

Abstract
Metabolic profiling of urine presents challenges due to the extensive random variation of metabolite concentrations, and to dilution resulting from changes in the overall urine volume. Thus statistical analysis methods play a particularly important role, however appropriate choices of these methods are not straightforward. Here we investigate constant and variance-stabilization normalization of raw and peak picked spectra, for use with exploratory analysis (principal component analysis) and confirmatory analysis (ordinary and Empirical Bayes t-test) in 1H NMR-based metabolic profiling of urine. We compare the performance of these methods using urine samples spiked with known metabolites according to a Latin square design. We find that analysis of peak picked and log-transformed spectra is preferred, and that signal processing and statistical analysis steps are interdependent. While variance-stabilizing transformation is preferred in conjunction with principal component analysis, constant normalization is more appropriate for use with a t-test. Empirical Bayes t-test provides more reliable conclusions when the number of samples in each group is relatively small. Performance of these methods is illustrated using a clinical metabolomics experiment on patients with type 1 diabetes to evaluate the effect of insulin deprivation.
Keywords: Metabolomics, Metabolite profiling, NMR spectroscopy, Normalization, Moderated t-test, Logarithmic transformation, Urine, Diabetes
Metabolomics, as well as the related fields of metabolite profiling and metabonomics, is an indispensable complement to genomics and proteomics in studies of complex biological mechanisms,1, 2 and in discovery of biomarkers of disease.3, 4 Metabolic profiling of urine is particularly attractive because urine collection is non-invasive, and urine contains metabolic signatures of many biochemical pathways.5, 6 However, urine metabolic profiling is hindered by a great biological variability. Factors such as diseases, drugs, toxins, and diet can cause changes in individual metabolites as well as the overall urinary volume, which consequently affects the observed metabolite concentrations. Therefore, urine profiling is quite challenging, and requires robust statistical methods that distinguish true changes in concentrations from random variation.
Nuclear magnetic resonance (NMR) spectroscopy is highly reproducible, and is therefore a method of choice for studies of samples with high biological variability and strong dilution effects.7-9 Nevertheless, technological variations such as baseline distortion and peak shifting can be introduced due to instrument drifts, matrix effect (solvent, pH and ion strength), the existence of macro molecule signals, imperfections in water suppression, shimming and acquisition parameters, and sample handling prior to the measurements. These artifacts are exacerbated when spectra are acquired in multiple batches or labs. Thus, statistical methods are needed to account for both biological and technological variation.
The goal of this paper is to emphasize the importance of choosing particular statistical analysis methods for analysis of urine 1H NMR spectra. We clarify the use of the methods theoretically, evaluate their performance empirically using the experimental spike-in dataset, and illustrate their application on a clinical experiment.
Statistical methods for analysis of NMR spectra consist of (1) signal processing, specifically baseline correction, feature detection and quantification, and normalization and scaling, and (2) exploratory and confirmatory analysis of the quantified features. A variety of methods have been proposed, all making different assumptions about the properties of the data. When the assumptions are inappropriate, application of the methods will result in false discoveries and inaccurate interpretations10.
Signal processing: baseline correction
Baseline distortions can be corrected in frequency domain using methods such as polynomial fit,11 robust stepwise estimation procedure,12 and statistically motivated Lowess fit.13 The latter two improve the accuracy at the expense of computational complexity.
Signal processing: feature detection and quantification
This step can be approached from three different perspectives. The first retains the intensities of all features in the raw spectra, however, this yields a large number of redundant features, and quantitative comparisons can be negatively affected by horizontal peak shifts. The second approach involves binning, where the spectra are partitioned into bins of constant or adaptive width,14, 15 and the resulting features are quantified by integration of the signal in a bin. The approach partially accounts for the peak shifting by reducing spectral resolution;2 however it also leads to inaccurate peak quantification when neighboring peaks are assigned to the same bin, or when a same peak is split across bins. The last approach involves computational peak picking,16 combined with peak alignment17 that explicitly accounts for the peak shifting. The features are quantified by either peak area (often after smoothing), or by peak's apex. The approach can improve the accuracy of quantification, but is more sensitive to specific modeling assumptions and implementations.
Signal processing: normalization and scaling
The next step transforms feature abundances to ensure that they are comparable across runs, and possibly scales the abundances to make them comparable across features.2 All methods make assumptions regarding various aspects of the biological system, and the choice has a great impact on the subsequent results.2, 7, 18
The simplest strategy is constant normalization, i.e. division by a constant such as the total sum of intensity,2 or by the integral of a single or multiple peaks corresponding to internal standard compounds, e.g. creatinine.2, 19 Specifically, the total sum normalization assumes that the total dissolved metabolites are invariable, however this is not always the case in practice. Creatinine normalization, on the other hand, originates from clinical chemistry and uses the fact that creatinine clearance in an organism is fairly constant. However, urinary creatinine concentrations can vary significantly with reference to, e.g., age, gender and time of urine collection.20-22 Therefore, quantitative metabolite profiling relative to creatinine peak area may yield inaccurate results. A series of alternative signal-equalizing methods have been proposed, which include the minimum rank normalization,23 subspace time-domain normalization,24 probabilistic quotient normalization,25 and histogram matching normalization.26
Scaling is motivated by the fact that, similarly to the genomic27 and proteomic28 data, a sizable fraction of metabolomic NMR error is proportional to peak intensity.13, 29 However comparable variances are often required by subsequent methods such as principal component analysis (PCA). The discrepancy between peak variances can be partially alleviated by a logarithm transform, which acts as a global scaling procedure. Alternatively, scaling can be achieved by subtracting the mean of the log-transformed abundances from each feature, and by dividing by its standard deviation.
