

Abstract
The last decade has witnessed remarkable progress in the understanding of the mammalian cannabinoid system, from the cloning of the endogenous cannabinoid receptor to the discovery of new pharmacologic compounds acting on this receptor. Current and planned studies in humans include compounds with effects ranging from direct antagonists to inhibitors of reuptake and breakdown. This progress has been accompanied by a much greater understanding of the role of the cannabinoid system in modulating the neural circuitry that mediates anxiety and fear responses. This review focuses on the neural circuitry and pharmacology of the cannabinoid system as it relates to the acquisition, expression, and extinction of conditioned fear as a model of human anxiety. Preclinical studies suggest that these may provide important emerging targets for new treatments of anxiety disorders.
INTRODUCTION
Pathologic feelings of fear and anxiety are defining features of many devastating psychiatric illnesses, including posttraumatic stress disorder and specific phobias, and are major contributors to the morbidity associated with many other common psychiatric illnesses, ranging from depression to schizophrenia. As an important contributor to such a wide variety of psychopathology, the dysregulation of human fear constitutes a major burden to public health and well-being.
With respect to pharmacologic strategies for the treatment of anxiety, the most commonly used drugs include selective serotonin reuptake inhibitors (SSRIs)1–3 as well as agents that act to enhance the actions of γ-aminobutyric acid (GABA), usually through agonist-like activity at central GABAA receptors.4 Benzodiazepines and barbiturates, which act to enhance GABAergic neurotransmission, have been widely used and are generally recognized to be effective at reducing the expression of fear and anxiety in treated humans as well as most animal models of anxiety. Indeed, many animal models of anxiety are designed and validated using benzodiazepine-mediated anxiolysis as a readout. This being said, very little is known about the effect of benzodiazepines on extinction, with some research5 suggesting that benzodiazepines may even impair extinction of clinical fear. While effective, available anxiolytics often engender a number of undesirable side effects, including impaired arousal, amnestic effects, tolerance, dependence, and abuse liabilities.
Recently, a more detailed understanding of the neural circuitry involved in the formation, expression, and experience-dependent inhibition of mammalian fear responses has yielded a number of potentially useful therapeutic targets.6,7 It is hoped that new treatments aimed at these new drug targets will allow the development of anxiolytics with fewer side effects. This review will briefly discuss the literature examining the role of the endocannabinoid system in the learning, expression, and learned inhibition of the mammalian fear response. Furthermore, as agonists, antagonists, and reuptake inhibitors of the endocannabinoid neurotransmitter system are all being pursued for clinical use,8–15 we will briefly comment on how currently available studies from the animal literature may inform future clinical directions.
FEAR, ANXIETY, AND THE AMYGDALA
The neural mechanisms controlling fear and anxiety have been intensively studied in laboratory and clinical settings. These studies have elucidated several structures within the limbic system as key players in the production of both normal and pathologic fear, including the hippocampus, prefrontal cortex, and the amygdala. Among these structures, the amygdala has the most well-established role in the production of fear states in a variety of different animal species, ranging from mice to humans.16,17
The amygdala is an almond-shaped nuclear structure located within the temporal lobe. It can be subdivided into three major nuclei: the basolateral nuclear complex, the central nucleus, and the medial nucleus. Notably, these nuclei of the amygdala can be differentiated on the basis of their connectivity, the types of neurons they contain, and, finally on their roles in the production of behavioral states. Work in a variety of animal models has identified the central nucleus of the amygdala (CeA) as the major output of the amygdalar circuit, on the basis of its robust connectivity to other brain regions involved in the production of fear responses and on the basis of animal behavior following lesions of the CeA.16–19 In contrast, the basolateral complex of the amygdala (BLA) seems to be critical component in the learning of conditioned fear responses, and accordingly receives a wide array of sensory input, both from subcortical and cortical structures. From a behavioral perspective, it has been widely observed16–19 that animals with lesions to the BLA (especially dorsal lesions including the lateral nucleus) can express fear normally, but have profound deficits in learning new fear responses in a number of different conditioning tasks. This has led to the view that experience-dependent alterations in the neural circuitry of the BLA allow an animal to learn which sensory information should lead to the production of a fear response.
More recent studies have implicated the BLA in the extinction of a fear response through the repeated presentation of the conditioned stimulus in the absence of the unconditioned stimulus in previously fear conditioned animals. Additionally, these studies strongly suggest that extinction learning, while it also seems to rely on the BLA, is a form of learning that is distinct and parallel to fear learning.20–22
Organization of the Endocannabinoid Neurotransmitter System
At the current time, there are two known cannabinoid receptor subtypes: cannabinoid-type 1 (CB1) receptor, which are widely expressed throughout the peripheral and centra1 nervous system (CNS), and cannabinoid-type 2 receptor which show high levels of expression within the immune and enteric nervous systems as well as in glial cells of the CNS. Mounting evidence suggests that a third cannabinoid receptor subtype exists in the CNS.23–25 As the psychoactive effects of cannabinoid receptor activation have been attributed to centrally expressed receptors, and given that the vast majority of cannabinoid-type 2 receptor expression seems to be in the immune and enteric systems, we will focus largely on the centrally expressed CB1 receptor.
The CB1 receptor is a member of a large family of G-protein coupled receptors containing seven membrane-spanning domains. The CB1 receptor is coupled to the G-proteins, Gi/o, and activation of the receptor leads to the activation of a diverse array of intracellular signals, which can vary by cell type. In the cerebellum, hippocampus, cortex, amygdala, and virtually every other brain region studied, the majority of neuronal CB1 receptors appear to be expressed pre-synaptically. Here, they are thought to be activated by retrograde transmission of endogenous cannabinoids (eCBs) from the postsynaptic site (Figure 1).
FIGURE 1. Organization of the cannabinoid system.
