

Summary
Considerable biochemical evidence suggests that the protein kinase C (PKC) signaling cascade may be a convergent point for the actions of anti-manic agents, and that excessive PKC activation can disrupt prefrontal cortical regulation of thinking and behavior. To date, however, brain protein targets of PKC’s anti-manic effects have not been fully identified. Here we showed that PKC activity was enhanced in the prefrontal cortex of animals treated with the psychostimulant amphetamine. Phosphorylation of MARCKS, a marker of PKC activity, was increased in the prefrontal cortex of animals treated with the psychostimulant amphetamine, as well as in sleep-deprived animals (another animal model of mania), but decreased in lithium-treated animals. The antidepressant imipramine, which shows pro-manic properties in patients with bipolar disorder (BPD), also enhanced phospho-MARCKS in prefrontal cortex in vivo. We further explored the functional targets of PKC in mania-associated behaviors. Neurogranin is a brain-specific, postsynaptically located PKC substrate. PKC phosphorylation of neurogranin was robustly increased by pro-manic manipulations and decreased by anti-manic agents. PKC phosphorylation of the NMDA receptor site NR1S896 and the AMPA receptor site GluA1T840 was also enhanced in the prefrontal cortex of animals treated with the antidepressant imipramine, as well as behaviorally sleep-deprived animals, in striking contrast to the reduced activity seen in lithium-treated animals. These results suggest that PKC may play an important role in regulating NMDA and AMPA receptor functions. The biochemical profile of the PKC pathway thus encompasses both pro- and anti-manic effects on behavior. These results suggest that PKC modulators or their intracellular targets may ultimately represent novel avenues for the development of new therapeutics for mood disorders.
Keywords: Protein kinase C (PKC), mania, lithium, neurogranin, NR1, GluA1
Introduction
Bipolar disorder (BPD) is a chronic, severe, often life-threatening illness (Goodwin and Jamison, 2007) that affects approximately 1-2% of the US population (Judd and Akiskal, 2003). Although genetic factors play a major role in the etiology of BPD, the biochemical abnormalities underlying the predisposition to, and the pathophysiology of, this disorder remain to be fully elucidated. Recently, accumulating data have implicated glutamatergically-mediated synaptic plasticity in the treatment of BPD (Coyle and Duman, 2003; Du et al., 2008; Du et al., 2003; Du et al., 2004a; Kugaya and Sanacora, 2005; Maeng et al., 2008; Nestler and Carlezon, 2006; Sanacora et al., 2008; Young, 2007). Given that alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartic acid (NMDA) receptor trafficking is critical for regulating various forms of synaptic plasticity in the CNS, its potential involvement in the pathophysiology or treatment of certain neuropsychiatric disorders is an emerging area of interest (Barbon et al., 2006; Brebner et al., 2005; Carlezon and Nestler, 2002; Chao et al., 2002; Peineau et al., 2007; Pittaluga et al., 2007; Self, 2002; Snyder et al., 2000).
In recent years, research has increasingly investigated the role of intracellular signaling cascades in the pathophysiology and treatment of BPD (Manji et al., 2003; Zarate et al., 2006). One signaling cascade that has been studied with considerable interest is the protein kinase C (PKC) signaling pathway. It has a heterogeneous distribution in brain, and plays a major role in the regulation of neuronal excitability, neurotransmitter release, and long-term alterations in gene expression and plasticity (Huang, 1990; MacDonald et al., 2001; Nogues, 1997; Ramakers et al., 1997). A growing body of data has also implicated dysregulation of the PKC signaling cascade in the pathophysiology of BPD (Hahn and Friedman, 1999; Jope et al., 1996; Mathews et al., 1997; Turecki et al., 1998; Wang and Friedman, 1996). Specifically, PKC overactivity has been associated with motoric hyperactivity, risk-taking behavior, and excessive hedonic drive, all of which are cardinal symptoms of mania. Notably, the current mainstays in the treatment of mania—the structurally dissimilar anti-manic agents lithium (a monovalent cation) and valproate (VPA, a small fatty acid)—indirectly inhibit PKC (Chen et al., 2000a). In addition, clinical studies demonstrate that the PKC inhibitor tamoxifen has anti-manic efficacy in human manic subjects (Yildiz et al., 2008; Zarate et al., 2007) Overactive PKC signaling in the prefrontal cortex may thus explain many of the symptoms of mania. Notably, imaging studies have demonstrated marked under-activity of the right prefrontal cortex in mania (Arnsten and Manji, 2008); furthermore, severe dysfunction of the prefrontal cortex, particularly in the right hemisphere, is highly consistent with the symptoms of mania (Arnsten and Manji, 2008).
To date, however, the precise cellular targets of PKC that mediate these complex behavioral effects have not been fully elucidated. We therefore undertook a series of biochemical studies to investigate the potential role of PKC, and its targets, in the development and treatment of mania. Toward this end, we assessed the role of PKC phosphorylation of 1) NMDA and AMPA receptors; 2) neurogranin, which is a postsynaptically located and brain-specific PKC substrate; and 3) myristoylated alanine-rich C kinase substrate (MARCKS), an intracellular structural PKC substrate implicated in BPD, in a sleep deprivation model of mania, as well as in animals undergoing sustained treatment with the pro-manic agent amphetamine, the antidepressant imipramine, and the anti-manic agent lithium.
Methods
Treatment of animals with amphetamine, imipramine and lithium
All animal treatments, procedures, and care were approved by the National Institute of Mental Health (NIMH) Animal Care and Use Committee and followed the Guide for the Care and Use of Laboratory Animals (ISBN 0-309-05377-3). The mice were acclimated to the laboratory for a week before the experiment. Male C57/B6 mice (seven to eight weeks; Harlan, Indianapolis, IN) were housed three to four per cage in a 12 hour light/dark cycle and had access to water and food ad libitum. Amphetamine or imipramine-treated animals were given injections of imipramine (10 mg/kg in 0.3 ml of saline), amphetamine (2.5mg/kg in 0.3 ml of saline), or saline (twice daily, i.p.) for 10 days. For lithium treatment, C57BL/6 mice were fed with chow containing a low dose of lithium (Bio-Serv, Frenchtown, NJ, Li2CO3-1.2g /kg) for one week, followed by full dose lithium chow (Bio-Serv, Frenchtown NJ, Li2CO3-2.4g/kg) for 3 weeks. Control animals received control chow. Animals treated with lithium-containing chow were also provided with a bottle of saline in addition to drinking water. Blood samples were taken at the time of decapitation and analyzed for lithium concentration (0.89±0.25mM). After drug treatment, the prefrontal cortices from mouse brains were removed immediately after decapitation, immersed in liquid nitrogen, placed in labeled Eppendorf tubes on dry ice, and then stored and kept frozen at −80°C until processing.
Sleep deprivation
Male Wistar-Kyoto rats (Harlan, 11-12 week old at time of surgery) were used. Under deep isoflurane anesthesia (1.5-2% volume) to avoid damage to the left cortical hemisphere (used for the molecular studies), epidural screw electrodes were implanted over the right parietal and occipital cortex, and cerebellum (reference electrode). Electrodes were fixed to the skull with dental cement. Two stainless steel wires (diameter 0.4 mm) were inserted into the neck muscles to record the electromyogram (EMG).
After surgery, all rats were housed individually in transparent Plexiglas cages (36.5 × 25 × 46 cm), and kept in sound-proof recording boxes for the duration of the experiment. Lighting and temperature were kept constant (12 hour light/dark cycle, lights on at 10 am, 23 ± 1°C; food and water were available ad libitum and replaced daily at 10 am). Seven to ten days were allowed for recovery after surgery, and experiments were started only after the sleep/wake cycle had fully normalized. The rats were connected by means of a flexible cable to a commutator (Airflyte, Bayonne, NJ) and recorded continuously for two to four weeks using a Grass electroencephalograph (mod. 15LT, Astro-Med Inc., West Warwick, RI). Video recordings were performed continuously with infrared cameras (OptiView Technologies, Inc, Potomac Falls, VA) and stored in real time (AVerMedia Technologies, Inc, Milpitas, CA).
