

Abstract
Background
The effects of hypercapnic acidosis on the diaphragm and its recovery to normocapnia have been poorly evaluated. We studied diaphragmatic contractility facing acute variations of PaCO2 and evaluated the contractile function at 60 min after normocapnia recovery.
Methods
Thirteen piglets weighing 15–20 kg were anesthetized, ventilated and separated into two groups: a control group (n= 5) evaluated in normocapnia (time-control experiments) and a hypercapnia group (n= 8) in which animals were acutely and shortly exposed to five consecutive ranges of PaCO2 (40, 50, 70, 90 and 110 mmHg). Then CO2 insufflation was stopped. Diaphragmatic contractility was assessed by measuring transdiaphragmatic pressure (Pdi) variations obtained after bilateral transjugulary phrenic pacing at increased frequencies (20–120 Hz). For each level of PaCO2, pressure-frequency curves were obtained in vivo by phrenic nerve pacing.
Results
In the hypercapnia group, mean (±SD) Pdi significantly decreased from 41 ± 3 to 29 ± 3 cmH2O (P<0.05) between the first (40 mmHg) and the fifth stages of capnia (116 mmHg) at the frequency of 100 Hz stimulation. The observed alteration of the contractile force was proportional to the level of PaCO2 (r2= 0.61, P<0.01). Normocapnia recuperation allowed a partial recovery of the diaphragmatic contractile force (80% of the baseline value) at 60 min after CO2 insufflation interruption.
Conclusion
A short exposure to respiratory acidosis decreased diaphragmatic contractility proportionally to the degree of hypercapnia and this alteration was only partially reversed at 60 min following exposure.
Keywords: Acute Disease; Animals; Animals, Newborn; Diaphragm; physiology; Hypercapnia; physiopathology; Muscle Contraction; physiology; Recovery of Function; physiology; Swine
Introduction
Acute respiratory acidosis occurs with acute respiratory failure, which can result from any sudden respiratory parenchymal (eg, pulmonary edema and massive pulmonary embolism)1–3, airways 4–10, pleural, chest wall, neuromuscular (eg, spinal cord injury), or central nervous system event (eg, drug overdose)11–13. Two main situations of hypercapnic acidosis are encountered in patients with acute respiratory failure in intensive care unit. Firstly, it is observed in weak or comatose patients with a “short and acute” exposure to hypercapnia 11–14. Secondly, it is observed in mechanically ventilated patients with a limited tidal volume and plateau pressure (“protective settings”) 4,15,16. Although the effects of hypercapnia have been well evaluated on hemodynamic 17–19 and pulmonary functions 4,20–22, few data have evaluated in vivo effects of both “short” and “prolonged” hypercapnia exposure on diaphragmatic function. To our knowledge, only two studies have evaluated its effect on the diaphragm in animals at rest and without a fatigue protocol preceding hypercapnia 23,24. These studies 23,24 reported a decrease in diaphragmatic force related to respiratory acidosis, but neither of them evaluated the recovery of contractile diaphragm properties following acute hypercapnia. Nevertheless, approaching the functional recovery of diaphragmatic weakness secondary to acute hypercapnia is important in terms of clinical applications, such as the weaning process. Therefore, our model, using piglets, was designed to evaluate, firstly, the effect of short hypercapnia on transdiaphragmatic pressure (Pdi) and, secondly, the immediate (one hour) recovery of Pdi following the return to normocapnia. Our hypothesis was that a short and acute exposure to hypercapnia will decrease the Pdi which will not return to its baseline value after one hour recovery of hypercapnia.
Materials and Methods
The study was approved by the Institutional Ethics Committee for Animals and was conducted in an authorized laboratory under the supervision of authorized researchers and according to the official Edict of the French Ministry of Agriculture.
Animal preparation and experimental procedures
Thirteen piglets (15–20 kg) were anesthetized with intravenous pentobarbital sodium (5–6 mg.kg−1), intubated with a cuffed endotracheal tube and mechanically ventilated (Onyx Plus®, Tyco, Saint Louis, MO), with a FiO2 of 0.35, a tidal volume between 8 and 10 mL. kg−1, 20 cycles per minute to obtain normocapnia and 4 cm H2O end-expiratory pressure. In this study, we used the same experimental design described in our previous study 25 (Fig. 1). Briefly, the piglets were maintained under anesthesia with continuous intravenous propofol (15–20 mg.kg−1.h−1) and ketamine (3–4 mg.kg−1.h−1). The piglets did not have spontaneous ventilatory activity, as assessed by the esophageal pressure curves. Although, hypercapnia is a strong ventilatory stimulus, the anesthesia could be adapted to inhibit this stimulus. So we could affirm that the piglets were not in asynchrony with the ventilator. Neuromuscular agents were not used. An oral gastric tube was placed. A vesicostomy was performed and a urine catheter placed for urine collection. A carotidal arterial line (PiCCO®, Pulsion, Munich, Germany) was inserted for the monitoring of heart rate, arterial blood pressure and cardiac output 26. At the end of the procedure, animals were killed by intravenous injection of potassium chloride.
Figure 1.