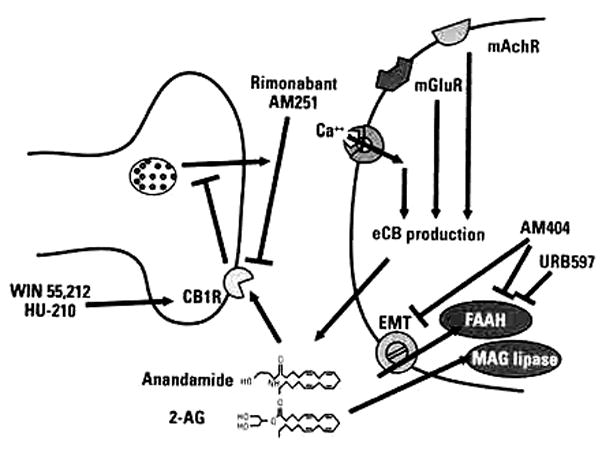
Increased intracellular calcium in the postsynaptic bouton is thought to be a major signal for the production of endocannabinoid transmitters (anandamide and 2-AG are shown). The mechanisms by which these local increases are mediated may vary by cell type and may involve NMDA receptors, mGluRs, mAChRs as well as G-protein induced increases in calcium release from intracellular stores. Reuptake and degradation of anandamide is thought to be a coupled process of transport across the membrane by the EMT, and degradation by FAAH, which degrades anandamide and related eCBs, and/or MAG lipase, which degrades 2-AG and related eCBs. Once they are produced in the postsynaptic cell, eCBs are thought to diffuse in a retrograde fashion to activate pre-synaptic CB1 receptors. Activation of CB1 receptors leads to decreases in transmitter release from the pre-synaptic terminal, by decreasing calcium influx through voltage-gated calcium channels, and by increasing inwardly-rectifying potassium currents. Several pharmacologic manipulators of eCB transmission are noted; including the CB1 agonists WIN 55,212-2 and HU-210, the CB1 antagonists rimonabant and AM251, and two inhibitors of eCB reuptake and/or breakdown (AM404 and URB597).
mAChRs=muscarinic acetylcholine receptors; mGluRs=metabotropic glutamate receptors; eCB=endogenous cannabinoid; CB1=cannabinoid-type 1; EMT=endocannabinoid membrane transporter; FAAH=fatty acid amide hydrolase; MAG=monoacyl-glycerol; 2-AG=2-arachidonoyl glycerol; NMDA=N/-methyl -D- asparte.
Chhatwal JP, Ressler KJ. CNS Spectr. Vol 12, No 3. 2007.
In addition to being retrograde neurotransmitters, eCBs differ from conventional neurotransmitters in a number of important ways. Unlike most neurotransmitters, eCBs seem to be produced on-demand (rather than stored in vesicles), by chemical modification of pre-existing lipid molecules. In accordance with their extreme lipophilicity, it is presumed that once produced, eCB neurotransmitters can freely cross the cell-membrane to activate their pre-synaptic receptors, rather than requiring the calcium-dependent docking and release machinery common to most classical neurotransmitters.
While the exact nature of the chemical signal that causes neurons and glia to produce eCBs is not known, there appears to be some consensus that increases in neural activity prompt the production of eCBs. Although more work is necessary, it seems that local increases in intracellular calcium concentrations within the synaptic bouton may be a likely mediator of this effect,26,27 and the activity of several key enzymes required for eCB synthesis are positively regulated by increasing intracellular calcium concentrations,28,29 It is thought that these increases in calcium may be a result of muscarinic acetylcholine receptor or metabotropic glutamate receptor activation.30–32
While a variety of eCB-like substances have been identified, two major classes of endocannabinoids, with distinct synthetic and degradative pathways have thus far been identified.33–35 The first class is exemplified by anandamide (the first eCB identified), which is thought to be formed by a calcium-sensitive, phospholipase D dependent hydrolysis of N-arachidonoyl phosphatidyl ethanolamine. Degradation of anandamide and related eCBs are thought to be mediated largely by the enzyme fatty acid amide hydrolase (FAAH). In contrast, 2-arachidonoyl glycerol (2-AG) is synthesized via phospholipase C-mediated hydrolysis of phosphatidylinositol, and can serve as a precursor to the synthesis of other lipid signaling molecules, in addition to serving as a neurotransmitter. While both anandamide and 2-AG are substrates for an as yet un-cloned endocannabinoid membrane transporter, 2-AG degradation seems to be largely dependent on monoacylglycerol lipase), and not FAAH.36
Once activated, the CB1 receptor leads to a decrease in the excitability of the pre-synaptic terminal in at least three ways, including the closure of calcium (N- and P/Q type) channels, increases in G-protein coupled inwardly rectifying potassium currents, and decreases in cyclic adenosine monophosphate-dependent sodium conductance. Together, these mechanisms are thought to indicate that CB1 receptor activation leads to decreases in neurotransmitter release from the pre-synaptic terminal.37,38 CB1 receptor activation is thought to be critical for several forms of synaptic plasticity, including long-term depression and depolarization-induced suppression of excitation and inhibition.
Endocannabinoids and Memory
δ-9 tetrahydrocannabinol, the main psychoactive substance in marijuana, and a potent, naturally occurring CB1 receptor agonist,39 is among the most widely used illicit drugs in human history. In addition to its ostensible euphoric effects, human marijuana use has been linked to a staggering array of behavioral effects, ranging from impairments of motor function to impulsivity to panicogenisis.40–44
There exists a large and somewhat inconsistent literature linking CB1 agonist administration to deficits in memory formation. Without going into any appreciable detail on the controversies in the human literature, there appears to be general consensus that acute and chronic CB1 agonist exposure can lead to profound memory deficits, especially in tasks involving trace-conditioning or short-term declarative memory tasks (ie, tasks that require short-term working memory45–46).
Studies of learning and memory in laboratory animals have generally suggested similar memory deficits in rodents, with a number of studies showing that CB1 agonists can impair performance in several hippocampal- and prefrontal cortex-dependent learning tasks, including non-match to sample tasks,47–49 the radial-arm maze,50 and the Morris-Water maze,51,52 It has also been reported53 that pre-training administration WIN 55,212-2, a potent, synthetic CB1 agonist, leads to profound deficits in the acquisition of contextual fear in rodents. Importantly, the authors of this same study also demonstrated CB1 agonist administration did not disrupt the formation of cued fear conditioning. Accordingly, although a great deal of work remains to be done, it is tempting to speculate that CB1 agonists are more disruptive in hippocampal and prefrontal cortex-dependent memory tasks, as opposed to amygdala-dependent tasks. More broadly, it may be the case that the neural processes underlying emotional memory formation (such as fear and extinction memories) and non-emotional memories are differentially sensitive to cannabinoid receptor activation.