EEG and EMG signals were amplified (amplification factor 2000), and filtered [attenuation 50 % amplitude (− 6 dB)] as follows: EEG: high-pass filter at 0.1 Hz, low-pass filter at 30 Hz; EMG: high-pass filter at 10 Hz, low-pass filter at 100 Hz. All signals were sampled and stored at 128 Hz resolution. Sleep stages were scored off-line by visual inspection of 4-s epochs (SleepSign, Kissei Comtec, Nagano, Japan). Wakefulness was characterized by a low-voltage, high-frequency EEG pattern and phasic EMG-activity. Non-rapid eye movement (NREM) sleep was characterized by the occurrence of high-amplitude s low waves, spindles, and low tonic EMG activity. In contrast, during REM sleep, the EEG was similar to that during waking, but only heartbeats and occasional twitches were evident in the EMG signal. Vigilance state could always be determined. All animal procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Facilities were reviewed and approved by the IACUC of the University of Wisconsin-Madison, and were inspected and accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Each day from 10 to 10:30 am all rats were gently handled and exposed to a new object to become familiar with the sleep deprivation procedure (these objects were not reused). Two groups of rats were used. Sleeping rats (S) were sacrificed during the light hours (around 2.30 pm) at the end of a long period of sleep (at least 45 minutes, interrupted by periods of waking no longer than two minutes) after spending at least 75% of the previous four hours asleep. Sleep deprived (SD) rats were sacrificed during the light period at the same time of day as S rats (around 2.30pm) after four hours of continuous wakefulness by exposure to novel objects. Novel objects included nesting and bedding material from other rat cages, wooden blocks, small rubber balls, plastic, metallic, wooden, or paper boxes and tubes of different shapes and colors. The boxes often had holes through which the rat could reach for a palatable food pellet or a paper towel. During the sleep deprivation experiment, a new object was introduced into the cage whenever the rat appeared drowsy and slow waves became apparent on the EEG. The rats were never disturbed when they were spontaneously awake, feeding, or drinking.
Rats were deeply anesthetized with isoflurane (within one minute) and decapitated. The head was cooled in liquid nitrogen and the whole brain was removed. Left cerebral cortex was dissected and divided into three regions (frontal, parietal, occipital). Samples were immediately frozen on dry ice and stored at − 80°C.
After a one-week accommodation period, the lithium-treated mice received lithium-containing chow (custom-produced by Bio-Serve, Frenchtown, NJ ). Drug-containing chow and control chow were identical, with the exception of the added drug, and were produced at both a low and regular concentration (1.2 and 2.4 g/kg, respectively). These doses of lithium have been used extensively by our group and others and have been found to lead to serum drug levels similar to those achieved therapeutically in the treatment of BPD (Yuan et al., 1999). Mice were initially treated for one week at the lower dose (to acclimatize them to the diet and reduce risks of side effects), followed by three weeks of the higher-dose treatment. The lithium experiment included 12 control and 12 experimental animals, all provided with an extra bottle of saline and daily bedding changes to minimize the effects of lithium-induced polyuria (a well-known side effect of lithium).
All mice were weighed and then killed by decapitation during the morning hours. Trunk blood was collected, and drug serum levels were performed by Medtox Laboratories (St. Paul, MN). Only animals with drug levels within the therapeutic range were used for additional studies. In general, ~80% of animals achieved therapeutic levels of lithium. Serum lithium blood levels of the animals used for additional analyses were 0.80 ± 0.13 mEq/L. Hippocampal tissue was dissected immediately after decapitation. Brain specimens were frozen rapidly in liquid nitrogen and stored at −80°C until further analysis.
Preparation of crude homogenates, cytosolic, and membrane fractions
The frontal cortices and hippocampi from adult mouse brains (three to four months old) were removed immediately after decapitation, immersed in liquid nitrogen, placed in labelled Eppendorf (Westbury, NY) tubes on dry ice and then stored and kept frozen at −80 C until processing. Samples were sonicated in Homogenization Buffer A (50 mM Tris-Cl, pH 7.5, containing 2 mM dithiothreitol, 2 mM EDTA, 2 mM EGTA, 50 μM 4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride, 50 mM KF, 50 nM okadaic acid, 1mM sodium orthovanidate, 5mM sodium pyrophosphate, 0.1% NP-40, and 5 μg/ml each of leupeptin, aprotinin, chymostatin, and pepstatin A), spun in the Eppendorf 5810R centrifuge (Westbury, NY) for 20 minutes at 4°C at 20,000 rcf, and the clear homogenate was used as total protein.
For the PKC assay and determination of phosphorylated and total myristoylated alanine-rich C kinase substrate (MARCKS) levels in the frontal cortex (see below), samples were separated into membranous and cytosolic fractions, because translocation of this structural and signalling protein into different cellular compartments offers insight into activity states. These samples were sonicated in Homogenization Buffer B (50 mM Tris-Cl, pH 7.5, containing 2 mM dithiothreitol, 2 mM EDTA, 2 mM EGTA, 50 μM 4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride, 50 mM KF, 50 nM okadaic acid, 1mM sodium orthovanidate, 5mM sodium pyrophosphate, and 5 μg/ml of leupeptin) centrifuged at 100,000 × g for 30 minutes at 4°C in an Optima™ Max Ultracentrifuge (Beckman Coulter, Fullerton, CA) to yield the cytosol fraction, and aliquots of the supernatant were stored at −70°C until experimentation. The pellet fractions were re-suspended in Homogenizing Buffer B containing 0.1% Nonidet P-40, sonicated, and centrifuged again. Aliquots of the resulting supernatants were taken as the membrane fractions and stored at −80°C until further analysis.
PKC activity assay
PKC activity was determined in both the cytosolic and membrane fractions as previously described (Wu et al., 2002). Briefly, measurement was at 30°C for five minutes in a mixture (25 μl) containing 30 mM Tris-Cl (pH 7.5), 6 mM MgCl2, 120 μM [-32P] ATP, 1.0 mg/ml bovine serum albumin, 20 μM Ng-(28-43) peptide substrate, 0.1 mg/ml phosphatidylserine, 0.02 mg/ml 1,2-dioleoylglycerol, 0.4 mMCaCl2 or 1.5 mM EGTA, and 1 μg protein of tissue extract. Reactions were stopped by tubes being placed at 99°C for five minutes, then allowed to cool for 10 minutes. Tubes were centrifuged with an Eppendorf centrifuge 5415D (Westbury, NY) for three minutes and 5 μl of reaction mixture was blotted on 4 × 4 cm Whatman P81 chromatography paper. Blotted paper was allowed to dry for 30 minutes and then washed in a 7% phosphoric acid solution three times for 10 minutes each. The paper was placed on aluminum foil and allowed to dry overnight. 32P-labeled peptide substrates blotted on chromatography paper were measured in a scintillation counter. All reactions were run in duplicate or triplicate. Activities were converted to picomoles of 32P- incorporated into peptide/μg of protein/min. These values were then expressed as percent of controls to allow comparison.