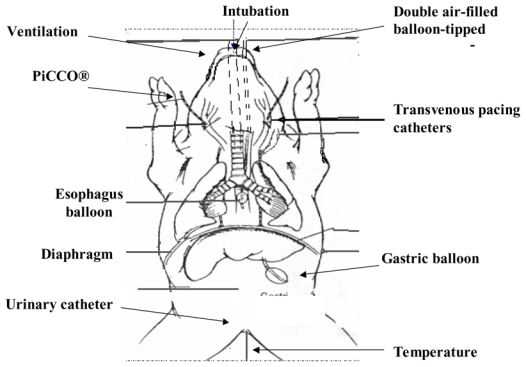
Design of the Study
Animals were separated into two groups (Fig. 2): 1) a control group (n= 5) mechanically ventilated in normocapnia without any intervention in which studied variables have been measured each hour over the same time period (6-h) as in the hypercapnic group (time-control experiments) and 2) a hypercapnic group (n= 8) acutely and shortly exposed to five consecutive ranges of PaCO2 in the first part of the study and evaluated at 60 min after normocapnia recovery in the second part of the study (see below). The two groups received the same care except for the capnia levels.
Figure 2.
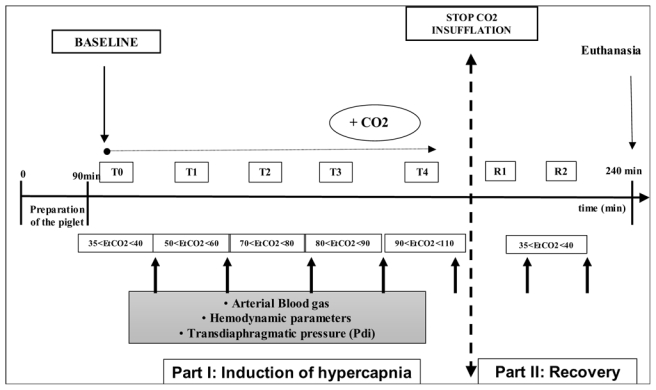
First part of the study: induction of hypercapnia
Five consecutives ranges of capnia were studied: 35–45, 50–60, 60–70, 90–100 and 110–120 mm Hg. Each level of PaCO2 (arterial pressure of carbon dioxide) was obtained by insufflating CO2 (Aga medical, Bassens, France) into the inspiratory line of the ventilator. We did not change the respiratory rate or the tidal volume. PaCO2 levels were checked by a capnograph (Deltatrac, Datex-Ohmeda, Helsinki, Finland) and then verified by arterial blood gases (iSTAT®, Abbott, Illinois). Steady state at all PaCO2 levels was verified by the constancy of end-tidal CO2 for at least 5 min. After obtaining end-tidal CO2 steady state, we performed a hemodynamic evaluation. Arterial blood gases and Pdi measurements were performed after 15 min of additional exposure at this level of capnia. After measurements, the inspiratory flow of CO2 was increased to the next level. We also calculated lung compliance as follows: tidal volume/(plateau pressure – esophageal pressure).
Second part of the study: recovery
After the end of the last studied period, the CO2 insufflation was discontinued. We then measured the Pdi at 30 and 60 min after the return to normocapnia, which was attained 5 to 10 min after stopping the insufflation.
In vivo measurement of transdiaphragmatic pressure (Pdi)
Bipolar transvenous pacing catheters were introduced via each internal jugular vein and adjusted to achieve stimulation of the phrenic nerve and subsequent contraction of the diaphragm. Double air-filled balloon-tipped catheters were placed transorally in the distal third of the esophagus and in the stomach for measurement of Pdi. Pdi was produced by supramaximal stimulation at frequencies of 20, 40, 60, 80, 100 and 120 Hz in a serial manner and recorded for a given PaCO2. Each train of impulses had a duration of 2000 ms. Each impulse in the train had duration of 150 ms. A pressure-frequency curve was obtained for each range of PaCO2 for each piglet at functional residual capacity, as described in our previous study 25.
Statistical analysis
After performing a Kolmogorov-Smirnov test to assess the normality of the variable distribution, we expressed data as mean ± SD. We performed separate analyses of hypercapnia and recovery phases of our study. Comparisons of several means were performed using repeated-measures analysis of variance and the Newman-Keuls test. The difference between the two groups was mainly analyzed by using the interaction between time and group. Comparison of two means was performed using a nonpaired Student t test. Correlation coefficients (r2) between PaCO2 and Pdi and CO were calculated. All P values were twotailed and a P value of less than 0.05 was required to rule out the null hypothesis. Statistical analysis was performed using SAS©/STAT software version 8.1 (SAS Institute, Cary, NC).
Results
No significant difference was observed between the control group and the hypercapnia group for all the studied baseline variables. In the hypercapnia group, each range of capnia was attained in the 8 piglets. “In the three firsts piglets, the CO2 insufflation was progressively stopped to avoid adverse cardiovascular effects contrary to the five others piglets in whom CO2 insufflation was stopped without steps. To avoid confusion we do not report the recovery phase in the three firsts piglets.”
The mean tidal volume was 9.2 ± 1.1 mL.kg−1 and the respiratory rate was 20 ± 2 breaths.min− 1. We did not change the respiratory settings throughout the study. The mean weight of the piglets was 17 ± 0.8 kg. The duration of the study was 240 ± 36 min.