Cannabinoids and Generalized Anxiety
Low doses of CB1 agonists, including both natural (ie, δ-9 tetrahydrocannabinol in cannabis sativa) and synthetic cannabinoids, have generally been observed as anxiolytic in various animal models of anxiety; however, higher doses of CB1 agonists have often been observed to be anxiogenic, with both findings appearing to be superficially consistent with the anecdotal history growing from human cannabis abuse8,54,55 Additionally, recent data from human studies of depression suggest that the endocannabinoid system may be involved in this common disorder that is often comorbid with anxiety,56 and CB1 antagonists are being explored as a potential treatment for depression.11
While it is not uncommon for a drug to have this sort of a biphasic response curve with respect to a complex behavioral state, such as anxiety, mounting evidence suggests that both endogenous and exogenous cannabinoid agonists also have a complex interaction with the steady-state levels of stress experienced by the organism.57–59 In this context, determination of a dose-response curve across multiple experimental paradigms is made more difficult, as identification of a “high” or “low” dose may depend on numerous experimental vagaries, such as which test is used, how the animals were handled prior to testing, and the extent to which the experimenter and behavioral apparatus are novel.
Studies emerging from the study of CB1 knockout mice generally suggest that disruption of eCB signaling leads to an anxiogenic phenotype (Table 1). Consistent with this, CB1 knockout mice show anxiogenic responses in the open-field test, light/dark box, social-interaction test, and elevated plus-maze.10,60,61 Similar results have been acquired using pharmacologic antagonists of the CB1-receptor, suggesting that the anxiety phenotype seen in CB1 knockouts is unlikely due to developmental consequences.10 Notably, however, increased anxiety has more consistently been seen with the more selective CB1 antagonist AM251, as opposed to the more commonly used CB1 antagonist rimonabant (compare Haller and colleagues61 and Haller and colleagues10).
TABLE 1.
The Effect of eCB Modulators on Animal Models of Anxiety
| Authors (Year) | Manipulation/Drug Type | Drug Name | Behavioral Paradigm | Observation |
|---|---|---|---|---|
| Rodriguez de Fonseca et al (1996)62 | CB1 agonist | HU-210 | Defensive withdrawal | Increased anxiety |
| Reich et al (2005)63 | CB1 antagonist | AM251 | Baseline freezing | Increased anxiety |
| Haller et al (2002, 2004)10,51 | CB1 antagonist | AM251 | Elevated plus-maze | Increased anxiety |
| Marsicano et al(2002)54 | CB1 gene knockout | N/A | Elevated plus-maze | No effect |
| Haller et al 2002, 2004)10,61 | CB1 gene knockout | N/A | Elevated plus-maze | Increased anxiety |
| Martin et al (2002)65 | C81 gene knockout | N/A | Light-dark box | Increased anxiety |
| Chhatwal et al (2005)66 | FAAH & reuptake inhibitor | AM404 | Baseline startle | No effect |
| Bortolato et al (2006)13 | FAAH & reuptake inhibitor | AM404 | Baseline startle | No effect |
| Bortolato et al (2006)13 | FAAH & reuptake inhibitor | AM404 | Defensive withdrawal | Decreased anxiety |
| Bortolato et al (2006)13 | FAAH & reuptake inhibitor | AM404 | Elevated plus-maze | Decreased anxiety |
| Bortolato et al (2006)13 | FAAH & reuptake inhibitor | AM404 | Ultrasonic vocalization | Decreased anxiety |
| Kathuria et al (2003)14 | FAAH inhibitor | URB597 | Elevated plus-maze | Decreased anxiety |
eCB=endogenous cannabinoid: CB1=cannabinoid-type 1: FAA=fatty acid amide hydrolase; N/A=not available.
Chhatwal JP, Ressler KJ. CNS Spectr. Vol 12. No 3. 2007.
Perhaps most intriguing from a therapeutic perspective, several recent reports suggest that prolonging the half-life of released endocannabinoids leads to anxiolysis in several rodent models. The first drug of this type to be studied in detail is AM404, which has been shown to be an inhibitor of both cannabinoid re-uptake through the endocannabinoid membrane transporter and the subsequent degradation of eCBs by FAAH. Systemic administration of AM404 to rats has been shown to increase levels of anandamide (but not 2-AG) in several brain regions and decrease numerous measures of anxiety in animal models, including decreased anxiety behavior in the defensive withdrawal test and the elevated-plus maze.13 Notably, similar reductions in anxiety were seen with two, more selective inhibitors of FAAH (URB532 and URB597),14 indicating that prolonging the activity of released anandamide may be a therapeutically useful goal in the treatment of anxiety. Coupled with their effects on basal anxiety, studies from our research group suggest that enhancing cannabinoid neurotransmission may also be useful adjuncts to the learned inhibition of fear (extinction), as we will discuss in some detail later in this article.56
Cannabinoids and Conditioned Fear
In addition to modulating basal anxiety states, recent evidence suggests that activation of cannabinoid receptors may play an important role in evoked stress responses. Conditioned fear, which is induced by pairing a neutral, conditioned stimulus (eg, a light, a tone, or a context) with an aversive stimulus (eg, a mild footshock or air blast), is an archetypal example of an evoked fear state, as presentation of the conditioned stimulus following pairing with an aversive stimulus leads to the production of a measurable fear response. These evoked fear responses have been used extensively in animal models to better understand both the mechanisms by which aversive memories are formed, and to model diseases such as post-traumatic stress disorder (PTSD) and specific phobia, where inciting cues lead to the production of pathologic fear states.
The process of contextual fear conditioning involves the pairing of a neutral training chamber (contextual conditioned stimulus) with an aversive, unconditioned stimulus (eg, a footshock), and the expression of contextual fear appears to be particularly susceptible to cannabinoid modulation. Like other learning processes, the formation fear memories (both cued and contextual) involves an initial, acquisition phase, where the animal teams to behavioral contingency between the conditioned stimulus and unconditioned stimulus, and a later, consolidation phase, where the nascent memory becomes more stable and is stored in a manner that allows for long-term memory retrieval.
