–256. doi: 10.1111/j.1471-4159.2009.05950.x. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161(5):1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, et al. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24(14):3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekirian TL, Saido TC, Markesbery WR, Russell MJ, et al. N-terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. J Neuropathol Exp Neurol. 1998;57(1):76–94. doi: 10.1097/00005072-199801000-00009. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Kitazawa M, LaFerla FM. Amyloid beta-peptide: the inside story. Curr Alzheimer Res. 2004;1(4):231–239. doi: 10.2174/1567205043332045. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, et al. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39(35):10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Kuchna I, Nowicki K, Frackowiak J, et al. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol. 2007;113(4):389–402. doi: 10.1007/s00401-006-0191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Feldmann N, et al. Intraneuronal APP/Abeta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol. 2002;12:275–286. doi: 10.1111/j.1750-3639.2002.tb00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Bayer TA. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide—the first step of a fatal cascade. J Neurochem. 2004;91(3):513–520. doi: 10.1111/j.1471-4159.2004.02737.x. [DOI] [PubMed] [Google Scholar]
- Wirths O, Weis J, Kayed R, Saido TC, et al. Age-dependent axonal degeneration in an Alzheimer mouse model. Neurobiol Aging. 2007;28:1689–1699. doi: 10.1016/j.neurobiolaging.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Wirths O, Breyhan H, Schäfer S, Roth C, et al. Deficits in working memory and motor performance in the APP/PS1ki mouse model for Alzheimer’s disease. Neurobiol Aging. 2008;29(6):891–901. doi: 10.1016/j.neurobiolaging.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Wirths O, Breyhan H, Cynis H, Schilling S, et al. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009;118(4):487–496. doi: 10.1007/s00401-009-0557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]