

Abstract
Mutations in LAMA2, the gene for the extracellular matrix protein laminin-α2, cause a severe muscular dystrophy termed congenital muscular dystrophy type-1A (MDC1A). MDC1A patients have accompanying CNS neural dysplasias and white matter abnormalities for which the underlying mechanisms remain unknown. Here, we report that in laminin-deficient mice, oligodendrocyte development was delayed such that oligodendrocyte progenitors accumulated inappropriately in adult brains. Conversely, laminin substrates were found to promote the transition of oligodendrocyte progenitors to newly formed oligodendrocytes. Laminin-enhanced differentiation was Src family kinase-dependent and resulted in the activation of the Src family kinase Fyn. In laminin-deficient brains, however, increased Fyn repression was accompanied by elevated levels of the Src family kinase negative regulatory proteins, Csk (C-terminal Src kinase), and its transmembrane adaptor, Cbp (Csk-binding protein). These findings indicate that laminin deficiencies delay oligodendrocyte maturation by causing dysregulation of signaling pathways critical for oligodendrocyte development, and suggest that a normal role for CNS laminin is to promote the development of oligodendrocyte progenitors into myelin-forming oligodendrocytes via modulation of Fyn regulatory molecules.
Introduction
Children born with mutations in LAMA2, the gene that encodes the α2-subunit of the extracellular matrix protein laminin, have defects in the size and shape of the forebrain and cerebellum along with white matter abnormalities (Chun et al., 2003; Colognato et al., 2005). These abnormalities accompany severe muscular dystrophy and are collectively termed congenital muscular dystrophy type-1A (MDC1A). In the peripheral nervous system, laminins and their receptors regulate the ability of Schwann cells to proliferate, survive, radially sort axons, and, ultimately, to myelinate (for review, see Court et al., 2006). In contrast, little is known about how laminins influence the development of oligodendroglia, the myelinating cells of the CNS. One study, however, has reported that α2-laminin-deficient mice have less forebrain myelin (Chun et al., 2003). While oligodendroglia lack basement membranes, structures in which laminin-mediated signal transduction has been defined classically, integrin and dystroglycan adhesion receptors have been implicated in oligodendrocyte laminin signaling using culture models (Baron et al., 2005; Colognato et al., 2007). Thus, one possibility to account for MDC1A CNS abnormalities is that laminin deficiencies disrupt signaling pathways that contribute to oligodendrocyte lineage progression and/or oligodendrocyte function. However, it is not known whether laminins modulate oligodendrocyte lineage progression in the brain, and if so, through what signaling pathways.
Insights into the influence of α2-laminins on oligodendrocyte development have, however, been provided by in vitro studies. For example, oligodendrocytes cultured on laminin show enhanced process extension and myelin membrane expansion (Buttery and ffrench-Constant, 1999; Howe, 2006). Laminins also enhance oligodendrocyte survival, at least in part by modulating receptor tyrosine kinase signaling (Frost et al., 1999; Colognato et al., 2002). Laminin has furthermore been shown to activate both integrin-linked kinase (ILK) and the Src family kinase Fyn in cultured oligodendrocytes (Chun et al., 2003; Colognato et al., 2004). The link between laminin and Fyn may be particularly relevant to oligodendrogenesis since Fyn is required for normal CNS myelination (Umemori et al., 1994; Krämer et al., 1999; Sperber et al., 2001). These studies, together with reports of CNS abnormalities in laminin-deficient mice and people, suggest the hypothesis that laminin-mediated signaling guides the onset of oligodendrocyte differentiation in the developing CNS.
Here, α2-laminin-deficient mice were used to investigate the in vivo influence of laminins on oligodendrogenesis. Laminin-deficient brains were found to have a developmental delay in oligodendrocyte maturation accompanied by an accumulation of oligodendrocyte progenitor cells (OPCs). We furthermore identified several signaling abnormalities to account for these delays. Laminin-deficient brains showed dysregulated Fyn and elevated levels of C-terminal Src kinase (Csk) and Csk-binding protein (Cbp), proteins that suppress Fyn activity. Laminin substrates were furthermore found to modulate Fyn regulation and to promote the transition of cultured oligodendrocyte progenitors to newly formed oligodendrocytes in a Fyn-dependent manner. These findings (1) indicate that the dysregulation of signaling pathways required for normal oligodendrocyte development may contribute to CNS abnormalities observed in MDC1A and (2) identify novel mechanisms by which laminins regulate gliogenesis in the developing brain.