Recent evidence suggests that manipulating cannabinoid neurotransmission can have effects on the acquisition and expression (but not the consolidation) of contextual fear conditioning (Table 2). Haller and colleagues61 have demonstrated that decreasing CB1-receptor activation through CB1 antagonist administration leads to decreases in the expression of contextual fear conditioning, while administration of CB1 agonists enhanced fear expression. In the same study, these authors also established that the knockout of the gene for the CB1 receptor also led to decreases in the expression of conditioned contextual fear, and to impairments in the initial acquisition of contextual fear responses. These results agreed with another recent study, in which it was observed that the administration of the CB1 antagonist AM251 also decreased the acquisition of contextual fear conditioning.68 However, while these studies suggest that increasing CB1 receptor activation leads to increases in fear acquisition and expression (and vice versa), some controversy still exists.53
TABLE 2.
The Effect of eCB Modulators on the Acquisition and Expression of Conditioned Fear
| Authors(Year) | Manipulation/Drug Type | Drug Name | Behavioral Paradigm | Observation |
|---|---|---|---|---|
| Mikics et al (2006)67 | CB1 agonist | WIN 55, 212-2 | Contextual fear conditioning | Increased expression of fear |
| Pamplona & Takahashi (2006)53 | CB1 agonist | WIN 55,212-2 | Contextual fear conditioning | No expression effect |
| Pamplona & Takahashi (2006)53 | CB1 agonist | WIN 55,212-2 | Contextual fear conditioning | Decreased fear learning |
| Mikics et al (2006)67 | CB1 antagonist | AM251 | Contextual fear conditioning | Decreased expression of fear |
| Arenos et al (2006)68 | CB1 antagonist | AM251 | Contextual fear conditioning | Decreased expression of fear |
| Arenos et al (2006)68 | CB1 antagonist | AM251 | Contextual fear conditioning | Decreased fear learning |
| Marsicano et al (2002)64 | CB1 antagonist | Rimonabant | Cued fear conditioning | No acquisition effect |
| Martin et al (2002)65 | CB1 gene knockout | N/A | Active avoidance (after footshock] | Increased avoidance |
| Mikics et al (2006)67 | CB1 gene knockout | N/A | Contextual fear conditioning | Decreased acquisition of fear |
| Mikics et al (2006)67 | CB1 gene knockout | N/A | Contextual fear conditioning | Decreased acquisition of fear |
| Marsicano et al (2002)64 | CB1 gene knockout | N/A | Cued fear conditioning | No acquisition effect |
eCB=endogenous cannabinoid; CB1=cannabinoid-type 1; N/A=not applicable.
Chhatwal JP, Ressler KJ, CNS Spectr. Vol 12, No 3 2007.
Contrasting with contextual conditioning, which seems to require both the amygdala and hippocampus, the process of cued fear conditioning seems to be largely mediated by the amygdala (ie, cued conditioning appears to be largely hippocampally independent). While there have been few direct studies of endocannabinoid modulation cued fear-conditioning, currently available results69 indicate that administration of CB1 antagonists or gene knockout of the CB1 gene do not lead to alterations in cued fear conditioning.
Taken together, these studies suggest decreasing the activation of CB1 receptors may decrease the expression of contextual fear memories, while not having a prominent impact on the learning or expression of amygdala-dependent cued fear conditioning.
Cannabinoids and the Extinction of Conditioned Fear
Extinction is a clinically relevant learning process by which an animal learns to inhibit a previously acquired fear response. Experimentally, inducing extinction is usually accomplished by repeated presentations of the feared cue or context in the absence of the aversive unconditioned stimulus used to generate the original fear memory. While we will not discuss these here, numerous lines of evidence21 suggest that extinction is a parallel and distinct form of learning than the process by which the original fear memory was formed. This evidence suggests that extinction is not simply an erasure of the original fear memory, prompting the possibility that there may be pharmacologic means by which the extinction learning process can be favored over the learning of undesirable, aversive associations.
From a clinical perspective, extinction represents a particularly interesting learning process, as many of the most effective treatments for PTSD, social phobia, specific phobias, and addiction make use of behavioral exposure therapy approaches, which ultimately seem to rely on extinction-like processes. This has lead to the suggestion that deficits in extinction learning may lead to the emergence of psychopathology following exposure to trauma, and may contribute to the persistence of inappropriate fear responses in a variety of different anxiety disorders. Interestingly, recent pre-clinical data suggests a possible mechanism by which exposure to uncontrollable stressors may lead to deficits in extinction learning.70
Consequently, the identification of pharmacologic strategies to enhance extinction in animal models may eventually be used in the treatment of human fear and anxiety. Indeed, Walker and colleagues71 recently identified D-cycloserine (DCS), a partial agonist of the N-methyl-D-asparte receptor, both as a pharmacologic means of enhancing extinction learning in rodents and as a means for enhancing the efficacy of behavioral exposure therapy for the treatment of acrophobia in humans.72 More recently, it has also been shown that DCS is also an effective adjuvant to exposure-based psychotherapy for social phobia.73 Ongoing studies are examining its efficacy in panic disorder, obsessive-compulsive disorder and PTSD. Together the results so far suggest that common mechanisms may underpin extinction of many different types of emotional memories.74
While the molecular mechanisms responsible for extinction have not been delineated well, recent studies64,66,75,76 suggest that, despite many similarities between the mechanisms of fear and extinction learning, activation of the cannabinoid system may be an important difference between these two forms of learning (Table 3). Strikingly, Marstcano and colleagues64 have elegantly shown that the antagonism of CB1 signaling, either by genetic or pharmacologic means, leads to profound deficits in extinction learning. This finding has since been replicated using several training paradigms, and in several strains of mice and in rats (Table 3), and firmly establishes that CB1 receptor activation is required for the process of extinction learning in rodents. Why CB1 activation seems to be involved with extinction (inhibition) of fear and not acquisition or expression of fear64,66 remains unknown. One possibility is the selective expression of CB1 receptors on specific cholecystokinin interneurons may lead to a “directional” effect of CB1 modulation on fear and anxiety.74 Alternatively, it is also possible that an optimal level of fear training and testing has not been arrived at to detect a subtle difference. Either way, it is clear that the learning of extinction is more sensitive to CB1 antagonism than the acquisition of fear. This CB1 dependency of extinction in rodent models of fear extinction (along with effects on generalized anxiety) suggests that CB1 antagonists, such as rimonabant, which will soon be available clinically, should be closely monitored for anxiety- and extinction-related side effects.
TABLE 3.