More recent approaches make an explicit use of the multiplicative structure of the data, and combine normalization and log-based scaling in a single model-based step. Examples are the generalized log transform developed for NMR-based metabolomics,30 and variance stabilizing normalization (VSN) originally developed for the analysis of gene expression microarrays.31, 32
Exploratory analysis
Exploratory analysis33 (e.g. PCA) aims at finding patterns in normalized data without using group labels. It detects samples with similar spectral characteristics in an unsupervised manner, and reduces the number of dimensions for visualization. The method assumes that the measurement error of all features is constant, and that the observed difference in variation is due to biological reasons. Therefore, the analysis will be negatively affected when some features have larger experimental noise.2, 34 Moreover, PCA lacks formal procedures of statistical inference, and cannot produce measures of classification accuracy such as sensitivity or specificity.
Confirmatory analysis
In contrast to exploratory analysis, confirmatory analysis makes an explicit use of group labels. Examples are per-feature Welch t-test which compares feature abundances between two groups of samples, and logistic regression which classifies samples based on the observed features. The methods produce formal statistical inference and measures of accuracy, and do not assume equal measurement error. However they require their own sets of assumptions. In particular, t-test assumes that measurement errors are normally distributed. The test is known to be robust to small deviations from Normality,35 but will produce inaccurate inference when the assumption is grossly violated.10 An alternative involves resampling inference procedures, which forgo the assumption of Normality at the expense of a diminished ability of finding true differences. Although resampling methods are more general they are not assumption-free, and selecting an appropriate procedure is not straightforward.36 Finally, when the number of samples in the experiment is small, inference based on the t-test is somewhat unstable. Empirical Bayes (or moderated) alternative to the t-test, originally developed for gene expression microarrays, combines the information regarding feature variability across all features, and improves both sensitivity and specificity of finding the true changes with small sample size.37
Evaluation
Given the diversity of assumptions made on signal processing and analysis methods, not all methods (either in isolation, or when used in combination) may be appropriate for NMR-based urine profiling. The methods are frequently evaluated using simulated or computer-edited data, however, such validations may not accurately represent real-life circumstances. We therefore designed a controlled experiment where metabolites were spiked in known concentrations into a background urine sample.38 Such spike-in experiments have been extensively used for calibration of signal processing and analysis of gene expression microarrays39 and mass spectrometry-based proteomics.40 Our experiment utilizes an efficient Latin square design,37 which allows one to compare the performance of statistical methods using a range of fold changes over a range of concentration baselines.
Methods
Chemicals
Sodium succinate dibasic, sodium fumarate dibasic, sodium 4-hydroxybenzoate, α-ketoglutaric acid sodium salt, 4-hydroxyphenylacetic acid, nicotinic acid sodium salt, and 3-(trimethylsilyl)propionic acid-d4 (TSP) sodium salt were from SigmaAldrich (St. Louis, MO, USA), and used without further purification.
Data Sets
The spike-in dataset is based on 6 samples made from the same urine sample and spiked with six compounds (spike-ins). The urine sample was collected from a healthy male volunteer. Six metabolites were spiked into urine according to the Latin square design (Table S1). The 6 samples were diluted using phosphate buffer (PH=7.4, final concentration of 0.2M) to prepare three sets of mixtures with three dilution levels : a set of 6 non-diluted samples, the same 6 samples diluted two-fold, and the same 6 samples diluted four-fold. Three replicates of each of the 18 samples were randomized, and used to acquire 54 NMR spectra.
1H NMR analysis was performed on a set of 7 urine samples from diabetic patients whose insulin treatment had been withdrawn for a period of 8 hours at Mayo Clinic CTSA Clinical Research Unit as previously reported41 along with 7 age and gender matched control urine samples from healthy individuals. The samples were collected at the Mayo Clinic medical center (Rochester, MN), and stored at -80 °C until they were shipped to Purdue over dry ice. The samples were again stored at -80 °C until they could be analyzed. All samples were collected, de-identified and analyzed in accordance with Internal Review Board approved procedures at both the Mayo Clinic and Purdue University. Spectral acquisition was performed without technical replication, and order of the samples was determined by randomization. This resulted in 14 NMR spectra.
NMR Spectroscopy
500 μl spiked urine was mixed with 50 μl phosphate buffer (PH=7.4, final concentration 0.2 M), 50 μl D2O containing 0.511 mM TSP. 550 μl of each solution was transferred to individual 5 mm NMR tubes. All 1H NMR experiments were carried out at 25°C on a Bruker DRX 500 MHz spectrometer equipped with an HCN 1H inverse detection probe with triple axis magnetic field gradients. 1H NMR spectra were acquired using the standard one-dimensional NOESY pulse sequence with water presaturation during the recycle delay of 3 s and a mixing time of 100 ms. Measurements for each sample were averaged over 32 transients using 32K time domain points, and Fourier transformed after multiplying by an exponential window function corresponding to a line broadening of 0.3 Hz, and the spectra were phased using the Bruker proprietary software XWINNMR version 3.5.