Materials and Methods
Animals.
Dystrophic dy/dy mice were shown to have a severe decrease in expression of the laminin α2 protein, but to date the precise genetic mutation is thought to lie outside the LAMA2 coding region and has not been identified, therefore genotyping is not possible (Sunada et al., 1994). However, dy/dy mice exhibit a severe dystrophic phenotype that is readily apparent by ∼2 weeks (weakness and hind leg paralysis). By 3 weeks, dy/dy mice show characteristic hindlimb muscle atrophy, weakness, paralysis, and sporadic contractures and were separated from their wild-type littermates (homozygous +/+ and dy/+). To ensure that all dy/dy mice were identified correctly, we only evaluated brains at 3 weeks of age and older. Many dy/dy mice die prematurely between 4 and 6 weeks, and therefore we restricted our analysis to mice at 6 weeks of age. Heterozygous 129P1/ReJ dy/+ mice were obtained from The Jackson Laboratory. All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Stony Brook University Institutional Animal Care and Use Committee.
Cell culture.
Disassociated neonatal cortices from postnatal day 0–2 rats were cultured (37°C, 7.5% CO2) in high glucose DMEM with 10% fetal calf serum (FCS) on poly-d-lysine (PDL)-coated flasks. Medium was changed every 3–4 d. By day 10–14, mixed glial cultures consisting of OPCs and microglia on an astrocyte monolayer were obtained. Purified OPCs were isolated from mixed glial cultures using a modification of the mechanical dissociation and differential adhesion method described by McCarthy and de Vellis (1980). To evaluate protein expression and protein phosphorylation in response to substrate conditions, OPCs were suspended in modified SATO's medium and added to Nunclon dishes coated with PDL or laminin-2 (human placental laminin, Sigma). To coat dishes, PDL or laminin-2 at 10 μg/ml were incubated for 4 h at 37°C. Following coating, surfaces were blocked with 10 μg/ml heat-inactivated, endotoxin-free BSA for 30 min at 37°C and washed with PBS. At indicated time points, cells were lysed in extraction buffer [1% SDS, 20 mm Tris pH 7.4 containing protease and phosphatase inhibitor cocktails (Calbiochem) added immediately before use] and heated to 95°C for 5 min.
Antibodies.
The following antibodies were used for immunocytochemistry: rat monoclonal IgG against laminin α2 subunit, clone 4H82 (Sigma); rabbit polyclonal IgG against laminin-1 (Sigma); mouse monoclonal IgG against neurofilament (NF) (Sigma); rat monoclonal IgG against PDGFRα (BD Biosciences); rat monoclonal IgG against myelin basic protein (MBP) (Serotec); mouse monoclonal IgG against APC (CC1) (Calbiochem). FITC-, Texas Red-, Cy3-, and biotin-conjugated donkey antibodies against rat, rabbit, and mouse IgG were used as secondary antibodies (Jackson ImmunoResearch). Texas Red-conjugated streptavidin (Jackson ImmunoResearch) was used to detect biotinylated secondary antibodies. The following antibodies were used for Western blotting: rabbit polyclonal IgG against NG2 (a generous gift from Dr. Joel Levine, Stony Brook University, Stony Brook, NY); mouse monoclonal IgG against CNP (Sigma); rat monoclonal IgG against MBP (Serotec); mouse monoclonal IgG against Kip1/p27 (BD Transduction Laboratories); rabbit monoclonal IgG against the quaking homolog KH domain RNA-binding protein (QKI) (Bethyl Laboratories); rabbit polyclonal IgG against cleaved caspase-3 (Asp175) (Cell Signaling Technology); rabbit polyclonal IgG against Fyn (Santa Cruz Biotechnology); rabbit polyclonal IgG against phospho-Tyr418 of Src family kinases (Calbiochem); rabbit polyclonal IgG against phospho-Tyr529 of Src family kinases (Calbiochem); rabbit polyclonal IgG against Csk (C-20) (Santa Cruz Biotechnology); rabbit polyclonal IgG against Cbp (generously provided by Dr. Jon Lindquist, Otto-von-Guericke University, Magdeburg, Germany); rabbit polyclonal IgG against phospho-Tyr317-Cbp (Tyr314-Cbp in mouse, raised against phospho-peptide KEISAMpYSS); and mouse monoclonal IgG against β-actin (Sigma).