Hemodynamic variables and gas exchange
The hemodynamic and gas exchange variables are reported in table 1. The baseline hemodynamic and gas exchange data of the two groups were not significantly different and did not change significantly in the control group during the 4 hours of the time-control experiment in normocapnia ventilation without any intervention. In the hypercapnia group, we observed a progressive increase in the heart rate and arterial blood pressure. There was a significant correlation between PaCO2 increase and cardiac output increase (r2= 0.54, P < 0.01). In the second part of the study (recovery) the hemodynamic variables returned to baseline values. The variables of oxygenation were maintained normal during the entire procedure.
Table 1.
Hemodynamic and respiratory variables for the five ranges of capnia and the two ranges of recovery obtained in the hypercapnia group and in the control group over the same time period
| Hypercapnia induction |
Between-group comparisons | Recovery phase |
Between-Group comparisons | ||||||
|---|---|---|---|---|---|---|---|---|---|
| T0 | T1 | T2 | T3 | T4 | R1 | R2 | |||
| PaCO2 (mmHg) | |||||||||
| Control | 39 ± 6 | 39 ± 6 | 41 ± 5 | 40 ± 6 | 39 ± 6 | - | 39 ± 6 | 41 ± 5 | - |
| Hypercapnia | 40 ± 7 | 54 ± 6 | 68 ± 9 | 95 ± 18 | 116 ± 16 | 43 ± 7 | 43 ± 5 | ||
| HR(c/min) | |||||||||
| Control | 113 ± 8 | 107 ± 6 | 110 ± 9 | 101 ± 11 | 107 ± 12 | p<0.05 | 101 ± 9 | 99 ± 12 | NS |
| Hypercapnia | 120 ± 4 | 123 ± 11 | 137 ± 9 | 171 ± 13*† | 191 ± 11*† | 128 ± 14 | 125 ± 14 | ||
| SBP (mmHg) | |||||||||
| Control | 90 ± 9 | 88 ± 7 | 92 ± 7 | 90 ± 8 | 93 ± 12 | p<0.05 | 88 ± 9 | 91 ± 7 | NS |
| Hypercapnia | 95 ± 7 | 122 ± 3† | 126 ± 4† | 144 ± 15*† | 132 ± 15*† | 99 ± 4 | 101 ± 5 | ||
| MBP (mmHg) | |||||||||
| Control | 72 ± 4 | 70 ± 6 | 78 ± 6 | 68 ± 5 | 72 ± 7 | p<0.05 | 71 ± 5 | 73 ± 5 | NS |
| Hypercapnia | 83 ± 7 | 105 ± 2† | 108 ± 3† | 123 ± 13*† | 112 ± 15*† | 87 ± 7 | 89 ± 2 | ||
| CO (l/min) | |||||||||
| Control | 2.8 ± 0.4 | 3.0 ± 0.2 | 2.7 ± 0.4 | 2.8 ± 0.4 | 3.1 ± 0.3 | p<0.05 | 3.0 ± 0.2 | 2.9 ± 0.5 | NS |
| Hypercapnia | 3.0 ± 0.2 | 3.5 ± 0.3 | 3.7 ± 0.5 | 4.3 ± 0.4† | 5.1 ± 0.6*† | 3.5 ± 0.7 | 3.6 ± 0.7 | ||
| pH | |||||||||
| Control | 7.45 ± 0.02 | 7.48 ± 0.03 | 7.44 ± 0.02 | 7.46 ± 0.03 | 7.47 ± 0.03 | p<0.05 | 7.46 ± 0.03 | 7.44 ± 0.03 | NS |
| Hypercapnia | 7.46 ± 0.03 | 7.34 ± 0.03* | 7.27 ± 0.05*† | 7.03 ± 0.08*† | 6.98 ± 0.01 *† | 7.41 ± 0.06 | 7.42 ± 0.07 | ||
| PaO2 (mmHg) | |||||||||
| Control | 142 ± 23 | 164 ± 27 | 151 ± 19 | 160 ± 22 | 146 ± 31 | NS | 154 ± 24 | 152 ± 21 | NS |
| Hypercapnia | 123 ± 32 | 151 ± 55 | 148 ± 63 | 145 ± 51 | 120 ± 69† | 144 ± 36 | 141 ± 39 | ||
| SaO2(%) | |||||||||
| Control | 99 ± 1 | 99 ± 1 | 99 ± 1 | 99 ± 1 | 99 ± 2 | NS | 99 ± 1 | 99 ± 1 | NS |
| Hypercapnia | 98 ± 1 | 99 ± 2 | 99 ± 1 | 99 ± 1 | 99 ± 2 | 99 ± 1 | 99 ± 2 | ||
| Bicarbonate (mmol/l) | |||||||||
| Control | 28 ± 2 | 29 ± 3 | 28 ± 1 | 29 ± 2 | 29 ± 2 | NS | 28 ± 3 | 28 ± 2 | NS |
| Hypercapnia | 30 ± 1 | 30 ± 3 | 31 ± 2 | 31 ± 3 | 30 ± 4 | 29 ± 5 | 30 ± 5 | ||
| EtCO2 (mmHg) | |||||||||
| Control | 39 ± 5 | 38 ± 4 | 39 ± 3 | 38 ± 5 | 37 ± 5 | p<0.05 | 39 ± 4 | 38 ± 5 | NS |
| Hypercapnia | 39 ± 6 | 56 ± 8† | 77 ± 4† | 90 ± 7† | 105 ± 13† | 44 ± 4 | 43 ± 5 | ||
| Lung compliance (ml/cmH2O) | |||||||||
| Control | 79 ± 7 | 82 ± 7 | 77 ± 9 | 79 ± 7 | 77 ± 6 | NS | 78 ± 7 | 83 ± 5 | NS |
| Hypercapnia | 74 ± 4 | 74 ± 5 | 74 ± 4 | 72 ± 5 | 73 ± 4 | 74 ± 5 | 73 ± 4 | ||
Data are mean ± SD.