Statistical Model for the Observed Spectra
We adopt the view that the observed spectra have an additive noise background and a multiplicative signal.13, 27-29 The model for a single spectrum can be described mathematically as
| (1) |
where ω denotes values of chemical shifts on the ppm scale, f(ω) is the signal intensity at the chemical shift ω, B(ω) is a random quantity due to baseline distortion and background noise, N is the fixed normalization factor, μ(ω) is the fixed signal of our primary interest, and η(ω) is the random deviation from the signal due to the biological and instrumental variation. In this notation, the goal of baseline correction is to remove the effect of nuisance factors B(ω), the goal of feature detection is to determine the values of chemical shifts ω′ that correspond to the underlying metabolites, and peak quantification determines baseline-corrected intensity f′(ω′). The goal of normalization is to remove the effect of the multiplicative factor N, and logarithm transform is applied to the normalized data to account for the multiplicative effect. Once these steps are performed, μ(ω′)+η(ω′) is reported for subsequent statistical analyses. Finally, the goal of the exploratory and confirmatory analysis is to best account for the non-systematic variation η(ω′) when making conclusions regarding differences in the signal μ(ω′) between groups.
Baseline correction
For all the analyses below, we utilized the approach by Li,42 and estimated the baseline effect B(ω) using statistically motivated locally weighted scatterplot smoothing (lowess) regression.
Feature detection and quantification
We compare two approaches The first utilized raw baseline-corrected spectra. The second employed an in-house two-step peak alignment procedure where a rough spectral alignment was first performed using the signal of the reference TSP. Locations of the peaks ω′ in the spectra were then determined using a routine similar to the one in ref.43, which calculates a mean of all spectra, and determines peak locations based on the mean spectrum profile. The results were subjected to a refined alignment using the dynamic time warping algorithm, the algorithm is described in more details in the Supporting Information.44 Finally, background-corrected intensities f′(ω′) were determined from the apex of the picked peaks ω′. The procedures were implemented in matlab and R.
Normalization and scaling
Creatinine and total sum normalization define N=1/C, where C is the intensity of the creatinine peak for creatinine normalization, and the total sum of intensities of all peaks for the total sum normalization. The signal processing combined with constant normalization and logarithm transform can be described mathematically as
| (2) |
where ω′ are chemical shifts and f′(ω′) are baseline-corrected intensities of picked peaks, and C is the normalization factor. Peak intensities normalized this way are typically characterized by feature-specific variances of noise η(ω), where peaks of low intensity are more variable than high-intensity peaks. We compare the performance of the log-transformed data to the traditional centering and scaling.
We also investigate the performance of VSN.32 It takes as input the baseline-corrected intensities of peaks f′(ω′), and applies the transformation
| (3) |
where a and b are parameters estimated from the data by maximum likelihood as part of the procedure. The transformation is related to the logarithm transform by the relationship
| (4) |
It coincides with the logarithm for large intensities, but is approximately linear for low intensities, and smoothly interpolates in between. In contrast with constant normalization, VSN enforces a constant average variance of noise η(ω), i.e. a variance that does not depend on the height of the peak. The analysis was implemented in R, and the VSN normalization utilized the vsn package in R-based project Bioconductor27.
Exploratory statistical analysis
Exploratory analysis was exemplified by PCA implemented in R.
Confirmatory statistical analysis
Confirmatory analysis was exemplified by two procedures. First, the two-sample Welch t-test was conducted for each normalized peak across sample groups, using the feature-specific test statistic
| (5) |
where S1,2 is the pooled estimate of variance for that feature, and n1 and n2 are the number of spectra in each of the two compared group.
The second confirmatory analysis method is the Empirical Bayes (or moderated) t-test.45 The moderated t-test has the same interpretation as an ordinary t-test, however it takes advantage of the large number of tests that are conducted in parallel for all features. It replaces S1,2 (eq. 5) by , where S0 and d0 are estimated from the entire set of peaks and dg = n1 + n2 − 2, effectively borrowing the information from all features to aid variance estimation of an individual peak.
The p-values resulting from both regular and moderated t-test were adjusted for multiple comparisons using the Benjamini and Hochberg procedure controlling for the false discovery rate (FDR),46 and the overall FDR level was set to 5%. Both analyses were implemented in R, using limma and multtest packages in R-based project Bioconductor.27
Analysis of experimental data sets
The spike-in dataset was used to evaluate the performance of the statistical analysis procedures. For the exploratory analysis, performance of the methods was evaluated according to the PCA scores and loadings plots. A successful normalization should eliminate systematic differences between dilutions and between individual spectra. For confirmatory analysis, we evaluated the sensitivity and specificity of detecting true changes in concentrations of the spiked metabolites by comparing pairs of the mixtures. Changes in the intensity of peaks from spiked compounds were examined for five baseline concentrations, and five fold changes. Detection of these changes is considered true positive discoveries, while detections of changes in peaks from background urine metabolites are considered false positives. Optimal methods will maximize the true positive discoveries while controlling the false positive rate.
We illustrate the performance of the methods using the clinical diabetes experiment where the true status of metabolites is unknown. We evaluate the practical difference between methods by comparing the scores and loadings plots, as well as by comparing the number and type of differentially abundant peaks between the two groups.