The Effect of eCB Modulators on the Extinction of Conditioned Fear
| Authors (Year) | Manipulation/Drug Type | Drug Name | Behavioral Paradigm | Measure of Fear | Observation |
|---|---|---|---|---|---|
| Marsicano et al (2002)64 | CB1 gene knockout | N/A | Extinction of conditioned fear (cue) | Conditioned freezing | Impairment of extinction |
| Chhatwal et al (2005)66 | CB1 agonist | WIN 55,212-2 | Extinction of conditioned fear (cue) | Fear-potentiated startle | No effect |
| Suzuki et al (2004)76 | CB1 antagonist | Rimonabant | Extinction of conditioned fear (context) | Conditioned freezing | Impairment of extinction |
| Marsicano et al (2002)64 | CB1 antagonist | Rimonabant | Extinction of conditioned fear (cue) | Conditioned freezing | Impairment of extinction |
| Chhatwal et al (2005)66 | CB1 antagonist | Rimonabant | Extinction of conditioned fear (cue) | Fear-potentiated startle | Impairment of extinction |
| Chhatwal et al (2005)66 | FAAH inhibitor, eCB reuptake inhibitor | AM404 | Extinction of conditioned fear (cue) | Fear-potentiated startle | Enhancement of extinction |
eCB=endogenous cannabinoid; CB1=cannabinoid-type 1; N/A=not applicable; FAAH=fatty acid amide hydrolase.
Chhatwal JP, Ressler KJ.
Conversely, this same CB1 dependency of extinction suggests that there may be means of manipulating the eCB system to increase extinction learning, and building on this notion, our group has recently tested whether increasing CB1 receptor activation through pharmacologic means would augment extinction in rodents. In initial studies, Chhatwal and colleagues66 used pre-extinction training administration of the synthetic CB1 agonist WIN 55,212-2 to attempt to enhance extinction, but found that this treatment was ineffective at altering learning. This led them to hypothesize that administration of full agonists at the CB1 receptor may lead to compensatory changes decreasing the efficacy of CB1-mediated signaling and/or that the CB1 receptor needed to be activated in a temporally-specific way during extinction training. To address these concerns, they next tested whether AM404, a drug that should act to increase the efficacy of released eCBs by decreasing their reuptake and breakdown, may be able to enhance extinction. Chhatwal and colleagues66 observed that AM404 given prior to extinction-training could enhance learning in rodents, as evidenced by the observation that animals receiving AM404 prior to extinction training showed less conditioned fear when tested off-drug (Figure 2). Furthermore, they also observed that animals who received AM404 prior to extinction also seemed more resistant to shock-induced reinstatement of fear, an animal model of stress-induced relapse.66 Notably, AM404 and similar compounds(Table 1) have been shown to be anxtolytic, raising the intriguing possibility that enhancers of eCB transmission may reduce the expression of fear and anxiety. This is an interesting finding, since the learning and behavioral literature in general suggests that agents that enhance extinction are often anxiogenic.77–78 Therefore, the anxiolytic nature of transient CB1 enhancers are especially attractive sense they also potentially facilitate the learned inhibition of fear proceeding through extinction-like processes, and avoid the well-known amnesic side effects of many currently available anxiolytics.
FIGURE 2. Administration of an inhibitor of eCB reuptake and breakdown enhances extinction66.
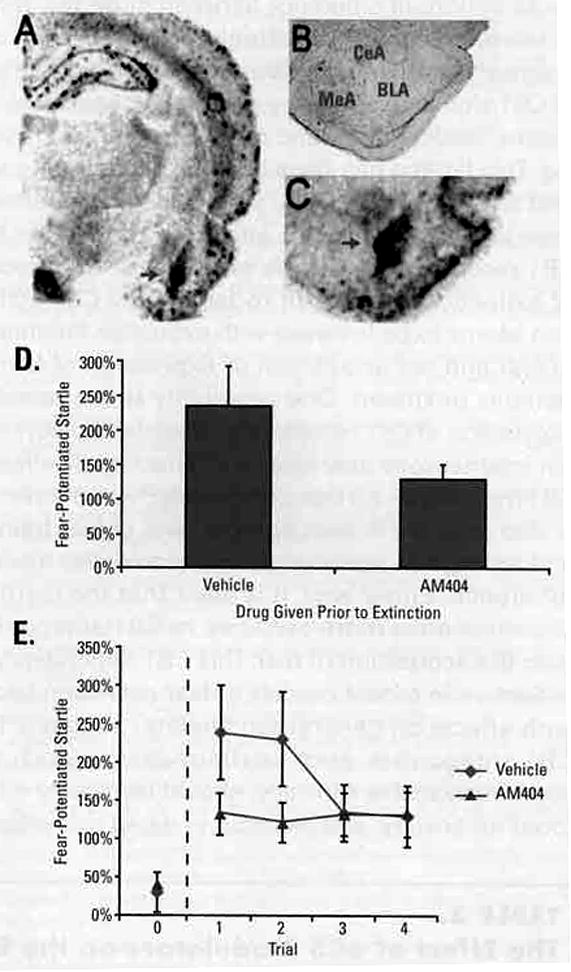
(A–C) CB1 mRNA expression is very high within the BLA, which is thought to be an important locus of associative emotional learning), with much lower levels seen in the CeA and MeA. (D) Animals receiving 10 mg/kg AM404 prior to extinction training show significantly reduced levels of conditioned fear when tested drug-free. This suggests that AM404 administration enhances extinction learning, and facilitates the learned inhibition of fear. (E) Following extinction, animals were tested for the resilience: of their extinction memories by presentation of a 0.4 mA footshock (dashed line) in the absence of the light CS. While all animals showed some re-emergence of fear following footshock, animals that received AM404 prior to extinction showed less fear than vehicle treated controls, suggesting that, in addition to enhancing extinction, AM404 administration may render extinction more resistant to shock-induced reinstatement, an animal model of stress-induced relapse.
Chhatwal JP, Davis M. Maguschak KA, Ressler KJ, Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. Reproduced with permission by Nature Publishing Group. Copyright (2005).
*P< .05 compared with vehicle group.
CB1=cannabinoid-type 1; CeA=central nuclei of the amygdala: BLA=basolateral amygdala; MeA=medial nuclei of the amygdala; CS=conditioned stimulus.
Chhatwal JP, Ressler KJ. CNS Spectr. Val 12, No 3. 2007.