Western blotting analysis
Aliquots of crude whole-cell homogenates, membrane fractions, or cytosol fractions taken from the frontal cortices of mice were used to determine phosphorylated and total protein. Protein concentrations were determined using the Bio-Rad (Hercules, CA) protein assay kit, and the linearity of the protein concentration for immunoblotting was ascertained by resolution of selected concentrations of protein. Equal amounts of proteins were subjected to 10% SDS-PAGE gels and separated by electrophoresis. Proteins were then electrophoretically transferred to nitrocellulose membranes. Nonspecific binding on the nitrocellulose was blocked with Tris Buffered Saline plus Tween 20 (TBST), 10% nonfat dry milk, and then incubated with anti-Neurogranin antibody (Upstate Laboratories, Syracuse, NY), anti-phospho-neurogranin (Ser 36) (Upstate Laboratories, Syracuse, NY), anti-actin antibody (Chemicon, Temecula, CA) anti-ATPase (Na+K) (Abcam, Cambridge, UK), anti-MARCKS antibody (Calbiochem, Gibbstown, NJ); anti-Phospho-MARCKS (Ser 152/156) antibody (Cell Signaling Technology, Danvers, MA), anti-phospho-NR1S986 (Upstate Laboratories, Syracuse, NY), anti-NR1(Chemicon, Temecula, CA), anti-phospho-GluA1T840 (Abcam, Cambridge, MA), anti-phospho-GluAS831 (Affinity BioReagents, Golden, CO), and anti-GluA1 (Chemicon, Temecula, CA). The secondary antibodies were horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (Cell Signaling Technology, Danvers, MA). The ECL plus kit (GE Healthcare, Piscataway, NJ) was used as a detection system. Similarly, membrane and cytosol fractions from the frontal cortices of mice were used to determine levels of phosphorylated and non-phosphorylated MARCKS with specific antibodies. Notably, in these experiments, nitrocellulose membranes were first probed with anti-phospho-MARCKS, anti-phospho-neurogranin, anti-phospho-GluA1, and anti-phospho-NR1 antibodies and then stripped with stripping buffer and re-probed with the anti-non-phospho-antibody. The bottom part of the same membrane was used for Western blot analysis of actin to assure that an equal amount of protein was loaded into the wells. Quantification of the immunoblots was performed by densitometric scanning of the x-ray film using a Kodak Image Station 440 CF and Kodak 1D Image Analysis Software (Eastman Kodak, Rochester, NY).
Results
PKC activity was enhanced in prefrontal cortex after treatment with amphetamine
Elevation of PKC activity/isozymes has been found in both postmortem brain tissue and peripheral cells in BPD patients (Arnsten and Manji, 2008), and activation of PKC in the frontal cortex has been associated with manic-like cognitive changes in both rodents and non-human primates (Birnbaum et al., 2004). Basal frontal cortical PKC activity was therefore investigated in the mice that received pro-manic amphetamine treatment. Chronic treatment with the psychostimulant amphetamine significantly increased membrane PKC activity by 194.5% (Figure 1). PKC activity in cytosol fractions from the frontal cortices was also increased by 172.4%, following treatment with amphetamine (Figure 1).
Figure 1. The psychostimulant amphetamine enhanced PKC activity in both membrane fraction and cytosol fraction of forebrain in vivo.
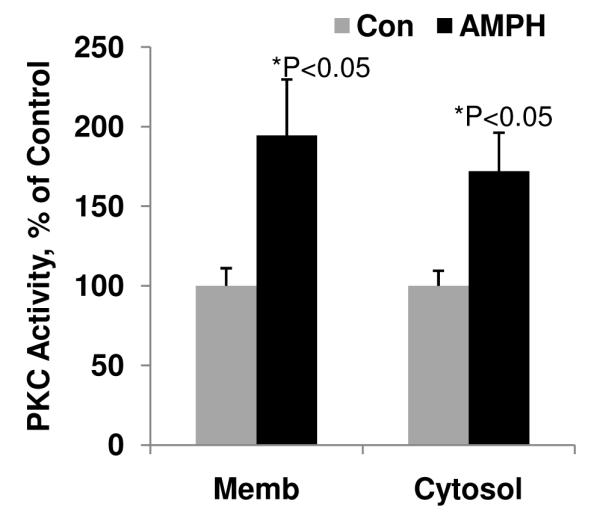
Mice were treated with amphetamine (AMPH, 5mg/kg, i.p.) for 10 days. Prefrontal cortex (Fcx) was dissected and fractionated into membranous and cytosolic fractions. PKC activity was determined with neurogranin (Ng) peptide as a substrate. (Student’s t-test, * p<0.05, N=5-8).
PKC phosphorylation of MARCKS is enhanced in prefrontal cortex of amphetamine-treated, sleep deprived, and imipramine-treated animals, but reduced in lithium-treated animals
Phosphorylation of MARCKS in membrane fractions from frontal cortex has been used as an indicator of PKC activity in brain tissue (Bar-Am et al., 2004; Pandey et al., 2003; Yamauchi et al., 1998). MARCKS belongs to a family of proteins involved in calcium regulation and cytoskeletal restructuring. Seminal studies by Lenox and associates demonstrated that MARCKS is a target for lithium’s actions (Lenox et al., 1996). The functional domain of MARCKS is bound to CaM, phosphorylated by PKC, and its activation contributes to binding of this protein to the cellular membrane.
The psychostimulant amphetamine evokes manic-like behavior in animal models at the dose used in these experiments, and sleep deprivation is also a well- established animal model for mania. We found that chronic treatment with the psychostimulant amphetamine significantly increased phospho-MARCKS (pMARCKS) levels in the membrane fraction by 197.2% (Figure 2). Nitrocellulose membranes for Western blot analysis of pMARCKS were re-probed with an anti-MARCKS antibody. The MARCKS levels in membrane fraction of frontal cortex from amphetamine-treated mice increased significantly by 151% (Figure 2). The pMARCKS and MARCKS levels in the cytosol fractions remained unchanged (Fig.2). Actin, used as a loading control for Western blot analyses for all membrane and cytosol fractions, was unaltered under these treatment conditions (Figure 2). In sleep deprived animals, we also found that pMARCKS levels were significantly increased in the membrane fraction of the prefrontal cortex up to 159% (Figure 3). The total MARCKS membrane fraction was also increased, suggesting increased translocation of MARCKS after PKC phosphorylation (Figure 3).
Figure 2. Phosphorylation of MARCKS at a PKC site in membranous fraction from the forebrain of amphetamine-treated animals.

(A) pMARCKS, MARCKS, and actin levels in membrane fraction from the forebrain of amphetamine (AMPH)-treated animals.
(B) pMARCKS, MARCKS, and actin levels in cytosol fraction from the forebrain of amphetamine (AMPH)-treated animals.
Data are presented as mean±SE (Student’s t-test, * p<0.05, N=4-8).
Figure 3. PKC phosphorylation of MARCKS in a sleepdeprivation (SD) animal model of mania.

Western blot analysis was performed to determine the pMARCKS or MARCKS in the membrane fractions. (N=8 animals per group, Student’s t-test, *p<0.05) Data are presented as mean±SE.
Antidepressant agents (particularly tricyclic antidepressant agents) can trigger manic episodes in individuals with BPD (Boerlin et al., 1998; Goldberg et al., 1999). Because the psychostimulant amphetamine and sleep deprivation enhanced pMARCKS in prefrontal cortex, we next investigated whether pMARCKS, as an indicator of PKC activation, is enhanced in prefrontal cortex of animals treated with the antidepressant imipramine. We found that pMARCKS levels were significantly increased in the membrane fraction of prefrontal cortex up to 181%, which is almost identical to the result obtained through treatment with amphetamine or sleep-deprivation (Figure 4). The total MARCKS membrane fraction was also increased, suggesting increased translocation of MARCKS after PKC phosphorylation (Figure 4). In addition, the PKC activity in membrane and cytosol fractions from prefrontal cortex of imipramine-treated animals were also significantly increased by 154% and 124% respectively, confirming the finding in phosphorylation of MARCKS (data not shown).
Figure 4. The antidepressant imipramine significantly enhanced pMARCKS in membrane fraction in prefrontal cortex.

Western blot analysis was performed to determine the pMARCKS or MARCKS in the membrane fractions (N=4-8 animals per group, Student’s t-test, *p<0.05). Data are presented as mean±SE.
PKC phosphorylation of MARCKS by lithium
Lithium has been used as an anti-manic agent for more than 50 years. We thus used lithium to investigate how anti-manic agents affect PKC activity in prefrontal cortex. We found that, in animals chronically treated with lithium for four weeks, pMARCKS levels were significantly decreased in the membrane fraction from the prefrontal cortex by 33% (Figure 5). The total membrane MARCKS levels showed a non-significant trend to decrease (Figure 5).