Results
Spike-in data set
The raw spectra before and after baseline correction are provided in Figure S1. Figure S2 shows that the coefficient of variation of raw baseline-corrected spectra is roughly constant for all signal intensities in this dataset, indicating that a multiplicative model of the measurement error is appropriate. Figure S3 shows the boxplots of raw peak intensities determined from the spectra, and illustrates that peak intensities of the 6 mixtures have a fairly similar distribution when a dilution level is fixed. This indicates that baseline correction and peak quantification performed well.
The three dilution types in Figure S3 have clearly distinct patterns of abundance, as seen from the differences between the peak intensity quantiles. These differences are artifacts of the dilution process, and should be normalized in order to avoid mistaking them for biological signals. Total sum, creatinine and VSN normalizations all account for the dilutions by setting the medians of peak intensities to a comparable level. VSN normalization produces the narrowest range of normalized intensities, and results in no outlying low-intensity peaks.
Figure 1 illustrates the ability of the experiment to quantify changes in concentrations of the spiked metabolites for a series of fold changes, starting from a baseline of 800 uM. As can be seen, observed fold changes agree with the expectation for all normalization methods. Metabolites with the absolute log fold changes of 2 and above typically produce observed changes that are beyond random variation. The results are consistent with the spike-in experiments obtained, e.g. for gene expression microarrays.39 VSN produces the narrowest range of variation.
Figure 1.
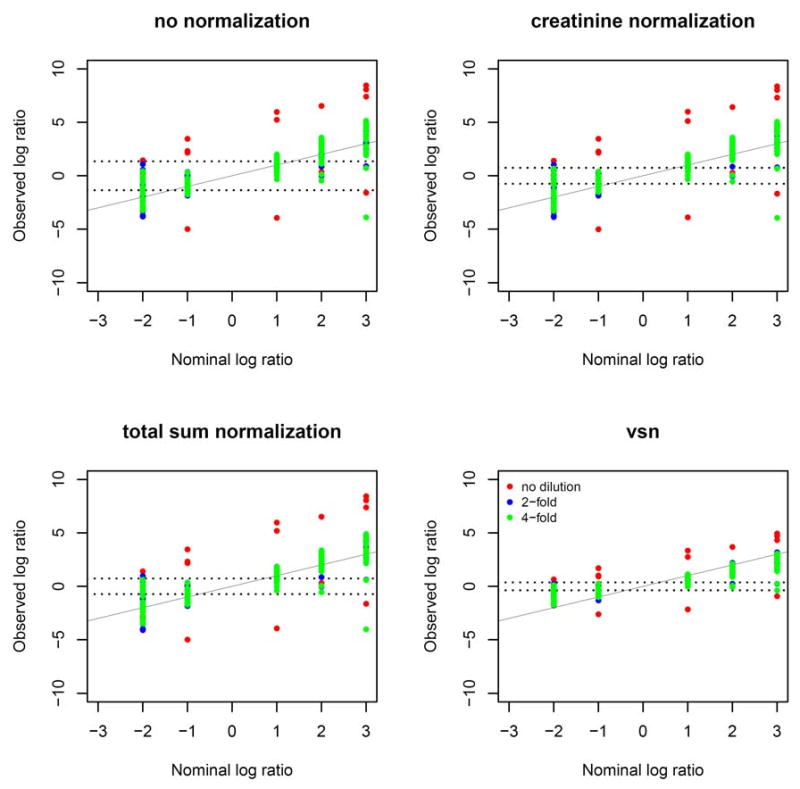
Observed log fold changes are plotted against nominal log fold changes for all metabolites, separately for each dilution. The fold changes are taken with respect to a baseline concentration of 800uM. The solid line represents the expected pattern. The dotted lines denote the 75th quantile of standard deviations of the background metabolites. Colors indicate dilution types.
Exploratory analysis: effect of normalization
Figure 2 shows PCA score plots of peak-picked and log-transformed data. The plot obtained from non-normalized intensities clearly reveals the structure of the experiment. The first principal component can be interpreted as the effect of the dilution (i.e. nuisance variation), and explains 59.7% of the overall variation between samples. The second principal component can be interpreted as the effect of the mixture type (i.e. the variation of interest), but explains only 19.6% of the overall variation. Thus the nuisance dilution-induced variation is thus three times bigger than the spike-induced variation of interest.
Figure 2.

PCA score plots for the 54 spectra in the spike-in dataset. X and Y axes are the first and the second principal components, respectively. Colors indicate the six mixture types, and shapes indicate the three dilution levels. VSN gives rise to the best performance in minimizing the dilution effect.
VSN shows the best performance in maximizing between-group separation while minimizing the variation within groups, and the first two principal components explain the largest proportion of total variation. With all normalization methods, two-fold and four-fold diluted samples are clustered more closely together for each mixture type than the undiluted samples. We discuss possible reasons for this in the next section.
Exploratory analysis: effect of feature quantification and scaling
PCA score plots of raw spectra, which make no use of peak picking and log transform, are shown in Figure S4A. Here PCA fails separating mixture types in clear stand-alone groups (as compared, e.g. to the VSN normalization in Figure 2), and the first two principal components explain a smaller percentage of total variation. The same procedure combined with auto scaling fails distinguishing between mixture types (Figure S4B), and PCA applied to peak picked data on the original scale also produces inferior results (Figure S5). This indicates that a combination of peak picking and (a generalized) log transform is preferred for PCA.