In the hypercapnia group, each range of capnia was attained in the 8 piglets. The recovery phase was attained in only 5 piglets.
HR = heart rate, SBP = systolic blood pressure, MBP = mean blood pressure, CO = cardiac output, T0 = first level of PaCO2 (baseline-normocapnia), T1 to T4: = successive levels of increase PaCO2; R1 = first recovery step at 30 min after return to normocapnia; R2 = second recovery step 60 min after return to normocapnia. Statistical comparison was not performed for PaCO2. P values for between group comparisons refer to interaction between time and group.
P < 0.05 vs baseline value.
P < 0.05 vs control group. NS = not significant.
In vivo measurement of transdiaphragmatic pressure
The baseline pressure-frequency curves of the two groups were not significantly different (Pdi at 20 Hz 25.5 ± 1.9 vs 26.9 ± 2.1 cmH2O, NS; and at 100 Hz: 40.5 ± 2.7 vs 42.8 ± 3.4 cmH2O, NS) and remained unchanged in the control group after 4 hours in normocapnia ventilation without any intervention (Pdi at 20 Hz: 26.9 ± 2.1 vs 25.2 ± 2.3 cmH2O, NS; and at 100 Hz: 42.8 ± 3.4 vs 38.9 ± 4.3, NS), indicating that anesthetic agents used did not significantly modify the diaphragmatic function.
The pressure-frequency curves in the hypercapnia group, for the 5 ranges of capnia, are represented in figure 3. Pdi was significantly lower (P < 0.05) at all stimulation frequencies between the third, the fourth and the fifth levels of capnia and the initial value of Pdi (normocapnia). We found a significant correlation between Pdi decrease and PaCO2 increase (r2= 0.61, P < 0.01);
Figure 3.
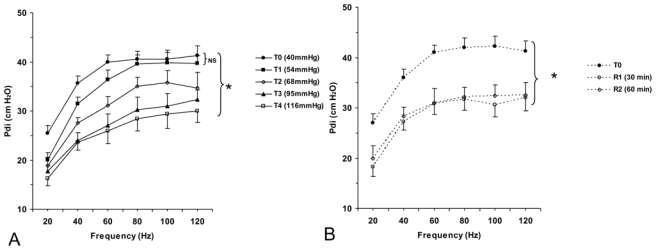
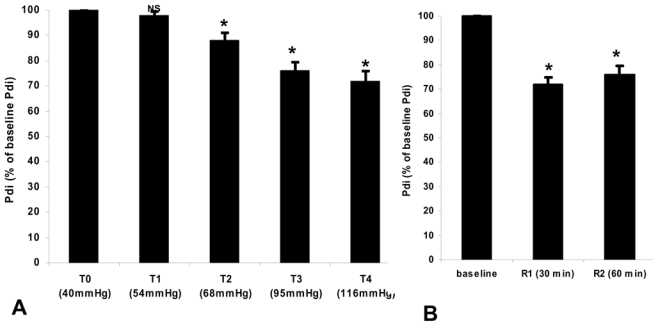
In the hypercapnia group, 30 and 60 min after the return to normocapnia, Pdi increased but did not return to its baseline value. Pdi reached only 80–85% of initial values and was significantly different from baseline values (P < 0.05) (figure 4).
Figure 4.
The lung compliance values throughout the study varied from 72±5 to 83±5 ml/cmH2O with no significant changes between steps, demonstrating that mechanical properties of the respiratory system remained stable during all the procedure (table 1).
DISCUSSION
Our main results are firstly, a short exposure to acute hypercapnic acidosis decreases contractile force (25–30%) of the diaphragm proportionally to the cumulative effects of increasing levels of hypercapnia and secondly, the recovery of this alteration is incomplete 60 min after the return to normocapnia.
In accordance with similar findings reported in the literature, respiratory acidosis appears to alter diaphragmatic contractile force 23,24,27. In the present study, we found that Pdi decreased significantly from 41 ± 3 to 29 ± 3 cm H2O between the first (40 mm Hg) and the fifth stage of capnia (116 mm Hg) at the frequency of 100 Hz stimulation (Fig. 3,4). To our knowledge, Schnader et al. 23 were the first to evaluate the effect of different ranges of hypercapnia on diaphragm force in dogs. They evaluated seven levels of PaCO2 by breathing various inspiratory gas mixtures and reducing the tidal volume and respiratory rate in six mechanically ventilated dogs. After direct stimulation of the phrenic nerve, they showed an alteration of the contractile force proportional to the level of capnia, as we found in the present study for similar levels of PaCO2 (Fig. 3). However, the reduction of tidal volume, which resulted in a decreased functional residual capacity in the Schnader’s trial 23 may have contributed to the deterioration to the length-force relation of the diaphragm since variations of pulmonary volume change the orientation of the diaphragmatic fibers thereby changing their contractile properties 28. To prevent any change of the length-force relationship we used only different inspiratory gas mixtures of CO2 to induce hypercapnia. Moreover, the stability of transpulmonary pressures suggests that lung volumes were similar throughout the study. Indeed, the obtained lung compliance values throughout the study varied from 72±5 to 83±5 ml/cmH2O, with no significant difference between steps (table 1).