Western blot analysis.
Cerebral cortices and cerebella were collected from 3- and 6-week old dy/dy and wild-type littermates. Following addition of extraction buffer (1 ml buffer per 200 mg tissue), tissues were incubated at 95°C for 10 min with occasional trituration. Homogenates were then centrifuged at 14,000 rpm to remove insoluble material. Protein concentration was determined (detergent compatible protein assay, Bio-Rad) and the lysates were boiled for 5 min in Laemmli solubilizing buffer (LSB) with 4% β-mercaptoethanol (βME). Proteins were separated by SDS-PAGE using 7.5, 10, 12, or 15% acrylamide minigels and transferred onto 0.45 μm nitrocellulose. Membranes were blocked in 0.1% Tween 20, and Tris buffered saline (TBS-T) containing either 4% bovine serum albumin (BSA) or 1% nonfat milk (blocking buffer) for 1 h, followed by primary antibodies in blocking buffer overnight at 4°C. Membranes were washed in TBS-T, incubated for 1 h in HRP-conjugated secondary antibodies (Amersham) diluted 1:3000 in blocking buffer, washed in TBS-T, and then developed using enhanced chemiluminescence (Amersham). Relative densitometries were determined using the NIH ImageJ Processing and Analysis Program.
Fluorescent immunocytochemistry using fresh frozen sections.
Tissues were embedded in Tissue-Tek OCT (Sakura Finetek) and cryosectioned to a thickness of 25 μm. Fresh frozen sections were fixed in methanol for 5 min at −20°C. After washing in PBS, sections were blocked for 1 h in PBS containing 10% donkey serum (DS; Sigma), followed by overnight incubation with primary antibodies in block buffer at 4°C. Sections were then washed in PBS and incubated in Texas Red-, CY3-, or FITC-conjugated secondary antibodies for 1 h, then washed again in PBS. Sections were incubated in 10 μg/ml DAPI in PBS for 10 min to counterstain nuclei and mounted using Slowfade Gold antifade reagent (Invitrogen).
Fluorescent immunocytochemistry using perfused tissue.
Mice were anesthetized with Avertin and perfused intracardially with a saline solution, followed by 4% paraformaldehyde (PFA) in PBS. Brains were harvested, postfixed for 1 h in PFA, and incubated overnight in 30% sucrose at 4°C. Tissues were embedded in Tissue-Tek OCT (Sakura Finetek) and cryosectioned to a thickness of 25 μm. For MBP immunocytochemistry, sections were postfixed in ethanol: acetic acid (95:5) for 20 min at −20°C. After quick washes in PBS, sections were incubated in 10% DS with 0.05% Triton X-100 for 1 h before labeling with anti-MBP primary antibodies. FITC-conjugated secondary antibodies were incubated in 10% DS for 1 h.
Alternatively, 50 μm floating sections were prepared. Briefly, cryosections were placed in chilled tissue stock solution (30% glycerol: 30% ethylene glycol: 10% 0.2 m phosphate buffer) to remove the remaining OCT. For immunodetection of OPCs, floating sections were postfixed for 15 min in 4% PFA then washed 3 times in PBS with 0.2% Triton X-100 (PBST). After blocking in 1% DS in PBST for 30 min, the sections were incubated in anti-PDGFRα primary antibody overnight at room temperature. Sections were then washed once with PBST and incubated in fluorescent secondary antibody for 1 h. Finally, floating sections were washed 3 times with PBST, followed by 3 washes with 0.1 m phosphate buffer. All sections were incubated in 10 μg/ml DAPI in PBS for 10 min to visualize nuclei before finally being mounted with SlowFade Gold antifade reagent.