Confirmatory analysis: effect of normalization
Confirmatory data analysis was performed on two sets of pair-wise comparisons of the mixtures. Set A mimics urine-like comparisons, where samples from one disease group can be systematically more diluted than samples from the other group. The set contains 90 pairwise comparisons between mixtures. Set B mimics blood-like comparisons between samples. It contains 45 pair-wise comparisons of mixtures, where all samples have the same level of dilution.
Figure 3A displays the average false positive rate (FPR) of comparisons in set A versus the number of differentially abundant peaks. The false positive rates of a regular t-test applied to the non-normalized peak intensities are well above the values of FPR after normalization. Creatinine normalization leads to the lowest FPR curve among all normalization methods, indicating its superior performance. Regardless of the normalization method, the moderated t-test tends to outperform ordinary t-test.
Figure 3.
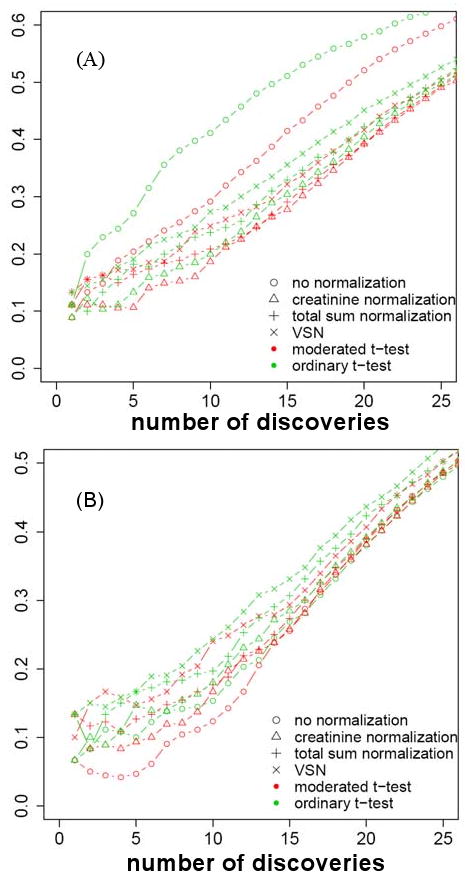
False positive rate (FPR) for detecting differentially abundant peaks. X-axis: number of detected differentially abundant peaks. Y-axis: average false positive rate, calculated over all pairs of mixtures in a comparison set. Shapes indicate normalization types. Colors indicate ordinary and moderated t-tests. (A) 90 urine-like pairwise comparisons of mixture types. Samples from different mixtures also differ in dilution. (B) 45 blood-like comparisons of mixture types. Samples from different mixtures have identical dilution.
Figure 3B displays a similar plot for the blood-like comparison set. The overall false positive rate is systematically lower for all normalization methods and for both t-tests in absence of dilution. Although non-normalized peak intensities appear to minimize FPR in the plot, we do not recommend forgoing the normalization. In this dataset all compounds were spiked to the same background urine sample, and the mixtures contain no true biological replication. In absence of dilution, normalization appears to over-correct the differences between very similar spectra, and this will not be the case in a real dataset. As in the previous case, the moderated t-test outperforms the regular t-test for this set of comparisons.
Confirmatory analysis: effect of feature quantification and scaling
Figure S6 plots results of all pairwise comparisons of the 6 mixtures after creatinine normalization for the spiked metabolites, using raw versus peak picked spectra. Working with raw spectra increases the total number of tests, and therefore requires a stronger adjustment for multiple comparisons. As can be seen from Figure S6, this results in systematically larger p-values (i.e. systematically smaller values on the −log scale) of tests for differential abundance in spiked metabolites.
The assumption of Normality underlying t-test was investigated using quantile-quantile plots for a randomly selected set of background peaks. The results (not shown for lack of space) indicate that some deviations from Normality exist, however the deviations are rarely severe. Log transformation improves the overall sensitivity and the specificity of the t-tests (Figure S7).
Clinical diabetes data set
Since overall urine volumes, as well as the concentrations of individual metabolites, vary dramatically in urine samples between diabetic patients and healthy controls,5 diabetes is an excellent test case for studies on normalization and data exploration. The application of NMR spectroscopy has already proved successful in numerous studies of diabetes.5, 8, 47-50 Therefore, selection of an appropriate processing method may be important for further optimizing the sensitivity and the reproducibility of the results. Due to the nature of the disease, the glucose region was removed from the raw spectra prior to normalization and analysis. Figures 4 and S8 show results of PCA on peak picked and log transformed data. Although all normalization methods clearly separate diabetic patients from controls, they produce different patterns on both scores and loading plots. Figure 5 displays the number of differentially abundant peaks detected when using different normalization methods, and using ordinary and moderated t-test at the estimated FDR of 5%. Different normalization algorithms share a large portion of detected features arising from metabolites such as 3-hydroxybutyrate, acetone and acetoacetate. However, conclusions regarding the differential abundance of metabolites such as dimethylamine and formate depended on the normalization procedure, indicating that the choice can have important implications for interpretation of the results. The total sum normalization produces noticeably more differentiating features than creatinine and VSN, however the true proportion of false positives in this dataset is unknown. The moderated t-test produces comparable results to the ordinary t-test in this dataset.
Figure 4.