Yanos et al. 24 compared the effects of an acute metabolic and respiratory acidosis (at the same level: pH = 7.05) on diaphragmatic contractile properties in ventilated dogs. As in Schnader’s 23 and our present studies, the authors found a decrease in diaphragmatic force due to respiratory acidosis, whereas they did not observe a significant alteration with a metabolic acidosis. They explained the discrepancy between the results observed with the two types of acidosis 24 by a faster decrease in intracellular pH (pHi) when hypercapnia was the cause of acidosis.
The results of our trial confirm that even a short and acute exposure to hypercapnia acidosis alters the contractile property of the diaphragm and thus increases the respiratory muscle work. Thus, this diaphragm contractile property alteration may potentiate the rapid deterioration that occurs during acute exacerbation of respiratory status in these patients. In addition, four human trials 29–32 studied the effect of hypercapnia on diaphragmatic force in healthy subjects. They all conserved spontaneous ventilation and performed physical exercise in a CO2-added atmosphere. They found conflicting results about the effect of CO2 on the diaphragm, but these works are difficult to transpose to the intensive care practice because the conditions of the studies were not similar (studied population, lower level of hypercapnia, duration of the procedure, hyperventilation as a consequence of the inhalation of CO2, level of neuromuscular drive).
Reduced muscle contractility during hypercapnia is thought to be a result of the decrease in pHi 6 which has been demonstrated using 31P nuclear magnetic resonance 33. Increased binding of calcium to the sarcoplasmic reticulum, decreased affinity of troponin for calcium, and reduction in the rate of glycolysis, and hence ATP resynthesis, have been suggested as possible mechanisms underlying the reduction in contractility. Some authors 23,30 postulated that decreased force production during hypercapnia was caused by a secondary fall in pHi which would decrease the binding of Calcium to troponin. However, none of these two experimental studies 23,24 evaluated the recovery upon the return to normocapnia.
Our results show that after a short and acute exposure to hypercapnic acidosis the Pdi does not recover its baseline value one hour after discontinuing the CO2 insufflation (Fig. 3, 4). This alteration was not completely reversed when normocapnia was restored, the recovery of the diaphragmatic contractile force reaching only 80% of its initial value 60 minutes after discontinuing the CO2 insufflation (Fig. 3, 4). This may play a role in the recovery of decompensated chronically obstructive or asthmatic patients who may not immediately recover their diaphragmatic force after an acute respiratory failure episode. We investigated the healthy piglet diaphragm, but caution must be employed when the extrapolating these results to an altered diaphragm 34.
Only one human study 30 evaluated the recovery period after short (2 min) exposure to moderate hypercapnia (end-tidal CO2 was increase from 5.5% to 8.9%) in seven healthy subjects who spontaneously hyperventilated. Although the model of hypercapnia used is different from ours, the results of the present study are in accordance with those reported by Rafferty et al. 30. Indeed, these authors 30 showed that hypercapnia induced a significant decrease in twitch Pdi and found that 60 minutes after the discontinuation of CO2, the Pdi reached only 87% of its baseline value. In contrast, Rafferty et al. 30 observed that the recovery of diaphragmatic force was complete 90 min after the discontinuation of CO2. Diaphragm muscle activity is an important determinant of diaphragmatic blood flow. To our knowledge, the effects of increased cardiac output (CO) due to hypercapnia upon the diaphragmatic blood flow and its consequences upon the contractile properties have never been reported. We found an increase in CO, heart rate and blood pressure (Table 1) with hypercapnia. Among the trials which studied the effects of hypercapnia on diaphragmatic function, only one 24 evaluated cardiac output variations. In their trial, Yanos et al. 24 studied only one level of acidosis and did not find a significant increase in CO, both for metabolic and respiratory acidosis. Our results are in accordance with those reported by Yanos et al. 24, at the same level of pH (Table 1), but CO increased significantly for a PaCO2 > 95 mm Hg. Kendrick et al. 33 demonstrated a linear relationship between hypercapnia and diaphragmatic blood flow. Although, we did not evaluate the pH and/or PaCO2 intra and extracellular variations in the diaphragm, we can speculate that these mechanisms may explain in part our results. Greatly increasing blood flow acts to maintain diaphragm contractility by preventing the build-up of metabolic by-products in the intra or extracellular milieu 34. Therefore, increasing arterial PaCO2 leads to an increase in CO and hence diaphragm blood flow, which in turn acts to reduce any direct effects of CO2 upon diaphragm contractility, possibly explaining the absence of an effect of acute hypercapnia on the diaphragm in some human studies 29–32. Despite having a stimulatory effect either on ventilation and cardiac output, hypercapnia may have a deleterious effect on diaphragmatic contractile function by a combinative effect of acidosis-induced calcium sequestration into the sarcoplasmic reticulum 23 and promoting oxidative stress in striated muscles 35.