Figure 5. Lithium (Li) significantly attenuated PKC phosphorylation of MARCKS.
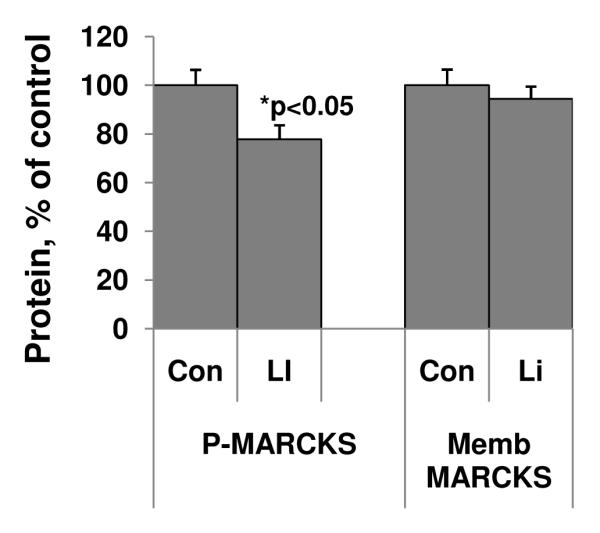
C57/B6 mice were treated with lithium for four weeks. Western blot analysis was performed to determine the pMARCKS and MARCKS in the membrane fractions (N=14, Student’s t-test, *p<0.05). Data are presented as mean±SE.
PKC phosphorylation of presynaptic neurogranin as a target in manic related animal models
Neurogranin is a key postsynaptic protein that regulates long-term potentiation (LTP). We therefore investigated the biochemical effects of the psychostimulant amphetamine and sleepdeprivation on neurogranin levels. The pro-manic effects of these treatments are generally observed in a similar time-frame to that used in the biochemical studies. Chronic administration of the psychostimulant amphetamine or sleep deprivation in animals significantly increased levels of phosphorylated neurogranin in the frontal cortex up to 199% and 173%, respectively, as compared to control animals (Figure 6A). We also examined the phosphorylation of neurogranin in the imipramine-treated animal model. Phosphorylation of neurogranin was 189%, higher in these animals compared with the control group (Figure 6A). Total neurogranin levels in amphetamine-treated, sleep deprived, or imipramine-treated animals remained unchanged (Figure 6B).
Figure 6. The presynaptic protein neurogranin is a target of PKC in pro- and anti-manic animal models.

(A) Phosphorylation of neurogranin of prefrontal cortex from mania related animal models.
Western-blot analysis of phosphorylated and non-phosphorylated neurogranin levels in crude homogenates from the frontal cortex of animals undergoing sustained treatment with amphetamine (AMPH), sleep deprivation (SD), imipramine (IMI), and lithium (Li).
(B) Lack of effect on total neurogranin levels in the frontal cortex in all animal models.
For all experimental groups, N=6-8, Student’s t-test, *p<0.05. Data are presented as mean±SE.
To determine if levels of neurogranin were also altered by anti-manic agents, chronic treatment with lithium was undertaken. As predicted, and in marked contrast to the effects observed with amphetamine or imipramine, chronic lithium reduced phosphorylated levels of neurogranin in the frontal cortex by 40% (Figure 6A). Total neurogranin levels after lithium treatment remained unchanged (Figure 6B).
PKC phosphorylation of NMDA NR1 receptors as a functional regulator in pro-manic or anti-manic animal models
Recent work has shown that AMPA/NMDA receptor throughput may contribute to the pathophysiology and treatment of mood disorders (Du et al., 2004b; Zarate and Manji, 2008). Therefore, we also assessed PKC phosphorylation of NMDA receptors at the NR1S896 site, which is involved in NR1 trafficking from the endoplasmic reticulum (ER) to the membrane of functional synapses (Sanchez-Perez and Felipo, 2005). In sleep-deprived animals, phosphorylation of NR1S896 was also significantly higher (162%; Figure 7A). However, although the psychostimulant amphetamine showed a trend to increase the phosphorylation of NR1S896, this increase did not reach statistical significance. Interestingly, total NR1 levels were decreased after amphetamine treatment (Figure 7B). NMDA NR1S896 receptor phosphorylation was significantly enhanced by the antidepressant imipramine up to 128%. In contrast, chronic treatment with lithium inhibited the phosphorylation of NR1S896 by 44.4% (Figure 7A). Total NR1 changes were not significant for sleep-deprived, imipramine-treated and lithium-treated groups (Figure 7B).
Figure 7. Phosphorylation of NMDA receptor NR1S896 at PKC site in mania-associated animal models.

Total protein samples from the prefrontal cortex of amphetamine (AMPH)-, imipramine (IMI)-, or lithium (Li)- treated animals or sleep-deprived (SD) animals were subjected to Western blot analysis with anti-phospho-NR1S896 or anti-NR1 antibodies. Data are presented as mean±SE (**p<0.01 Student’s t-test. N=8-20 for each group).
(A) Phospho-NR1S896 levels in mania related animal models..
(B) Total NR-1 levels in mania related animal models..
PKC phosphorylation of AMPA receptors at region GluA1T840 and GluAS831as a molecular target in pro- and anti-manic animal models
AMPA receptors have been implicated in the treatment of BPD (Du et al., 2008; Du et al., 2004c; Du et al., 2007). We therefore investigated the phosphorylation of GluA1T840 (PKC site) and GluA1S831 (PKC /CAMKII site) in these animal models of mania. Treatment with the psychostimulant amphetamine, the antidepressant imipramine, and sleep deprivation all significantly enhanced phosphorylation of GluA1 at T840 up to 132%, 129 %, and 122%, respectively (Figure 8A). In contrast, levels of AMPA receptor GluA1T840 (a PKC site) were decreased in lithium-treated animals by 38.4% (Figure 8A). Similarly, GluA1S831 (a CAMKII/PKC site) was significantly increased in amphetamine-treated, and imipramine-treated animals to168% and 147%, respectively (Figure 8B). Consistent with previous study (Vyazovskiy et al., 2008), phosphorylation of GluA1S831 had a very strong trend to increase by 150% in sleep-deprived animals (N=8 for each group, student’s t test, p=0.08, Figure 8B). However, lithium treatment did not significantly affect on GluA1S831, which is also consistent with previous finding (Du et al., 2004a). Changes in total GluA1 levels were not significant in any of the experimental groups (Figure 8C).
Figure 8. AMPA GluA1T840 and GluAS831 phosphorylation regulated by PKC in mania-associated animal models.

Total protein samples from the prefrontal cortex of sleep deprived animals or amphetamine (AMPH)-, imipramine (IMI)-, or lithium (Li)-treated animals were subjected to Western blot analysis with anti-phospho-GluA1T840, anti-phospho-GluA1S831, or anti-GluA1 antibodies. Data are presented as mean±SE (*p<0.05 Student’s t-test. N=8 animals for each group).
(A) Phospho-GluA1T840 levels in mania-related animal models.
(B) Phospho-GluA1S831 levels in mania related animal models
(C) Total GluA1 levels in mania related animal models.
Discussion
Accumulating clinical, preclinical, and genetic data have implicated overactive PKC intracellular signaling in mania (Einat et al., 2004). However, the functional targets of PKC have not been identified. We therefore undertook this series of studies to ascertain whether PKC modulation of these critical substrates is involved in the pathophysiology and treatment of manic-like behaviors. We found that 1) PKC activity in the prefrontal cortex was enhanced by treatment with the psychostimulant amphetamine; 2) phosphorylation of MARCKS, another indicator for PKC activity in vivo, was increased in the prefrontal cortex in two animal models of mania: amphetamine treatment and sleep deprivation; MARCKS phosphorylation was decreased in animals treated with the anti-manic agent lithium; 3) phosphorylation of neurogranin, a selective postsynaptic PKC molecular target, was similarly increased in these two animal models of mania and reduced after treatment with lithium; and 4) phosphorylation of the NMDA receptor NR1S896 and GluA1T840 was enhanced in sleep deprived animals as well as those treated with the antidepressant imipramine; treatment with lithium reduced the phosphorylation.