PCA scores plots for the diabetes data set. X and Y axes indicate the first and the second principal components, respectively. Black dots indicate samples from diabetic patients. Open circles indicate samples from healthy controls. The choice of normalization procedure impacts the appearance of the PCA score plots.
Figure 5.

Venn diagrams of differentially abundant peaks detected for the diabetic urine data set at the false discovery rate of 5%. Total sum normalization produces the highest number of differentiating peaks, while the choice of ordinary or moderated t-test has little impact on this data set.
Discussion
Results of the spike-in experiment and of the clinical dataset lead us to the following conclusions.
Conclusion I: Different signal processing and normalization methods produce different results
For exploratory data analysis such as PCA, samples cluster differently on the scores plot after different signal processing and normalization methods. For confirmatory data analysis with the t-test, signal processing and normalization affect both the identity and the error rates of the differentially abundant metabolites. Consequently, the selection of a robust normalization method is quite important.
Conclusion II: Working with peak picked and log-transformed data improves the accuracy of the results
Peak picked data combined with the log transform result in a stronger difference between sample groups with PCA, and improved sensitivity and specificity of the t-tests, for two reasons. First, peak picking reduces the overall number of features. For PCA, this simplifies the problem by looking for patterns in the lower-dimensional data. For t-test, this reduces the conservative effect of correction for multiple testing. The conclusions are conditional on the correctly performed peak detection, quantification and alignment, and these procedures should be carefully implemented and calibrated. Second, (a generalized) log transform explicitly takes into account the multiplicative structure of the data, and translates it into additive signal. As can be seen from the Figure S9A, dependence of the measurement error on the mean signal is much weaker on the log scale. However lower-abundant peaks tend to have larger variation on the log scale. This is preferred to autoscaling, which standardizes each feature by feature-specific standard deviation, and is therefore more likely to remove the true biological signal. Log transform can also help satisfy the assumption of Normality and constant variance of a feature across groups required by the t-test. As we discuss next, these properties of the data, combined with appropriate normalization, improve the accuracy of the results.
Conclusion III: Optimal normalization depends on the subsequent statistical analysis
On the basis of the spike-in dataset, VSN was preferred in combination with PCA, and a constant normalization is preferred in combination with the t-test. This result is not surprising given that PCA and t-test are based on different distributional assumptions, and the normalization methods enforce different distributional properties on the normalized peaks. To illustrate the point, Figures S9B∼C and S10 demonstrate that an increased variance of lower-abundant peaks on the log scale persists after creatinine and total sum normalizations. The relationship has important implications for PCA, which considers all peaks simultaneously and reflects the patterns of largest variation. Therefore, PCA combined with constant normalization is unduly influenced by noisy and low-abundant peaks. This fact is further demonstrated by Figure S11, which shows that features with low average intensities have systematically stronger loadings, regardless of their spike-in status, for the case of constant normalization.
VSN, on the other hand, is designed to enforce roughly equal variances on all peaks, regardless of their mean intensity. Since the same transformation is applied to all features simultaneously, it is more likely to reflect the true biological variation than scaling, which standardizes each feature separately by the feature-specific standard deviation. Figure S11 shows that with VSN the loadings of the spike-in metabolites tend to be stronger than the loadings of the background peaks, and this is true for all mean intensities of peaks.
The situation is reversed for the t-test, which is performed separately for each feature, and does not require the assumption of constant variance of all features. When using VSN in conjunction with a t-test the variance of each feature is estimated twice, once as part of the normalization procedure, and once as part of the test. This double estimation results in overfitting, and is responsible for the inferior performance of VSN.
Conclusion IV: The moderated t-test is preferred to the ordinary t-test for analysis of small data sets
Despite a relatively small number of peaks in an NMR metabolomic experiment as compared to gene expression microrrays, the moderated t-test minimizes the FPR regardless of normalization. While the ordinary t-test calculates the variance of each peak separately, Empirical Bayes analysis combines peak-specific variance with a “summary” variance over all peaks (Equation 5). Since we typically observe several hundred peaks, the “summary” variance over all peaks is more reliable, and results in a more accurate outcome of the test.
Additional comments
In the course of our experiment we also found that, when using PCA, two-fold and four-fold diluted samples were clustered more closely together than the undiluted samples, and this pattern was particularly strong for constant normalization. The pattern can be attributed to the peaks that exhibit non-linear changes in intensity with increasing dilution.
It is generally assumed that dilution proportionally affects concentrations of all metabolites and of the spectral intensities. This has been used in previous normalization studies in which the dilution factors were computationally synthesized.26 Here, we experimentally diluted spiked urine samples, and observed that dilution can potentially induce non-linear variations in some peaks. Figure S12 shows raw NMR spectra and peak integral values for several such non-linearly behaving peaks in the spike-in dataset for one mixture at three dilution levels. Expected peak integral values are also plotted for the three dilution levels. One can see that the selected peaks have bigger or smaller integral than expected after dilution (Figure S12A∼C). The integrals of such peaks are relatively small compared to most peaks in the spectrum (Figure S1), but this unusual behavior can give rise to large PC1 loadings (Figure S11). This pattern differs from the majority of the metabolite peaks in the spectra, which generally decrease in intensity with dilution, as expected (Figure S12D). Due to the fact that urine composition is extremely complex, the reason for such non-linear behavior is not clear at present. Upon dilution, even under careful buffer control, factors such as pH, ion strength, macromolecular or paramagnetic metal binding could all vary, and thus affect the physicochemical behavior of molecules (small or large) in urine. When such non-linearities are present, they create a confounding effect, i.e. cannot be distinguished from the true biological differences. This confounding will adversely affect all the normalization procedures. We will further investigate this result in our future work.