Moreover, although in the hypercapnia group the anesthesia level was adapted to blunt the respiratory stimulus induced by hypercapnia, we did not observe any significant drop in cardiac output. In addition, the anaesthetic drug dose did not differ significantly between the two groups through the study period. However, Fujii et al. 36 reported in dogs that either a low dose (1.5 mg.kg−1.h−1) or a large dose (6.0 mg.kg−1.h−1) of propofol infusion decreased Pdi at low frequency (20 Hz) but at not any change at a higher frequency (120 Hz). Nishina et al. 37 reported that little or large concentrations of ketamine and of propofol had no effect on diaphragm contractility under non fatigued and fatigued conditions. Regarding the hemodynamic literature related to hypercapnia 17,19,20,38 and the studies which evaluated the effects of anesthetic drugs on diaphragm function 36,37,39, we can assume that the observed alteration of diaphragmatic function in the current study was mainly due to hypercapnia rather than hemodynamic variations and/or anesthetic drugs.
Some remarks must be included to assess the limitations of our study. Firstly, although piglet respiratory muscles are similar to those of humans, this study was limited because we studied the effects of hypercapnia acidosis on healthy diaphragm muscles. Although the evidence for the capacity of hypercapnic acidosis to induce diaphragm dysfunction in animal models is convincing, it is considerably more difficult to obtain conclusive proof of diaphragmatic dysfunction in mechanically-ventilated critically ill patients or exhausted patients in acute respiratory failure. It is known that prolonged mechanical ventilation induces diaphragm alteration 25,40,41. The duration of experiments in the present study did not exceeded 6-h, which is not enough to induce a diaphragm dysfunction. Indeed, in a previous work of our team 25 and in the Radell et al. trial 42, the diaphragmatic pressure was preserved until 24h after controlled mechanical ventilation in piglets. Secondly, the periods of hypercapnia have not been randomized with a washout period of normocapnia, and then a cumulative effect cannot be discarded. However, in the clinical situation, the evolution of hypercapnia acidosis is generally a gradual process, similar to what we tried to reproduce in the design of our present study. Thirdly, we evaluated the effect of “short and acute” hypercapnia acidosis but we cannot extrapolate the effects of prolonged and/or moderate hypercapnia (> 24 hrs) on diaphragmatic contractile function. ARDS patients are frequently ventilated in moderate hypercapnia during several days. In case of septic origin, the diaphragmatic alteration is due both to the infection and the prolonged mechanical ventilation.
In conclusion, our study showed an alteration in diaphragmatic contractile properties proportional to the severity of the respiratory acidosis and the absence of total recovery one hour after discontinuing the CO2 insufflation. This alteration may play a role in weak patients with acute respiratory failure whose diaphragm is frequently ineffective despite an intensive workload. Further studies are needed to better evaluate the evolution of the diaphragm function after a prolonged “protective ventilation” period with mild to moderate hypercapnia and its impact upon clinical outcomes related to the weaning process.
Acknowledgments
The authors are grateful to Pr Peter Dodek (St. Paul’s Hospital and University of British Columbia, Vancouver, British Columbia, Canada) for his very useful comments, Patrick McSweeny (Biomedical Engineer, Fisher-Paykell, Courtaboeuf, France) for his English editing and Pr Bruno Riou (University Pierre et Marie Curie, Paris, France) for his statistic helpful.
Footnotes
Presented in part at the 2006 Annual Meeting of American Society of Anaesthesiology (Chicago, IL, October 14–18th, 2006)
The study was solely supported by departmental sources. No conflict of interest was declared.
References
- 1.Bouchama A, Curley W, Al-Dossary S, Elguindi A. Refractory hypercapnia complicating massive pulmonary embolism. Am Rev Respir Dis. 1988;138:466–8. doi: 10.1164/ajrccm/138.2.466. [DOI] [PubMed] [Google Scholar]
- 2.Hypercapnia complicating massive pulmonary embolism. Crit Care Med. 1985;13:249–50. doi: 10.1097/00003246-198504000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg SK, Lipschutz JB, Fein AM, Lippmann ML. Hypercapnia complicating massive pulmonary embolism. Crit Care Med. 1984;12:686–8. doi: 10.1097/00003246-198408000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Hickling KG. Permissive hypercapnia. Respir Care Clin N Am. 2002;8:155–69. doi: 10.1016/s1078-5337(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 5.Adnet F, Plaisance P, Borron SW, Levy A, Payen D. Prolonged severe hypercapnia complicating near fatal asthma in a 35-year-old woman. Intensive Care Med. 1998;24:1335–8. doi: 10.1007/s001340050772. [DOI] [PubMed] [Google Scholar]