CONCLUSION
The last decade has witnessed an enormous amount of progress in the understanding of the molecular biology, physiology, pharmacology, and behavioral neuroscience underlying the endogenous cannabinoid system. These receptors and their ligands have ubiquitous roles ranging from appetite and pain response to modulation of fear and anxiety. A burgeoning understanding of their roles in regulating the extinction of fear responses may lead to a particularly important role in translation of the preclinical research to novel treatments of anxiety disorders.
Needs Assessment
Current and planned studies in humans include compounds that modulate the endogenous cannabinoid system. However, most clinicians are not yet aware of the progress in this field. This review focuses on the neural circuitry and pharmacology of the cannabinoid system as it relates to the preclinical models of human anxiety.
Learning Objectives
At the end of this activity, the participant should be able to:
Discuss the preclinical data examining the role of cannabinoid-type 1 (CB1) modulators in anxiety.
Understand the difference between CB1 agonists and CB1 reuptake inhibitors.
Better understand the role of endogenous cannabinoids in extinction of conditioned fear
Target Audience: Neurologists and psychiatrists
CME Accreditation Statement
This activity has been planned and implemented in accordance with the Essentials and Standards of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the Mount Sinai School of Medicine and MBL Communications, Inc. The Mount Sinai School of Medicine is accredited by the ACCME lo provide continuing medical education for physicians.
Credit Designation
The Mount Sinai School of Medicine designates this educational activity for a maximum of 3 AMA PRA Category 1 Credits?”. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Faculty Disclosure Policy Statement
It is the policy of the Mount Sinai School of Medicine to ensure objectivity, balance, independence, transparency, and scientific rigor in all CME-sponsored educational activities. All faculty participating in the planning or implementation of a sponsored activity are expected to disclose to the audience any relevant financial relationships and to assist in resolving any conflict at interest that may arise from the relationship. Presenters must also make a meaningful disclosure to the audience of their discussions of unlabeled or unapproved drugs or devices. This information will be available as part of the course material.
This activity has been peer-reviewed and approved by Eric Hollander, MD, chair at the Mount Sinai School of Medicine. Review date February 13.2007 Dr. Hollander does not have an affiliation with or financial interest in any organization that might pose a conflict of interest.
To Receive Credit for This Activity
Read the three CME-designated articles, reflect on the information presented, and complete the CME quiz. To obtain credits, you Should score 70% or better. Early submission of this posttest is encouraged to measure outcomes for this CME activity. Please submit this posttest by March 1, 2009 to be eligible for credit. The estimated time to complete all three articles and the quiz is 3 hours. Release date: March 2007. Termination date: March 2009.
Acknowledgments
Funding/Support: This work was supported in part by National Institutes of Health grants MH069884 and DA019624 awarded to Dr. Ressler, MH070218 awarded to Dr. Chhatwal, and a National Alliance for Research on Schizophrenia and Depression grant awarded to Dr. Ressler from the Burroughs Wellcome Fund. Dr. Chhatwal is supported by a National Research Service Award Fellowship. Drs. Chhatwal and Ressler are also supported by the National Institute of Health/National Center for Research Resources base grant P51RR000165 awarded to the Yerkes National Primates Research Center and the Center for Behavioral Neuroscience National Science Foundation agreement IBN-987675.
Footnotes
Disclosures: Dr. Chhatwal does not have an affiliation with or financial interest in any organization that might pose a conflict of interest. Dr. Ressler has received grant/research support from the Burroughs Wellcome Foundation, Lundbeck, the National Alliance for Research on Schizophrenia and Depression, the National Institute of Mental Health, the National Institute on Drug Abuse, and is on the advisory board of Tikvah Therapeutics with regard to N-methyl-D-aspartate based therapeutics.
References
- 1.Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for post traumatic stress disorder (PTSD) Cochrane Database Syst Rev. 2006;1:CD002795. doi: 10.1002/14651858.CD002795.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davidson JR. Pharmacologic treatment of acute and chronic stress following trauma: 2006. J Clin Psychiatry. 2006;67(suppl 2):34–39. [PubMed] [Google Scholar]
- 3.Holmes A, Heilig M, Rupniak NM, Steckler T, Griebel G. Neutopeptide systems as novel therapeutic targets for depression and anxiety disorders. Trends Pharmacol Sci. 2003;24:580–538. doi: 10.1016/j.tips.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Roy-Byrne PP. The GABA-benzodiazepine receptor complex: structure, function, and role in anxiety. J Clin Psychiatry. 2005;66(suppl 2):14–20. [PubMed] [Google Scholar]
- 5.Otto MW, Bruce SE, Deckersbach T. Benzodiazepine use, cognitive impairment, and cognitive-behavioral therapy for anxiety disorders: issues in the treatment of a patient in need. J Clin Psychiatry. 2005;66(suppl 2):34–38. [PubMed] [Google Scholar]
- 6.Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- 7.Davis M, Myers KM, Chhatwal J, Ressler KJ. Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx. 2006;3:82–96. doi: 10.1016/j.nurx.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter AC, Felder CC. The endocannabinoid nervous system: unique opportunities for therapeutic intervention. Pharmacol Ther. 2001;90:45–60. doi: 10.1016/s0163-7258(01)00130-9. [DOI] [PubMed] [Google Scholar]
- 10.Haller J, Varga B, Ledent C, Freund TF. CB1 cannabinoid receptors mediate anxiolytic affects: convergent genetic and pharmacological evidence with CB1-specific agents. Behav Pharmacol. 2004;15:299–304. doi: 10.1097/01.fbp.0000135704.56422.40. [DOI] [PubMed] [Google Scholar]