Oligodendrocyte cell density and survival.
To determine whether the laminin-α2 deficiency in dy/dy mice affects oligodendrocyte cell density, fresh frozen cortical, cerebellar, and brainstem sections (25 μm) were fixed in 4% PFA for 15 min at room temperature. Following washes with PBS, sections were incubated in 10% DS with 0.5% Triton X-100 for 1 h at room temperature. Immunocytochemistry using anti-APC (CC1; a marker expressed in mature oligodendrocytes) or anti-PDGFRα primary antibody was performed overnight at 4°C. Sections were washed 4 times in PBS, then incubated in Cy3-conjugated secondary antibody for 1 h, before washing again in PBS. In the cerebral cortex, CC1(+) oligodendrocytes and PDGFRα(+) OPCs were counted in the corpus callosum, lateral septum, and anterior commissure. Along the corpus callosum, oligodendroglia found at the fornix and at fixed distances of 0.8, 1.0, and 1.7 mm from the midline were scored in matching sections of wild-type and dy/dy brains. CC1(+) and PDGFRα(+) cell densities were also determined in the proximal white matter of the cerebellum, white matter of cerebellar folia, and ventral regions of the brainstem.
To identify apoptotic oligodendroglia, the CC1- and PDGFRα-immunostained sections were subjected to a TUNEL assay according to the manufacturer's instructions (Apoptag). Sections were then counterstained with 10 μg/ml DAPI and mounted with SlowFade Gold antifade reagent. TUNEL-positive cells per unit area, as well as the percentage of TUNEL-positive, CC1-positive cells (or TUNEL-positive, PDGFRα-positive cells) were determined.
Fluorescence microscopy and image acquisition.
Sections were visualized using a Zeiss Axioplan inverted fluorescence microscope fitted with 10× eyepiece magnification using 5× [0.16 numerical aperture (NA)], 10× (0.3 NA), 20× (0.5 NA), 40× (0.75 NA), and 63× (1.4 NA, oil) objectives. Images were acquired using a Zeiss Axiocam MRM digital camera and Zeiss AxioVision imaging software. For thick sections (50 μm) imaged using 63× oil immersion, Z-stacks of 0.25 μm increments were captured and maximal image projections were obtained.
Electron microscopy and morphometric analysis.
Mice were perfused with 4% paraformaldehyde/2.5% glutaraldehyde in 0.1 m phosphate buffered saline. The brains were postfixed overnight in the same fixative and then individual structures were cut in the appropriate plane on a Leica VT-1000 Vibratome at 50–60 μm. Vibratomed sections were processed using standard transmission electron microscopy techniques. Briefly, vibratomed sections were placed in 2% osmium tetroxide in 0.1 m phosphate buffer, washed in 0.1 m phosphate buffer and dehydrated in a graded series of ethyl alcohol. Sections were then vacuum infiltrated in Durcupan resin (Drurcupan ACM Epoxy, Electron Microscopy Sciences) overnight, flat embedded between two pieces of ACLAR film (Ted Pella) and placed in a 60°C oven for 48–72 h to polymerize. Areas to be sampled were blocked and ultrathin sections were cut at ∼70–80 nm on a Rechert-Jung Ultracut E ultra-microtome. Ultrathin sections were placed on formvar-coated copper slot grids and counterstained with uranyl acetate and lead citrate. Samples were viewed with a FEI Tecnai BioTwinG2 transmission electron microscope. Digital images were acquired with an AMT XR-60 CCD digital camera system and compiled and analyzed using Adobe Photoshop and ImageJ (NIH). The g-ratio of myelinated axons was determined by dividing the axon diameter by the myelin diameter. A minimum of 300 axons per genotype was measured for each region.