- 6.Kavanagh BP, Laffey JG. Hypercapnia: permissive and therapeutic. Minerva Anestesiol. 2006;72:567–76. [PubMed] [Google Scholar]
- 7.Mazzeo AT, Spada A, Pratico C, Lucanto T, Santamaria LB. Hypercapnia: what is the limit in paediatric patients? A case of near-fatal asthma successfully treated by multipharmacological approach. Paediatr Anaesth. 2004;14:596–603. doi: 10.1111/j.1460-9592.2004.01260.x. [DOI] [PubMed] [Google Scholar]
- 8.Mutlu GM, Factor P, Schwartz DE, Sznajder JI. Severe status asthmaticus: management with permissive hypercapnia and inhalation anesthesia. Crit Care Med. 2002;30:477–80. doi: 10.1097/00003246-200202000-00034. [DOI] [PubMed] [Google Scholar]
- 9.Simpson SQ. Oxygen-induced acute hypercapnia in chronic obstructive pulmonary disease: what’s the problem? Crit Care Med. 2002;30:258–9. doi: 10.1097/00003246-200201000-00045. [DOI] [PubMed] [Google Scholar]
- 10.Goldstein B, Shannon DC, Todres ID. Supercarbia in children: clinical course and outcome. Crit Care Med. 1990;18:166–8. doi: 10.1097/00003246-199002000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Gupta R, Haydock T. Severe hypercapnia caused by acute heroin overdose. Ann Emerg Med. 2004;43:665–6. doi: 10.1016/j.annemergmed.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 12.McCrimmon DR, Alheid GF. On the opiate trail of respiratory depression. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1274–5. doi: 10.1152/ajpregu.00428.2003. [DOI] [PubMed] [Google Scholar]
- 13.Han SR, Ho CS, Jin CH, Liu CC. Unexpected intraoperative hypercapnia due to undetected expiratory valve dysfunction--a case report. Acta Anaesthesiol Sin. 2003;41:215–8. [PubMed] [Google Scholar]
- 14.Potkin RT, Swenson ER. Resuscitation from severe acute hypercapnia. Determinants of tolerance and survival. Chest. 1992;102:1742–5. doi: 10.1378/chest.102.6.1742. [DOI] [PubMed] [Google Scholar]
- 15.Hickling KG, Walsh J, Henderson S, Jackson R. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: a prospective study. Crit Care Med. 1994;22:1568–78. doi: 10.1097/00003246-199422100-00011. [DOI] [PubMed] [Google Scholar]
- 16.Hickling KG, Wright T, Laubscher K, Town IG, Tie A, Graham P, Monteath J, A’Court G. Extreme hypoventilation reduces ventilator-induced lung injury during ventilation with low positive end-expiratory pressure in saline-lavaged rabbits. Crit Care Med. 1998;26:1690–7. doi: 10.1097/00003246-199810000-00024. [DOI] [PubMed] [Google Scholar]
- 17.Cullen DJ, Eger EI., 2nd Cardiovascular effects of carbon dioxide in man. anesthesiology. 1974;41:345–9. doi: 10.1097/00000542-197410000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Bouvet F, Dreyfuss D, Lebtahi R, Martet G, Le Guludec D, Saumon G. Noninvasive evaluation of acute capillary permeability changes during high-volume ventilation in rats with and without hypercapnic acidosis. Crit Care Med. 2005;33:155–60. doi: 10.1097/01.ccm.0000150657.02138.29. [DOI] [PubMed] [Google Scholar]
- 19.Cardenas VJ, Zwischenberger JB, Tao W, Nguyen PD, Schroeder T, Traber LD, Traber DL, Bidani A. Correction of blood pH attenuates changes in hemodynamics and organ blood flow during permissive hypercapnia. Crit Care Med. 1996;24:827–34. doi: 10.1097/00003246-199605000-00017. [DOI] [PubMed] [Google Scholar]
- 20.Hino JK, Short BL, Rais-Bahrami K, Seale WR. Cerebral blood flow and metabolism during and after prolonged hypercapnia in newborn lambs. Crit Care Med. 2000;28:3505–10. doi: 10.1097/00003246-200010000-00026. [DOI] [PubMed] [Google Scholar]
- 21.Joyce CJ, Hickling KG. Permissive hypercapnia and gas exchange in lungs with high Qs/Qt: a mathematical model. Br J Anaesth. 1996;77:678–83. doi: 10.1093/bja/77.5.678. [DOI] [PubMed] [Google Scholar]
- 22.Torbati D, Mangino MJ, Garcia E, Estrada M, Totapally BR, Wolfsdorf J. Acute hypercapnia increases the oxygen-carrying capacity of the blood in ventilated dogs. Crit Care Med. 1998;26:1863–7. doi: 10.1097/00003246-199811000-00030. [DOI] [PubMed] [Google Scholar]
- 23.Schnader JY, Juan G, Howell S, Fitzgerald R, Roussos C. Arterial CO2 partial pressure affects diaphragmatic function. J Appl Physiol. 1985;58:823–9. doi: 10.1152/jappl.1985.58.3.823. [DOI] [PubMed] [Google Scholar]