- 11.Witkin JM, Tzavara ET, Davis RJ, Li X, Nomikos GG. A therapeutic role for cannabinoid CB1 receptor antagonists in major depressive disorders. Trends Pharmacol Sci. 2005;28:603–617. doi: 10.1016/j.tips.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 12.La Rana G, Russo R, Campolongo P, et al. Modulation of neuropathic and inflammatory pain by the endocannabinoid transport inhibitor AM404 [N-(4-hydroxyphenyl)-eicosa-5,8,11,14-tetraenamide. J Pharmacol Exp Ther. 2006;317:1365–1371. doi: 10.1124/jpet.105.100792. [DOI] [PubMed] [Google Scholar]
- 13.Bortolato M, Campolongo P, Mangieri RA, et al. Anxiolytic-like properties of the anandamide transport inhibitor AM404. Neuropsychopharmacology. 2006;31:2652–2659. doi: 10.1038/sj.npp.1301061. [DOI] [PubMed] [Google Scholar]
- 14.Kathuria S, Gaetani S, Fegley D, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 15.Scheen AJ, Finer N, Hollander P, Jensen MD, Van Gaal LF. Efficacy and tolerability of rimonabant in overweight or obese patients with type 2 diabetes: a randomised controlled study. Lancet. 2006;368:1660–1672. doi: 10.1016/S0140-6736(06)69571-8. [DOI] [PubMed] [Google Scholar]
- 16.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 17.Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 18.Maren S. The amygdala, synaptic plasticity, and fear memory. Ann N Y Acad Sci. 2003;985:106–113. doi: 10.1111/j.1749-6632.2003.tb07075.x. [DOI] [PubMed] [Google Scholar]
- 19.Quirk GJ, Gehlert DR. Inhibition of the amygdala: key to pathological states? Ann N Acad Sci. 2003;985:263–272. doi: 10.1111/j.1749-6632.2003.tb07087.x. [DOI] [PubMed] [Google Scholar]
- 20.Rescorla R. Experimental Extinction. In: Mowrer R, Klein S, editors. Handbook of Contemporary Learning Theories. Mahwah, NJ: Lawrence Erlbaum Associates; 2001. pp. 113–154. [Google Scholar]
- 21.Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- 22.Walker DL, Davis M. The role of amygdala glutamate receptors in fear learning, fear- potentiated startle, and extinction. Pharmacol Biochem Behav. 2002;71:379–392. doi: 10.1016/s0091-3057(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 23.Baker D, Pryce G, Davies WL, Hiley CR. In silico patent searching reveals a new can nabinoid receptor. Trends Pharmacol Sci. 2006;27:1–4. doi: 10.1016/j.tips.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Begg M, Pacher P, Batkai S, et al. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Sawzdargo M, Nguyen T, Lee DK, et al. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res Mol Brain Res. 1999;64:193–198. doi: 10.1016/s0169-328x(98)00277-0. [DOI] [PubMed] [Google Scholar]
- 26.Isokawa M, Alger BE. Ryanodine receptor regulates endogenous cannabinoid mobilization in the hippocampus. J Neurophysiol. 2006;95:3001–3011. doi: 10.1152/jn.00975.2005. [DOI] [PubMed] [Google Scholar]
- 27.Rancz EA, Hausser M. Dendritic calcium spikes are tunable triggers of cannabinoid release and short-term synaptic plasticity in cerebellar Purkinje neurons. J Neurosci. 2006;26:5423–5437. doi: 10.1523/JNEUROSCI.5284-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci. 1996;16:3934–3942. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cadas H, di Tomaso E, Piomelli D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci. 1997;17:1226–1242. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, et al. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Ohno-Shosaku T, Hashimotodani Y, Maejima T, Kano M. Calcium signaling and synaptic modulation: regulation of endocannabinoid-mediated synaptic modulation by calcium. Cell Calcium. 2005;38:369–374. doi: 10.1016/j.ceca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Ohno-Shossku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 33.Mackie K. Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int J Obes (Lond) 2006;30(suppl 1):S19–S23. doi: 10.1038/sj.ijo.0803273. [DOI] [PubMed] [Google Scholar]
- 34.Iversen L. Pharmacology. Endogenous cannabinoids. Nature. 1994;372:619. doi: 10.1038/372619a0. [DOI] [PubMed] [Google Scholar]
- 35.Iversen L. Cannabis and the brain. Brain. 2003;126(pt 6):1252–1270. doi: 10.1093/brain/awg143. [DOI] [PubMed] [Google Scholar]
- 36.Gulyas AI, Cravatt BF, Bracey MH, et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 37.Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22:565–572. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- 38.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 39.Mechoulam R, Gaoni Y. A Total Synthesis of DI-Delta-1-Tetrahydrocannabinol, the active constituent of hashish. J Am Chem Soc. 1965;87:3273–3275. doi: 10.1021/ja01092a065. [DOI] [PubMed] [Google Scholar]
- 40.D’Souza DC, Kosten TR. Cannabinoid antagonists: a treatment in search of an illness. Arch Gen Psychiatry. 2001;58:330–331. doi: 10.1001/archpsyc.58.4.330. [DOI] [PubMed] [Google Scholar]
- 41.Reeve VC, Grant JD, Robertson W, Gillespie HK, Hollister LE. Plasma concentrations of delta-9-tetrahydrocannabinol and impaired motor function. Drug Alcohol Depend. 1983;11:167–175. doi: 10.1016/0376-8716(83)90077-7. [DOI] [PubMed] [Google Scholar]
- 42.McDonald J, Schleifer L, Richards JB, de Wit H. Effects of THC on behavioral measures of impulsivity in humans. Neuropsychopharmacology. 2003;28:1356–1365. doi: 10.1038/sj.npp.1300176. [DOI] [PubMed] [Google Scholar]
- 43.Yesavage JA, Leirer VO, Denari M, Hollister LE. Carry-over effects of marijuana intoxication on aircraft pilot performance a preliminary report- Am J Psychiatry. 1985;142:1325–1329. doi: 10.1176/ajp.142.11.1325. [DOI] [PubMed] [Google Scholar]
- 44.Dannon PN, Lowengrub K, Amiaz R, Grunhaus L, Kotler M. Comorbid cannabis use and panic disorder short term and long term follow-up study. Hum Psychopharmacol. 2004;19:97–101. doi: 10.1002/hup.560. [DOI] [PubMed] [Google Scholar]
- 45.Fride E. Endocannabinoids in the central nervous system: from neuronal networks to behavior. Curr Drug Targets CNS Neurol Disord. 2005;4:633–642. doi: 10.2174/156800705774933069. [DOI] [PubMed] [Google Scholar]
- 46.Heishman SJ, Arasteh K, Stitzer ML. Comparative effects of alcohol and marijuana on mood, memory, and performance. Pharmacol Biochem Behav. 1997;58:93–101. doi: 10.1016/s0091-3057(96)00456-x. [DOI] [PubMed] [Google Scholar]