Statistics.
Statistical significance of data sets was determined using the Student's two-tailed, paired t test. Raw values were then normalized to each wild-type littermate control (wild type of the pair set at 100%). Graphs depict the mean values, with error bars depicting SEM. Statistical analysis on overall g-ratio was performed using the Mann–Whitney rank sum test (SigmaStat). Student's t test was performed on binned g-ratios where normal distribution and equal variance was determined; mean g-ratio and SEM is depicted (SigmaStat).
Results
Laminin-deficient brains show delayed oligodendrocyte maturation
Human laminin deficiencies result in a variety of CNS developmental defects that may in part reflect altered gliogenesis. Indeed, hypomyelination has been reported in the corpus callosum and optic nerve of 5-week-old dy/dy mice, a dystrophic mouse model in which laminin α2 subunit expression is severely reduced but not absent (Chun et al., 2003). To determine whether laminins influence the progression of oligodendrogenesis, we evaluated both oligodendrocytes and oligodendrocyte progenitors in dy/dy brains at two different ages. First, we evaluated mature oligodendrocytes by counting the number of cells positive for CC1, a marker for oligodendrocyte cell bodies (Fig. 1). In 3-week-old corpus callosum, we found a significant reduction in the number of CC1(+) cells per mm2 in dy/dy animals compared with wild-type littermates (Fig. 1A,B; Table 1) (1100.1 ± 54.0 vs 1404.6 ± 103.0, respectively, n = 3, p = 0.0287). By 6 weeks, however, the density of CC1(+) cells in the corpus callosum of both wild-type and dy/dy animals had increased; these increases were greater in dy/dy animals such that by 6 weeks dy/dy and wild-type corpus callosum had similar levels of CC1(+) cells (Fig. 1A; Table 1) [1828.5 ± 268.6 CC1(+) cells per mm2 in wild-type animals; 1640.8 ± 234.5 in dy/dy littermates, n = 3, p = 0.2804]. This result suggested that, in the corpus callosum, oligodendrocyte differentiation is delayed, but not prevented.
Figure 1.

Delayed oligodendrocyte maturation in dy/dy brains. A, Mature CC1(+) oligodendrocytes per square millimeter in the corpus callosum were measured in wild-type (wt; white bars) and dy/dy (dy; black bars) littermates at 3 weeks (3W) and 6 weeks (6W) of age. Graphs depict mean (±SEM) counts obtained from 7 different areas of the corpus callosum (n = 3; *p < 0.05). B, Representative images of CC1(+) oligodendrocytes (green) in the corpus callosum of 3-week-old wild-type and dy/dy brains. Nuclei are visualized using DAPI (blue). Scale bar, 50 μm. C, Mature CC1(+) oligodendrocytes per mm2 in the proximal cerebellar white matter were measured in wild-type (white bars) and dy/dy (black bars) littermates at 3 weeks and 6 weeks of age. Graphs depict mean (±SEM) counts obtained from 6 different areas of the proximal cerebellar white matter (n = 5; *p < 0.05 for comparison between 3W wt and 6W wt). D, Mature CC1(+) oligodendrocytes per mm2 in the brainstem were measured in wild-type (white bars) and dy/dy (black bars) littermates at 3 weeks and 6 weeks of age. Graphs depict mean (±SEM) counts obtained from 3 different areas of the brainstem (n = 5; *p < 0.05 for comparisons between 3W wt and 6W wt, and, between 6W wt and 6W dy). E, Top panels, Representative images of MBP immunocytochemistry (green) in the corpus callosum of 3-week-old wild-type (wt) and dy/dy (dy) brains. Scale bar, 200 μm. Middle panels: representative images of MBP (green) and NF (red) immunocytochemistry in the corpus callosum of 3-week-old wild-type (wt') and dy/dy (dy') brains. Scale bar, 25 μm. Bottom panels, Representative images of MBP (green) and NF (red) immunocytochemistry in the corpus callosum of 3-week-old wild-type (wt“) and dy/dy (dy”) brains. Scale bar, 10 μm. F, Lysates from 3-week-old cerebral cortices were evaluated by Western blot to detect MBP. MBP isoforms 21.5, 18.5, 17.2 and 14.0 kDa are indicated. Actin blots are shown as a loading control.