- 24.Yanos J, Wood LD, Davis K, Keamy M., 3rd The effect of respiratory and lactic acidosis on diaphragm function. Am Rev Respir Dis. 1993;147:616–9. doi: 10.1164/ajrccm/147.3.616. [DOI] [PubMed] [Google Scholar]
- 25.Jaber S, Sebbane M, Koechlin C, Hayot M, Capdevila X, Eledjam JJ, Prefaut C, Ramonatxo M, Matecki S. Effects of short vs. prolonged mechanical ventilation on antioxidant systems in piglet diaphragm. Intensive Care Med. 2005;31:1427–33. doi: 10.1007/s00134-005-2694-1. [DOI] [PubMed] [Google Scholar]
- 26.Marx G, Sumpelmann R, Schuerholz T, Thorns E, Heine J, Vangerow B, Rueckoldt H. Cardiac output measurement by arterial thermodilution in piglets. Anesth Analg. 2000;90:57–8. doi: 10.1097/00000539-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 27.Xu X, Zhou J, Yang Q, Fang L, Xie Q, Shen Y. An in vitro rat diaphragmatic fatigue model induced by combined hypoxic and hypercapnic acidosis and the effect of salmeterol. Pharmacol Res. 2006;53:171–6. doi: 10.1016/j.phrs.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Polkey MI, Hamnegard CH, Hughes PD, Rafferty GF, Green M, Moxham J. Influence of acute lung volume change on contractile properties of human diaphragm. J Appl Physiol. 1998;85:1322–8. doi: 10.1152/jappl.1998.85.4.1322. [DOI] [PubMed] [Google Scholar]
- 29.Juan G, Calverley P, Talamo C, Schnader J, Roussos C. Effect of carbon dioxide on diaphragmatic function in human beings. N Engl J Med. 1984;310:874–9. doi: 10.1056/NEJM198404053101402. [DOI] [PubMed] [Google Scholar]
- 30.Rafferty GF, Lou Harris M, Polkey MI, Greenough A, Moxham J. Effect of hypercapnia on maximal voluntary ventilation and diaphragm fatigue in normal humans. Am J Respir Crit Care Med. 1999;160:1567–71. doi: 10.1164/ajrccm.160.5.9801114. [DOI] [PubMed] [Google Scholar]
- 31.Jonville S, Delpech N, Denjean A. Contribution of respiratory acidosis to diaphragmatic fatigue at exercise. Eur Respir J. 2002;19:1079–86. doi: 10.1183/09031936.02.00268202. [DOI] [PubMed] [Google Scholar]
- 32.Mador MJ, Wendel T, Kufel TJ. Effect of acute hypercapnia on diaphragmatic and limb muscle contractility. Am J Respir Crit Care Med. 1997;155:1590–5. doi: 10.1164/ajrccm.155.5.9154862. [DOI] [PubMed] [Google Scholar]
- 33.Kendrick JE, De Haan SJ, Parke JD. Regulation of blood flow to respiratory muscles during hypoxia and hypercapnia. Proc Soc Exp Biol Med. 1981;166:157–61. doi: 10.3181/00379727-166-41039. [DOI] [PubMed] [Google Scholar]
- 34.Barclay JK. A delivery-independent blood flow effect on skeletal muscle fatigue. J Appl Physiol. 1986;61:1084–90. doi: 10.1152/jappl.1986.61.3.1084. [DOI] [PubMed] [Google Scholar]
- 35.Arbogast S, Reid MB. Oxidant activity in skeletal muscle fibers is influenced by temperature, CO2 level, and muscle-derived nitric oxide. Am J Physiol Regul Integr Comp Physiol. 2004;287:R698–705. doi: 10.1152/ajpregu.00072.2004. [DOI] [PubMed] [Google Scholar]
- 36.Fujii Y, Hoshi T, Takahashi S, Toyooka H. Propofol decreases diaphragmatic contractility in dogs. Anesth Analg. 1999;89:1557–60. doi: 10.1097/00000539-199912000-00046. [DOI] [PubMed] [Google Scholar]
- 37.Nishina K, Mikawa K, Kodama S, Kagawa T, Uesugi T, Obara H. The effects of enflurane, isoflurane, and intravenous anesthetics on rat diaphragmatic function and fatigability. Anesth Analg. 2003;96:1674–78. doi: 10.1213/01.ANE.0000060455.94684.69. [DOI] [PubMed] [Google Scholar]
- 38.Fujii Y. Comparative effects of dopamine and dobutamine on hypercapnic depression of diaphragmatic contractility in dogs. Pulm Pharmacol Ther. 2004;17:289–92. doi: 10.1016/j.pupt.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Fujii Y, Hoshi T, Takahashi S, Toyooka H. The effect of sedative drugs on diaphragmatic contractility in dogs: propofol versus midazolam. Anesth Analg. 2000;91:1035–7. doi: 10.1097/00000539-200010000-00052. table of contents. [DOI] [PubMed] [Google Scholar]
- 40.Capdevila X, Lopez S, Bernard N, Rabischong E, Ramonatxo M, Martinazzo G, Prefaut C. Effects of controlled mechanical ventilation on respiratory muscle contractile properties in rabbits. Intensive Care Med. 2003;29:103–10. doi: 10.1007/s00134-002-1548-3. [DOI] [PubMed] [Google Scholar]
- 41.Bernard N, Matecki S, Py G, Lopez S, Mercier J, Capdevila X. Effects of prolonged mechanical ventilation on respiratory muscle ultrastructure and mitochondrial respiration in rabbits. Intensive Care Med. 2003;29:111–8. doi: 10.1007/s00134-002-1547-4. [DOI] [PubMed] [Google Scholar]
- 42.Radell PJ, Remahl SjG, Nichols DG, Eriksson LI. Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med. 2002;28:358–64. doi: 10.1007/s00134-002-1207-8. [DOI] [PubMed] [Google Scholar]