- 47.Mallet PE, Beninger RJ. The cannabinoid CBI receptor antagonist SR141716A attenuates the memory impairment produced by delta9-tetrahydrocannabinol or anandamide. Psychopharmacology (Berl) 1998;140:11–19. doi: 10.1007/s002130050733. [DOI] [PubMed] [Google Scholar]
- 48.Hampson RE, Deadwyler SA. Cannabinoids reveal the necessity of hippocampal neural encoding for short-term memory in rats. J Neurosci. 2000;20:8932–8942. doi: 10.1523/JNEUROSCI.20-23-08932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hampson RE, Hedberg T, Deadwyler SA. Differential information processing by hippocampal and subicular neurons. Ann N Y Acad Sci. 2000;911:151–165. doi: 10.1111/j.1749-6632.2000.tb06724.x. [DOI] [PubMed] [Google Scholar]
- 50.Braida D, Sala M. Cannabinoid-induced working memory impairment is reversed by a second generation cholinesterase inhibitor in rats. Neuroreport. 2000;11:2025–2029. doi: 10.1097/00001756-200006260-00044. [DOI] [PubMed] [Google Scholar]
- 51.Varvel SA, Lichtman AH. Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther. 2002;301:315–924. doi: 10.1124/jpet.301.3.915. [DOI] [PubMed] [Google Scholar]
- 52.Da S, Takahashi RN. SR 141716A prevents delta 9-tetrahydrocannabinol-induced spatial learning deficit in a Morris-type water maze in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:321–325. doi: 10.1016/s0278-5846(01)00275-5. [DOI] [PubMed] [Google Scholar]
- 53.Pamplona FA, Takahashi RN. WIN 55212-2 impairs contextual fear conditioning through the activation of CB1 cannabinoid receptors. Neurosci Lett. 2006;397:88–92. doi: 10.1016/j.neulet.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 54.Hollister LE. Health aspects of cannabis. Pharmacol Rev. 1986;38:1–20. [PubMed] [Google Scholar]
- 55.Viveros MP, Llorente R, Moreno E, Marco EM. Behavioral and neuroendocrine effects of cannabinoids in critical developmental periods. Behav Pharmacol. 2005;16:353–362. doi: 10.1097/00008877-200509000-00007. [DOI] [PubMed] [Google Scholar]
- 56.Vinod KY, Hungund BL. Role of the endocannabinoid system in depression and suicide. Trends Pharmacol Sci. 2006;27:533–545. doi: 10.1016/j.tips.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Kamprath K, Wotjak CT. Nonassociative learning processes determine expression and extinction of conditioned fear in mice. Learn Mem. 2004;11:770–786. doi: 10.1101/lm.86104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel S, Cravatt BF, Hillard CJ. Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology. 2005;30:497–507. doi: 10.1038/sj.npp.1300535. [DOI] [PubMed] [Google Scholar]
- 59.Genn RF, Tucci S, Marco EM, Viveros MP, File SE. Unconditioned and conditioned anxiogenic effects of the cannabinoid receptor agonist CP 55,940 in the social interaction test. Pharmacol Biochem Behav. 2004;77:567–573. doi: 10.1016/j.pbb.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 60.Uriguen L, Perez-Rial S, Ledent C, Palomo T, Manzanares J. Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology. 2004;46:966–973. doi: 10.1016/j.neuropharm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Haller J, Bakos N, Szirmay M, Ledent C, Freund TF. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci. 2002;16:1395–1398. doi: 10.1046/j.1460-9568.2002.02192.x. [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez de Fonseca F, Rubio P, Menzaghi F, et al. Corticotropin-releasing Factor (CRF) antagonist [D-Phe12,Nle21,38,C alpha MeLeu37]CRF attenuates the acute actions of the highly potent cannabinoid receptor agonist HU-210 on defensive-withdrawal behavior in rats. J Pharmacol Exp Ther. 1996;276:56–64. [PubMed] [Google Scholar]
- 63.Reich CG, Alger BE. Endocannabinoids modulate acquisition and extinction in trace fear conditioning. Paper presented at: Annual Meeting of the Society for Neuroscience; November 12–16, 2005; Washington, D.C. [Google Scholar]
- 64.Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C. Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem. 2002;80:448–456. doi: 10.1046/j.0022-3042.2001.00716.x. [DOI] [PubMed] [Google Scholar]
- 65.Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O. Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology (Berl) 2002;159:379–387. doi: 10.1007/s00213-001-0946-5. [DOI] [PubMed] [Google Scholar]
- 66.Chhatwal JP, Davis M, Maguschak KA, Ressler KJ. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- 67.Mikics E, Dombi T, Barsvari B, et al. The effects of cannabinoids on contextual conditioned fear in CB1 knockout and CD1 mice. Behav Pharmacol. 2006;17:223–230. doi: 10.1097/00008877-200605000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Arenos JD, Musty RE, Bucci DJ. Blockade of cannabinoid CB(1) receptors alters contextual learning and memory. Eur J Pharmacol. 2006;539:177–183. doi: 10.1016/j.ejphar.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 69.Marsicano G, Wotjak CT, Azad SC, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 70.Izquierdo A, Wellman CL, Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- 73.Hofmann SG, Meuret AE, Smits JA, et al. Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- 74.Hofmann SG, Pollack MH, Otto MW. Augmentation treatment of psychotherapy for anxiety disorders with D-cycloserine. CNS Drug Rev. 2006;12:208–217. doi: 10.1111/j.1527-3458.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chhatwal JP, Gutman AR, Maguschak K, Davis M, Ressler KJ. Endocannabinoid modulation of CCK2 receptor activation may be a key event in the extinction of conditioned fear. Paper presented at: Annual Meeting of the Society for Neuroscience; October 14–18, 2006; Atlanta, Ga. [Google Scholar]
- 76.Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rescorla RA. Extinction can be enhanced by a concurrent excitor. J Exp Psychol Anim Behav Process. 2000;26:251–260. doi: 10.1037//0097-7403.26.3.251. [DOI] [PubMed] [Google Scholar]
- 78.Cain CK, Blouin AM, Barad M. Andrenergic transmission facilitates extinction of conditional fear in mice. Learn Mem. 2004;11:179–187. doi: 10.1101/lm.71504. [DOI] [PMC free article] [PubMed] [Google Scholar]