PKC is implicated in the pathophysiology of BPD in humans
Previous studies (Wang and Friedman, 1996) reported increased PKC activity and translocation in postmortem brains of individuals diagnosed with BPD compared to controls; these effects were accompanied by elevated levels of selected PKC isozymes in the cortex of BPD subjects. It has also been shown that mood stabilizers decreased PKC in the platelets of patients with BPD (Pandey et al., 2007). Moreover, recent human genetic evidence has also identified polymorphisms that confer susceptibility to BPD in the gene encoding for DGKH (Baum et al., 2008), which regulates the formation of diacyl glycerol (an activator of PKC), IP3, and phospholipase C (the initial enzyme in the PKC cascade) (Turecki et al., 1998). In addition, a recent preclinical study from our group found that tamoxifen (a non-steroidal anti-estrogen and known PKC inhibitor (Baltuch et al., 1993)) significantly reduced amphetamine-induced hyperactivity in a large open field and attenuated amphetamine-induced phosphorylation of GAP-43 at the PKC site (Einat et al., 2004; Einat et al., 2007). Three independent clinical trials also found that tamoxifen significantly improved the symptoms of mania compared to placebo (Bebchuk et al., 2000; Yildiz et al., 2008; Zarate et al., 2007).
Overactive PKC signaling in the prefrontal cortex may explain many of the symptoms of mania (Arnsten and Manji, 2008). Functional imaging studies have shown reduced activity in the right prefrontal cortex during mania (Blumberg et al., 2003; Blumberg et al., 1999). In addition, structural imaging studies have further shown a loss of prefrontal volume in untreated patients with BPD (Blumberg et al., 2006).
Antidepressant agents (particularly tricyclic antidepressant agents) can trigger manic episodes in individuals with BPD (Boerlin et al., 1998; Goldberg et al., 1999). However, whether or not PKC activity is decreased in patients with depression or in animal models of depression remains unclear. Panday and colleagues found that MARCKS phosphorylation was significantly decreased in the membrane fraction of prefrontal cortex and hippocampus obtained from both depressed and non-depressed suicide subjects (Pandey et al., 2003; Pandey et al., 2004). However, in another study, PKC activity was increased in the platelets from drug-free patients with depression (Pandey et al., 1998). It is thus possible that this enhancement in PKC activity may contribute to the pro-manic effects of imipramine.
PKC is a target for the anti-manic agents lithium and VPA
Consistent evidence exists that lithium, at therapeutically relevant concentrations, exerts major effects on the PKC signaling cascade. Studies in rodents have demonstrated that chronic (but not acute) lithium treatment produces an isozyme-selective reduction in PKC and ε in the frontal cortex and hippocampus (Chen et al., 2000b; Li et al., 1993; Manji et al., 1993; Manji and Lenox, 1999). Furthermore, chronic lithium treatment has been shown to dramatically reduce the hippocampal levels of the protein MARCKS, which is one of the PKC substrates implicated in regulating long-term neuroplastic events (Lenox et al., 1992). Notably, the anti-manic agent VPA, which is structurally dissimilar to lithium, produces very similar effects to those of lithium on PKC α and ε isozymes and on MARCKS, one of the PKC substrates (Chen et al., 1994; Lenox and Hahn, 2000; Manji and Chen, 2000; Manji and Lenox, 1999, 2000; Watson et al., 1998).
PKC activity in animal models of mania
In this study, we used two animal models of mania. Several independent laboratories have demonstrated that both acute and chronic amphetamine treatment alter PKC activity, PKC’s relative cytosol to membrane distribution, and phosphorylation of GAP-43, which has been implicated in long-term alterations of presynaptic neurotransmitter release (Giambalvo, 1992a, b; Gnegy, 1993; Iwata et al., 1997). Sleep deprivation is another animal model of mania, and its effects can be prevented by lithium (Gessa et al., 1995). In synaptoneurosomes of rat cortex and hippocampus, GluA1-containing AMPA receptor levels and phosphorylation of GluA1S845 are high during wakefulness and low during sleep, suggesting a regulation of glutamate receptor function through phosphorylation (Vyazovskiy et al., 2008). In addition, in that study the sleep deprived group showed a nearly 50% increase in total GluA1 levels in synaptoneurosomes compared with the waking group (Vyazovskiy et al., 2008). The present study focused on the PKC targets that regulate synaptic plasticity and synaptic transmission. We found that PKC-phosphorylation of three important synaptic targets—phospho-neurogranin, phospho-NMDA, and phospho-AMPA—were related to manic behavior in four pro- and anti-manic animal models. Taken together, the evidence suggests that PKC phosphorylation may contribute to the pathophysiology of mania and the formation of manic behaviors.
The PKC target neurogranin is an important molecule regulating synaptic function
Neurogranin is a calcium-sensitive CaM-binding protein, whose CaM-binding affinity is attenuated by oxidation and phosphorylation by PKC (Giambalvo and Price, 2003; Huang et al., 2000). Binding of neurogranin to CaM can be restored by dephosphorylation of neurogranin with protein phosphatase I (PP1), protein phosphotase 2A (PP2A), and by the calcium-CaM dependent protein phosphatase 2B (Li et al., 2003). It is not only a major PKC target, but also an upstream regulator of three major signalling cascades—calcium/calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA), and PKC—that are thought to play important roles in the pathophysiology and treatment of severe mood disorders (Barbiero et al., 2007; Coyle and Duman, 2003; Manji and Chen, 2000; Popoli et al., 2001). Under manic conditions, increased phosphorylation of neurogranin by PKC (Figure 3) liberates calmodulin towards activation of CAMKII and CAMKIV, which is the key for the formation of LTP. However, we postulate that lithium treatment increases the binding of neurogranin with calmodulin and leads to lower activation of CaMKII.
Regulation of the NMDA receptor NR1S896 is related to the trafficking of NR1 to the synapses
Accumulating data have shown that the glutamatergic system is involved in the pathophysiology and treatment of mood disorders (Maeng and Zarate, 2007; Zarate et al., 2006). The NMDA receptor is present in the brain primarily as a tetramer, in which two subunits are obligatory glycine-binding NR1 subunits, while the remaining two are typically glutamate-binding NR2 subunits. NMDA receptors are targets for PKC and PKC-dependent tyrosine kinase cascades. Direct phosphorylation of NMDA receptors by PKC increases the sensitivity of the NMDA receptor to inactivation by intracellular calcium. In the hippocampus, PKC is a point of convergence for a variety of G-protein coupled and growth factor receptors that maintain control over NMDA receptor function and synaptic plasticity (McDonald et al., 2001). The phosphorylation of NR1 at S896 has been reported to be critical for NR1 receptor transportation to the synapses (Sanchez-Perez and Felipo, 2005). Therefore, enhanced NR1S896 phosphorylation in the prefrontal cortex would lead to the efficient transportation of NR1 to the synapses in this brain region.
AMPA GluA1T840 phosphorylation is significantly increased in animals treated with imipramine and sleep deprived animals, and decreased in animals treated with lithium
The AMPA subtypes of glutamate receptors appear to play a major role in regulating short- and long-term forms of synaptic plasticity (Lee, 2006). Regulation of GluA1 trafficking via phosphorylation of its PKA site (S845) has been shown to be important for the treatment of manic-like behaviors (Du et al., 2008; Du et al., 2004a). Notably, this trafficking is now known to depend, in large part, upon AMPA receptor subunit phosphorylation at serine, threonine, or tyrosine residues located on the intracellular C-terminal domain (reviewed in (Du et al., 2004a). Protein kinase signaling cascades, such as PKA, PKC, CaMKII, and tyrosine kinases (Trks; or Src Family Kinases) are involved in site-specific AMPA receptor phosphorylation (Banke et al., 2000; Barria et al., 1997; Boehm et al., 2006; Roche et al., 1996). Other glutamate (NMDA receptors) and G-protein coupled receptors (mGluRs, Dopamine Receptors DA1, serotonin receptors) can also regulate AMPA receptors through protein phosphorylation and/or dephosphorylation mechanisms (Du et al., 2004a; Zarate et al., 2003). A threonine-840 (T840) on the GluA1 subunit of AMPA receptors was identified as a novel phosphorylation site by PKC in vitro and is functionally relevant to synaptic plasticity (Lee et al., 2007). Here, we identified GluA1T840 as a target of PKC, conferring its effects on mood-associated behaviors (Figure 7).