In summary, signal processing and data analysis are key to the success of metabolic profiling experiments, in particular when involving highly variable samples such as urine. Although signal processing and data analysis are frequently considered as separate and independent steps, selection of a signal processing method should depend on the subsequent analysis steps, and should be selected to satisfy the assumptions necessary for the subsequent analysis most closely. In particular, peak picking and logarithm transform are beneficial. VSN transformation is a preferred normalization for exploratory analysis such as PCA, and creatinine normalization is preferred for t-test. The moderated t-test is superior to the regular t-test in experiments when the number of biological samples in each group is relatively small. Statistical methods evaluated on urine data in this work are also applicable to serum and plasma. Although the dilution effects are relatively small thanks to homeostasis of biological systems ensures stable blood content, they can be introduced experimentally when addition of buffer is needed to reach a measurable volume.
Supplementary Material
Acknowledgments
The authors thank the NIH (R01DK41973, UL1 RR24150) and Pfizer, Inc. for their generous support.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Van Dien S, Schilling CH. Molecular Systems Biology. 2006 doi: 10.1038/msb4100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craig A, Cloareo O, Holmes E, Nicholson JK, Lindon JC. Anal Chem. 2006;78:2262–2267. doi: 10.1021/ac0519312. [DOI] [PubMed] [Google Scholar]
- 3.Gowda GAN, Zhang SC, Gu HW, Asiago V, Shanaiah N, Raftery D. Expert Rev Mol Diagn. 2008;8:617–633. doi: 10.1586/14737159.8.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffin JL. Curr Opin Chem Biol. 2006;10:309–315. doi: 10.1016/j.cbpa.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 5.Zhang S, Gowda GAN, Asiago V, Shanaiah N, Barbas C, Raftery D. Analytical Biochemistry. 2008 doi: 10.1016/j.ab.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan ZZ, Gu HW, Talaty N, Chen HW, Shanaiah N, Hainline BE, Cooks RG, Raftery D. Analytical and Bioanalytical Chemistry. 2007;387:539–549. doi: 10.1007/s00216-006-0546-7. [DOI] [PubMed] [Google Scholar]
- 7.Schnackenberg KL, Sun J, Espandiari P, Holland DR, Hanig J, Beger DR. BMC Bioinformatics. 2007;8:S3. doi: 10.1186/1471-2105-8-S7-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, Cheeseman M, Nunez D, Sweatman BC, Haselden JN, Cox RD, Connor SC, Griffin JL. Physiological Genomics. 2007;29:99–108. doi: 10.1152/physiolgenomics.00194.2006. [DOI] [PubMed] [Google Scholar]
- 9.Odunsi K, Wollman RM, Ambrosone CB, Hutson A, McCann SE, Tammela J, Geisler JP, Miller G, Sellers T, Cliby W, Qian F, Keitz B, Intengan M, Lele S, Alderfer JL. International Journal of Cancer. 2005;113:782–788. doi: 10.1002/ijc.20651. [DOI] [PubMed] [Google Scholar]
- 10.Broadhurst DI, Kell DB. Metabolomics. 2006;2:171–196. [Google Scholar]
- 11.Gan F, Ruan GH, Mo JY. Chemometr Intell Lab. 2006;82:59–65. [Google Scholar]
- 12.Chang D, Banack CD, Shah SL. Journal of Magnetic Resonance. 2007;187:288–292. doi: 10.1016/j.jmr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Xi Y, Rocke MD. BMC Bioinformatics. 2008;9:324. doi: 10.1186/1471-2105-9-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson PE, Reo NV, DelRaso NJ, Doom TE, Raymer ML. Metabolomics. 2008;4:261–272. [Google Scholar]
- 15.De Meyer T, Sinnaeve D, Van Gasse B, Tsiporkova E, Rietzschel ER, De Buyzere ML, Gillebert TC, Bekaert S, Martins JC, Van Criekinge W. Anal Chem. 2008;80:3783–3790. doi: 10.1021/ac7025964. [DOI] [PubMed] [Google Scholar]
- 16.Ammann LP, Merritt M. Metabolomics. 2007;3:1–11. [Google Scholar]
- 17.Veselkov KA, Lindon JC, Ebbels TMD, Crockford D, Volynkin VV, Holmes E, Davies DB, Nicholson JK. Anal Chem. 2009;81:56–66. doi: 10.1021/ac8011544. [DOI] [PubMed] [Google Scholar]
- 18.Warrack BM, Hnatyshyn S, Ott KH, Reily MD, Sanders M, Zhang HY, Drexler DM. J Chromatogr B. 2009;877:547–552. doi: 10.1016/j.jchromb.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Viau C, Lafontaine M, Payan JP. International Archives of Occupational and Environmental Health. 2004;77:177–185. doi: 10.1007/s00420-003-0495-9. [DOI] [PubMed] [Google Scholar]