The animal models of mania used here—amphetamine-treated and sleep-deprived animals—are animal models of acute mania. During the occurrence of acute manic-like behaviors, we propose that there is increased neuronal activity. The phosphorylation of MARCKS, neurogranin, NMDA, and GluA1 should result in increased neuroplasticity, which may contribute to the pathophysiology of the manic episode.
In summary, biochemical data presented here indicate that abnormalities in the frontal cortical PKC signaling cascade may play an important role in the pathophysiology and treatment of BPD. Accumulating preclinical and clinical data suggest that PKC inhibitors may be potential therapies for BPD (Einat et al., 2007; Yildiz et al., 2008; Zarate et al., 2007). This work elucidates the mechanisms whereby PKC activation triggers and amplifies the functional cascade of many target proteins—including NMDA receptors, AMPA receptors, neurogranin, and MARCKS—to regulate pre- and post-synaptic functions in prefrontal cortex. These results suggest that modulation of either PKC or its functional targets may lead to the development of novel therapeutics for mood disorders.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health, and by NIMH (P20 MH077967 to CC) and NIH Director’s Pioneer award (to GT). We thank Ioline Henter for outstanding editorial assistance.
Footnotes
Conflict of Interest We would like to acknowledge the support of the Intramural Research Program of the National Institute of Mental Health. The author(s) declare that, except for income received from our primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnsten AFT, Manji HK. Mania: a rational neurobiology. Future Medicine. 2008;3:125–131. [Google Scholar]
- Baltuch GH, Couldwell WT, Villemure JG, Yong VW. Protein kinase C inhibitors suppress cell growth in established and low-passage glioma cell lines. A comparison between staurosporine and tamoxifen. Neurosurgery. 1993;33:495–501. doi: 10.1227/00006123-199309000-00021. discussion 501. [DOI] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Am O, Yogev-Falach M, Amit T, Sagi Y, Youdim MB. Regulation of protein kinase C by the anti-Parkinson drug, MAO-B inhibitor, rasagiline and its derivatives, in vivo. J Neurochem. 2004;89:1119–1125. doi: 10.1111/j.1471-4159.2004.02425.x. [DOI] [PubMed] [Google Scholar]
- Barbiero VS, Giambelli R, Musazzi L, Tiraboschi E, Tardito D, Perez J, Drago F, Racagni G, Popoli M. Chronic antidepressants induce redistribution and differential activation of alphaCaM kinase II between presynaptic compartments. Neuropsychopharmacology. 2007;32:2511–2519. doi: 10.1038/sj.npp.1301378. [DOI] [PubMed] [Google Scholar]
- Barbon A, Popoli M, La Via L, Moraschi S, Vallini I, Tardito D, Tiraboschi E, Musazzi L, Giambelli R, Gennarelli M, Racagni G, Barlati S. Regulation of editing and expression of glutamate alpha-amino-propionic-acid (AMPA)/kainate receptors by antidepressant drugs. Biol Psychiatry. 2006;59:713–720. doi: 10.1016/j.biopsych.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, Schulze TG, Cichon S, Rietschel M, Nothen MM, Georgi A, Schumacher J, Schwarz M, Jamra R. Abou, Hofels S, Propping P, Satagopan J, Detera-Wadleigh SD, Hardy J, McMahon FJ. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2008;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebchuk JM, Arfken CL, Dolan-Manji S, Murphy J, Hasanat K, Manji HK. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry. 2000;57:95–97. doi: 10.1001/archpsyc.57.1.95. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Yuan PX, Wang M, Vijayraghavan S, Bloom AK, Davis DJ, Gobeske KT, Sweatt JD, Manji HK, Arnsten AF. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 2004;306:882–824. doi: 10.1126/science.1100021. [DOI] [PubMed] [Google Scholar]
- Blumberg HP, Krystal JH, Bansal R, Martin A, Dziura J, Durkin K, Martin L, Gerard E, Charney DS, Peterson BS. Age, rapid-cycling, and pharmacotherapy effects on ventral prefrontal cortex in bipolar disorder: a cross-sectional study. Biol Psychiatry. 2006;59:611–618. doi: 10.1016/j.biopsych.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Blumberg HP, Leung HC, Skudlarski P, Lacadie CM, Fredericks CA, Harris BC, Charney DS, Gore JC, Krystal JH, Peterson BS. A functional magnetic resonance imaging study of bipolar disorder: state- and trait-related dysfunction in ventral prefrontal cortices. Arch Gen Psychiatry. 2003;60:601–609. doi: 10.1001/archpsyc.60.6.601. [DOI] [PubMed] [Google Scholar]
- Blumberg HP, Stern E, Ricketts S, Martinez D, de Asis J, White T, Epstein J, Isenberg N, McBride PA, Kemperman I, Emmerich S, Dhawan V, Eidelberg D, Kocsis JH, Silbersweig DA. Rostral and orbital prefrontal cortex dysfunction in the manic state of bipolar disorder. Am J Psychiatry. 1999;156:1986–1988. doi: 10.1176/ajp.156.12.1986. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Boerlin HL, Gitlin MJ, Zoellner LA, Hammen CL. Bipolar depression and antidepressant-induced mania: a naturalistic study. J Clin Psychiatry. 1998;59:374–379. doi: 10.4088/jcp.v59n0706. [DOI] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Nestler EJ. Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci. 2002;25:610–615. doi: 10.1016/s0166-2236(02)02289-0. [DOI] [PubMed] [Google Scholar]
- Chao SZ, Ariano MA, Peterson DA, Wolf ME. D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem. 2002;83:704–712. doi: 10.1046/j.1471-4159.2002.01164.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Manji HK, Hawver DB, Wright CB, Potter WZ. Chronic sodium valproate selectively decreases protein kinase C alpha and epsilon in vitro. J Neurochem. 1994;63:2361–2364. doi: 10.1046/j.1471-4159.1994.63062361.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Masana MI, Manji HK. Lithium regulates PKC-mediated intracellular cross-talk and gene expression in the CNS in vivo. Bipolar Disord. 2000a;2:217–236. doi: 10.1034/j.1399-5618.2000.20303.x. [DOI] [PubMed] [Google Scholar]
- Chen RH, Ding WV, McCormick F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J Biol Chem. 2000b;275:17894–17899. doi: 10.1074/jbc.M905336199. [DOI] [PubMed] [Google Scholar]
- Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, Ferrell RE, Middleton FA, Devlin B, Levitt P, Lewis DA, Nimgaonkar VL. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002;11:1373–1380. doi: 10.1093/hmg/11.12.1373. [DOI] [PubMed] [Google Scholar]
- Clark L, Manes F, Antoun N, Sahakian BJ, Robbins TW. The contributions of lesion laterality and lesion volume to decision-making impairment following frontal lobe damage. Neuropsychologia. 2003;41:1474–1483. doi: 10.1016/s0028-3932(03)00081-2. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Duman RS. Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron. 2003;38:157–160. doi: 10.1016/s0896-6273(03)00195-8. [DOI] [PubMed] [Google Scholar]
- Du J, Creson TK, Wu LJ, Ren M, Gray NA, Falke C, Wei Y, Wang Y, Blumenthal R, Machado-Vieira R, Yuan P, Chen G, Zhuo M, Manji HK. The role of hippocampal GluR1 and GluR2 receptors in manic-like behavior. J Neurosci. 2008;28:68–79. doi: 10.1523/JNEUROSCI.3080-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Gray NA, Falke C, Yuan P, Szabo S, Manji HK. Structurally dissimilar antimanic agents modulate synaptic plasticity by regulating AMPA glutamate receptor subunit GluR1 synaptic expression. Ann N Y Acad Sci. 2003;1003:378–380. doi: 10.1196/annals.1300.031. [DOI] [PubMed] [Google Scholar]
- Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST, Einat H, Manji HK. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004a;24:6578–6589. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Quiroz J, Yuan P, Zarate C, Manji HK. Bipolar disorder: involvement of signaling cascades and AMPA receptor trafficking at synapses. Neuron Glia Biol. 2004b;1:231–243. doi: 10.1017/S1740925X05000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Quiroz J, Yuan P, Zarate CAJ, Manji HK. Bipolar disorder: involment of signaling cascades and AMPA receptor trafficking at synapses. Neuron Glia Biolw. 2004c;1:231–243. doi: 10.1017/S1740925X05000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z, Falke C, Zarate CA, Manji HK. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32:793–802. doi: 10.1038/sj.npp.1301178. [DOI] [PubMed] [Google Scholar]
- Einat H, Chen G, Manji H. [Possible involvement of protein kinase C in the pathophysiology and treatment of bipolar disorder] Harefuah. 2004;143:420–425. 462. [PubMed] [Google Scholar]
- Einat H, Yuan P, Szabo ST, Dogra S, Manji HK. Protein kinase C inhibition by tamoxifen antagonizes manic-like behavior in rats: implications for the development of novel therapeutics for bipolar disorder. Neuropsychobiology. 2007;55:123–131. doi: 10.1159/000106054. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Pani L, Fadda P, Fratta W. Sleep deprivation in the rat: an animal model of mania. Eur Neuropsychopharmacol. 1995;5(Suppl):89–93. doi: 10.1016/0924-977x(95)00023-i. [DOI] [PubMed] [Google Scholar]
- Giambalvo CT. Protein kinase C and dopamine transport--1. Effects of amphetamine in vivo. Neuropharmacology. 1992a;31:1201–1210. doi: 10.1016/0028-3908(92)90048-t. [DOI] [PubMed] [Google Scholar]
- Giambalvo CT. Protein kinase C and dopamine transport--2. Effects of amphetamine in vitro. Neuropharmacology. 1992b;31:1211–1222. doi: 10.1016/0028-3908(92)90049-u. [DOI] [PubMed] [Google Scholar]
- Giambalvo CT, Price LH. Effects of fenfluramine and antidepressants on protein kinase C activity in rat cortical synaptoneurosomes. Synapse. 2003;50:212–222. doi: 10.1002/syn.10262. [DOI] [PubMed] [Google Scholar]
- Gnegy ME. Calmodulin in neurotransmitter and hormone action. Annu Rev Pharmacol Toxicol. 1993;33:45–70. doi: 10.1146/annurev.pa.33.040193.000401. [DOI] [PubMed] [Google Scholar]
- Goldberg JF, Garno JL, Leon AC, Kocsis JH, Portera L. A history of substance abuse complicates remission from acute mania in bipolar disorder. J Clin Psychiatry. 1999;60:733–740. doi: 10.4088/jcp.v60n1103. [DOI] [PubMed] [Google Scholar]
- Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar and Recurrent Unipolar Disorders. Oxford University Press; New York: 2007. [Google Scholar]
- Hahn CG, Friedman E. Abnormalities in protein kinase C signaling and the pathophysiology of bipolar disorder. Bipolar Disord. 1999;1:81–86. doi: 10.1034/j.1399-5618.1999.010204.x. [DOI] [PubMed] [Google Scholar]
- Huang KP. Role of protein kinase C in cellular regulation. Biofactors. 1990;2:171–178. [PubMed] [Google Scholar]
- Huang KP, Huang FL, Li J, Schuck P, McPhie P. Calcium-sensitive interaction between calmodulin and modified forms of rat brain neurogranin/RC3. Biochemistry. 2000;39:7291–7299. doi: 10.1021/bi000336l. [DOI] [PubMed] [Google Scholar]
- Iwata S, Hewlett GH, Gnegy ME. Amphetamine increases the phosphorylation of neuromodulin and synapsin I in rat striatal synaptosomes. Synapse. 1997;26:281–291. doi: 10.1002/(SICI)1098-2396(199707)26:3<281::AID-SYN9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Jope RS, Song L, Li PP, Young LT, Kish SJ, Pacheco MA, Warsh JJ. The phosphoinositide signal transduction system is impaired in bipolar affective disorder brain. J Neurochem. 1996;66:2402–2409. doi: 10.1046/j.1471-4159.1996.66062402.x. [DOI] [PubMed] [Google Scholar]
- Judd LL, Akiskal HS. The prevalence and disability of bipolar spectrum disorders in the US population: re-analysis of the ECA database taking into account subthreshold cases. J Affect Disord. 2003;73:123–131. doi: 10.1016/s0165-0327(02)00332-4. [DOI] [PubMed] [Google Scholar]
- Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr. 2005;10:808–819. doi: 10.1017/s1092852900010403. [DOI] [PubMed] [Google Scholar]
- Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther. 2006;112:810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Kameyama K, He K, Yu S, Rossetti L, Wilen D, Huganir RL. Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol Cell Neurosci. 2007;36:86–94. doi: 10.1016/j.mcn.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenox RH, Hahn CG. Overview of the mechanism of action of lithium in the brain: fifty-year update. J Clin Psychiatry. 2000;61(Suppl 9):5–15. [PubMed] [Google Scholar]
- Lenox RH, McNamara RK, Watterson JM, Watson DG. Myristoylated alanine-rich C kinase substrate (MARCKS): a molecular target for the therapeutic action of mood stabilizers in the brain? J Clin Psychiatry. 1996;57(Suppl 13):23–31. discussion 32-23. [PubMed] [Google Scholar]
- Lenox RH, Watson DG, Patel J, Ellis J. Chronic lithium administration alters a prominent PKC substrate in rat hippocampus. Brain Res. 1992;570:333–340. doi: 10.1016/0006-8993(92)90598-4. [DOI] [PubMed] [Google Scholar]
- Li HY, Li JF, Lu GW. [Neurogranin: a brain-specific protein] Sheng Li Ke Xue Jin Zhan. 2003;34:111–115. [PubMed] [Google Scholar]
- Li PP, Sibony D, Green MA, Warsh JJ. Lithium modulation of phosphoinositide signaling system in rat cortex: selective effect on phorbol ester binding. J Neurochem. 1993;61:1722–1730. doi: 10.1111/j.1471-4159.1993.tb09809.x. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Kotecha SA, Lu WY, Jackson MF. Convergence of PKC-dependent kinase signal cascades on NMDA receptors. Curr Drug Targets. 2001;2:299–312. doi: 10.2174/1389450013348452. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA., Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9:467–474. doi: 10.1007/s11920-007-0063-1. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Manji HK, Chen G. Post-receptor signaling pathways in the pathophysiology and treatment of mood disorders. Curr Psychiatry Rep. 2000;2:479–489. doi: 10.1007/s11920-000-0006-6. [DOI] [PubMed] [Google Scholar]
- Manji HK, Etcheberrigaray R, Chen G, Olds JL. Lithium decreases membrane-associated protein kinase C in hippocampus: selectivity for the alpha isozyme. J Neurochem. 1993;61:2303–2310. doi: 10.1111/j.1471-4159.1993.tb07474.x. [DOI] [PubMed] [Google Scholar]
- Manji HK, Lenox RH. Ziskind-Somerfeld Research Award. Protein kinase C signaling in the brain: molecular transduction of mood stabilization in the treatment of manic-depressive illness. Biol Psychiatry. 1999;46:1328–1351. doi: 10.1016/s0006-3223(99)00235-8. [DOI] [PubMed] [Google Scholar]
- Manji HK, Lenox RH. Signaling: cellular insights into the pathophysiology of bipolar disorder. Biol Psychiatry. 2000;48:518–530. doi: 10.1016/s0006-3223(00)00929-x. [DOI] [PubMed] [Google Scholar]
- Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, N AG, Zarate CA, Jr., Charney DS. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry. 2003;53:707–742. doi: 10.1016/s0006-3223(03)00117-3. [DOI] [PubMed] [Google Scholar]
- Mathews R, Li PP, Young LT, Kish SJ, Warsh JJ. Increased G alpha q/11 immunoreactivity in postmortem occipital cortex from patients with bipolar affective disorder. Biol Psychiatry. 1997;41:649–656. doi: 10.1016/S0006-3223(96)00113-8. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Chung HJ, Huganir RL. Identification of protein kinase C phosphorylation sites within the AMPA receptor GluR2 subunit. Neuropharmacology. 2001;41:672–679. doi: 10.1016/s0028-3908(01)00129-0. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Ni YG, Gold SJ, Iredale PA, Terwilliger RZ, Duman RS, Nestler EJ. Region-specific regulation of RGS4 (Regulator of G-protein-signaling protein type 4) in brain by stress and glucocorticoids: in vivo and in vitro studies. J Neuroscience. 1999;19:3674–3680. doi: 10.1523/JNEUROSCI.19-10-03674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogues X. Protein kinase C, learning and memory: a circular determinism between physiology and behaviour. Prog Neuropsychopharmacol Biol Psychiatry. 1997;21:507–529. doi: 10.1016/s0278-5846(97)00015-8. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Dwivedi Y, Kumari R, Janicak PG. Protein kinase C in platelets of depressed patients. Biol Psychiatry. 1998;44:909–911. doi: 10.1016/s0006-3223(97)00535-0. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Dwivedi Y, Ren X, Rizavi HS, Roberts RC, Conley RR. Cyclic AMP response element-binding protein in post-mortem brain of teenage suicide victims: specific decrease in the prefrontal cortex but not the hippocampus. Int J Neuropsychopharmacol. 2007;10:621–629. doi: 10.1017/S1461145706007231. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Dwivedi Y, Ren X, Rizavi HS, Roberts RC, Conley RR, Tamminga C. Altered expression and phosphorylation of myristoylated alanine-rich C kinase substrate (MARCKS) in postmortem brain of suicide victims with or without depression. J Psychiatr Res. 2003;37:421–432. doi: 10.1016/s0022-3956(03)00047-5. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Dwivedi Y, Rizavi HS, Ren X, Conley RR. Decreased catalytic activity and expression of protein kinase C isozymes in teenage suicide victims: a postmortem brain study. Arch Gen Psychiatry. 2004;61:685–693. doi: 10.1001/archpsyc.61.7.685. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Y.T. W, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Raiteri L, Longordo F, Luccini E, Barbiero VS, Racagni G, Popoli M, Raiteri M. Antidepressant treatments and function of glutamate ionotropic receptors mediating amine release in hippocampus. Neuropharmacology. 2007;53:27–36. doi: 10.1016/j.neuropharm.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Popoli M, Mori S, Brunello N, Perez J, Gennarelli M, Racagni G. Serine/threonine kinases as molecular targets of antidepressants: implications for pharmacological treatment and pathophysiology of affective disorders. Pharmacol Ther. 2001;89:149–170. doi: 10.1016/s0163-7258(00)00108-x. [DOI] [PubMed] [Google Scholar]
- Ramakers GM, Pasinelli P, Hens JJ, Gispen WH, De Graan PN. Protein kinase C in synaptic plasticity: changes in the in situ phosphorylation state of identified pre- and postsynaptic substrates. Prog Neuropsychopharmacol Biol Psychiatry. 1997;21:455–486. doi: 10.1016/s0278-5846(97)00013-4. [DOI] [PubMed] [Google Scholar]
- Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Perez AM, Felipo V. Serines 890 and 896 of the NMDA receptor subunit NR1 are differentially phosphorylated by protein kinase C isoforms. Neurochem Int. 2005;47:84–91. doi: 10.1016/j.neuint.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Self DW. Where’s the excitement in psychostimulant sensitization? Neuropsychopharmacology. 2002;26:14–17. doi: 10.1016/S0893-133X(01)00373-6. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecki G, Grof P, Cavazzoni P, Duffy A, Grof E, Ahrens B, Berghofer A, Muller-Oerlinghausen B, Dvorakova M, Libigerova E, Vojtechovsky M, Zvolsky P, Joober R, Nilsson A, Prochazka H, Licht RW, Rasmussen NA, Schou M, Vestergaard P, Holzinger A, Schumann C, Thau K, Rouleau GA, Alda M. Evidence for a role of phospholipase C-gamma1 in the pathogenesis of bipolar disorder. Mol Psychiatry. 1998;3:534–538. doi: 10.1038/sj.mp.4000447. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Wang HY, Friedman E. Enhanced protein kinase C activity and translocation in bipolar affective disorder brains. Biol Psychiatry. 1996;40:568–575. doi: 10.1016/0006-3223(95)00611-7. [DOI] [PubMed] [Google Scholar]
- Watson DG, Watterson JM, Lenox RH. Sodium valproate down-regulates the myristoylated alanine-rich C kinase substrate (MARCKS) in immortalized hippocampal cells: a property of protein kinase C-mediated mood stabilizers. J Pharmacol Exp Ther. 1998;285:307–316. [PubMed] [Google Scholar]
- Wilkins AJ, Shallice T, McCarthy R. Frontal lesions and sustained attention. Neuropsychologia. 1987;25:359–365. doi: 10.1016/0028-3932(87)90024-8. [DOI] [PubMed] [Google Scholar]
- Woods DL, Knight RT. Electrophysiologic evidence of increased distractibility after dorsolateral prefrontal lesions. Neurology. 1986;36:212–216. doi: 10.1212/wnl.36.2.212. [DOI] [PubMed] [Google Scholar]
- Wu J, Li J, Huang KP, Huang FL. Attenuation of protein kinase C and cAMP-dependent protein kinase signal transduction in the neurogranin knockout mouse. J Biol Chem. 2002;277:19498–19505. doi: 10.1074/jbc.M109082200. [DOI] [PubMed] [Google Scholar]
- Yamauchi E, Kiyonami R, Kanai M, Taniguchi H. Presence of conserved domains in the C-terminus of MARCKS, a major in vivo substrate of protein kinase C: application of ion trap mass spectrometry to the elucidation of protein structures. J Biochem. 1998;123:760–765. doi: 10.1093/oxfordjournals.jbchem.a022002. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Guleryuz S, Ankerst DP, Ongur D, Renshaw PF. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008;65:255–263. doi: 10.1001/archgenpsychiatry.2007.43. [DOI] [PubMed] [Google Scholar]
- Young LT. Is bipolar disorder a mitochondrial disease? Journal of Psychiatry and Neuroscience. 2007;32:160–161. [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Chen G, Manji HK. Lithium activates the c-Jun NH2-terminal kinases in vitro and in the CNS in vivo. J Neurochem. 1999;73:2299–2309. doi: 10.1046/j.1471-4159.1999.0732299.x. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Manji HK. The role of AMPA receptor modulation in the treatment of neuropsychiatric diseases. Exp Neurol. 2008;211:7–10. doi: 10.1016/j.expneurol.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr., Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry. 2006;59:1006–1020. doi: 10.1016/j.biopsych.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr., Singh JB, Carlson PJ, Quiroz J, Jolkovsky L, Luckenbaugh DA, Manji HK. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9:561–570. doi: 10.1111/j.1399-5618.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- Zarate CAJ, Du J, Quiroz J, Gray NA, Denicoff KD, Singh J, Charney D, Manji HK. Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann N Y Acad Sci. 2003;1003:273–291. doi: 10.1196/annals.1300.017. [DOI] [PubMed] [Google Scholar]