- 20.Saude EJ, Adamko D, Rowe BH, Marrie T, Sykes BD. Metabolomics. 2007;3:439–451. [Google Scholar]
- 21.Barr DB, Wilder LC, Caudill SP, Gonzalez AJ, Needham LL, Pirkle JL. Environmental Health Perspectives. 2005;113:192–200. doi: 10.1289/ehp.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu H, Pan Z, Xi B, Hainline BE, Shanaiah N, Asiago V, NaganaGowda GA, Raftery D. NMR in Biomedicine. 2008 ASAP. [Google Scholar]
- 23.Romano R, Santini MT, Indovina PL. Journal of Magnetic Resonance. 2000;146:89–99. doi: 10.1006/jmre.2000.2102. [DOI] [PubMed] [Google Scholar]
- 24.Lemmerling P, Vanhamme L, Romano R, Van Huffel S. Journal of Magnetic Resonance. 2002;157:190–199. doi: 10.1006/jmre.2002.2598. [DOI] [PubMed] [Google Scholar]
- 25.Dieterle F, Ross A, Schlotterbeck G, Senn H. Anal Chem. 2006;78:4281–4290. doi: 10.1021/ac051632c. [DOI] [PubMed] [Google Scholar]
- 26.Torgrip RJO, Aberg KM, Alm E, Schuppe-Koistinen I, Lindberg J. Metabolomics. 2008;4:114–121. [Google Scholar]
- 27.Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. 2005 [Google Scholar]
- 28.Cruz-Marcelo A, Guerra R, Vannucci M, Li Y, Lau CC, Man TK. Bioinformatics. 2008 August 11; doi: 10.1093/bioinformatics/btn398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karakach TK, Wentzell PD, Walter JA. Analytica Chimica Acta. 2009;636:163–174. doi: 10.1016/j.aca.2009.01.048. [DOI] [PubMed] [Google Scholar]
- 30.Parsons HM, Ludwig C, Gunther UL, Viant MR. BMC Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huber W, Heydebreck Av, Sültmann H, Poustka A, Vingron M. Bioinformatics. 2002;19:S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 32.Motakis ES, Nason GP, Fryzlewicz P, Rutter GA. Bioinformatics. 2006;22:2547–2553. doi: 10.1093/bioinformatics/btl412. [DOI] [PubMed] [Google Scholar]
- 33.Trygg J, Holmes E, Lundstedt T. J Proteome Res. 2007;6:469–479. doi: 10.1021/pr060594q. [DOI] [PubMed] [Google Scholar]
- 34.Halouska S, Powers R. Journal of Magnetic Resonance. 2006;178:88–95. doi: 10.1016/j.jmr.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Montgomery CD. Wiley. (5) 2000 [Google Scholar]
- 36.Mehta TS, Zakharkin SO, Gadbury GL, Allison DB. Physiological Genomics. 2006;28:24–32. doi: 10.1152/physiolgenomics.00095.2006. [DOI] [PubMed] [Google Scholar]
- 37.Hinkelmann K, Kempthorne O. Introduction to Experimental Design. (2nd) 2007;1 [Google Scholar]
- 38.Mehta T, Tanik M, Allison DB. Nature Genetics. 2004;36:943–947. doi: 10.1038/ng1422. [DOI] [PubMed] [Google Scholar]
- 39.Cope LM, Irizarry RA, Jaffee HA, Wu ZJ, Speed TP. Bioinformatics. 2004;20:323–331. doi: 10.1093/bioinformatics/btg410. [DOI] [PubMed] [Google Scholar]
- 40.Mueller L, Rinner O, Schmidt A, Letarte S, Bodenmiller B, Brusniak M, Vitek O, Aebersold R, Müller M. Proteomics. 2007;7:3470–3480. doi: 10.1002/pmic.200700057. [DOI] [PubMed] [Google Scholar]
- 41.Karakelides H, Asmann YW, Bigelow ML, Short KR, Dhatariya K, Coenen-Schimke J, Kahl J, Mukhopadhyay D, Nair KS. Diabetes. 2007;56:2683–2689. doi: 10.2337/db07-0378. [DOI] [PubMed] [Google Scholar]
- 42.Li X. PROcess: Ciphergen SELDI-TOF Processing R package version 1.12.0. 2005 [Google Scholar]
- 43.Morris JS, Coombes KR, Koomen J, Baggerly KA, Kobayashi R. Bioinformatics. 2005;21:1764–1775. doi: 10.1093/bioinformatics/bti254. [DOI] [PubMed] [Google Scholar]
- 44.Tomasi G, van den Berg F, Andersson C. Journal of Chemometrics. 2004;18:231–241. [Google Scholar]
- 45.Smyth KG. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 46.Benjamini Y, Hochberg Y. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 47.Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hodavance MS, Ralston SL, Pelczer I. Analytical and Bioanalytical Chemistry. 2007;387:533–537. doi: 10.1007/s00216-006-0979-z. [DOI] [PubMed] [Google Scholar]
- 49.Qiu Y, Rajagopalan D, Connor SC, Damian D, Zhu L, Handzel A, Hu GH, Amanullah A, Bao S, Woody N, MacLean D, Lee K, Vanderwall D, Ryan T. Metabolomics. 2008;4:337–346. [Google Scholar]
- 50.Makinen VP, Soininen P, Forsblom C, Parkkonen M, Ingman P, Kaski K, Groop PH, Ala-Korpela M, Grp FS. Molecular Systems Biology. 2008;4 doi: 10.1038/msb4100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.