Table 1.
CC1(+) cells per square millimeter
| Region | 3 weeks |
6 weeks |
3wk vs 6 wk |
||||||
|---|---|---|---|---|---|---|---|---|---|
| wt | dy | p value: wt vs dy | wt | dy | p value: wt vs dy | p value: wt | p value: dy | ||
| 1 | Corpus callosum | 1404.6 ± 103.0 | 1100.1 ± 54.0 | 0.0287* | 1828.5 ± 268.6 | 1640.8 ± 234.5 | 0.2804, n.s. | 0.3686, n.s. | 0.2017, n.s. |
| 2 | Lateral septum | 1217.7 ± 169.0 | 875.3 ± 144.3 | 0.3042, n.s. | 1713.8 ± 443.3 | 1315.2 ± 617.9 | 0.1183, n.s. | 0.4091, n.s. | 0.7103, n.s. |
| 3 | Anterior commissure | 1347.1 ± 32.7 | 1081.3 ± 18.6 | 0.0097** | 1769.3 ± 328.0 | 1558.8 ± 213.2 | 0.4976, n.s. | 0.2962, n.s. | 0.1707, n.s. |
| 4 | Cortex, all | 1359.9 ± 66.6 | 1061.9 ± 34.8 | 0.0139* | 1785.0 ± 286.4 | 1531.9 ± 183.9 | 0.2383, n.s. | 0.3466, n.s. | 0.1619, n.s. |
| 5 | Cerebellum, central | 978.2 ± 196.7 | 1228.9 ± 43.5 | 0.3438, n.s. | 1840.8 ± 70.5 | 1037.5 ± 199.7 | 0.1085, n.s. | 0.0074** | 0.3284, n.s. |
| 6 | Cerebellum, all | 1473.9 ± 76.3 | 1604.0 ± 82.2 | 0.0263* | 1840.8 ± 70.5 | 1389.6 ± 105.6 | 0.0473* | 0.0107* | 0.1471, n.s. |
| 7 | Brain stem | 988.5 ± 104.7 | 1084.6 ± 104.1 | 0.0026** | 1405.1 ± 64.9 | 1054.4 ± 47.6 | 0.0166* | 0.0159* | 0.9369, n.s. |
| 8 | Spinal cord | 1333.8 ± 103.4 | 1413.3 ± 52.8 | 0.7000, n.s. | |||||
| 9 | Optic nerve | 2923.1 ± 124.2 | 2245.5 ± 88.5 | 0.0335* | |||||
wt, Wild type; dy, dy/dy; wk, week; n.s., not significant; *p < 0.05;
**p < 0.01.
To determine whether oligodendrocyte differentiation was delayed in other brain regions we next evaluated CC1(+) cell density in cerebellum and brainstem (Fig. 1C,D). In the proximal white matter of 3-week-old cerebella we found similar levels of CC1(+) positive cells in wild-type and dy/dy littermates (Fig. 1C) (n = 5, p = 0.3438). In wild-type mice, however, between 3 weeks and 6 weeks the CC1(+) cell density significantly increased (Fig. 1C; Table 1) (978.2 ± 196.7 vs 1840.8 ± 70.5, n = 5, p = 0.0074), whereas in dy/dy mice CC1(+) cell density did not significantly change (Fig. 1C; Table 1) (n = 5; p = 0.3284). It should be noted that between 3 weeks and 6 weeks there was not a significant decrease in CC1(+) density in dy/dy cerebella; instead, oligodendrocytes in dy/dy cerebella failed to keep pace with the increase in density that was observed in wild-type animals. In brainstem (Fig. 1D), a similar phenomenon occurred: similar CC1(+) cell density was observed in wild-type and dy/dy brainstems at 3 weeks of age, but by 6 weeks, CC1(+) cell density had significantly increased in wild-type brainstem (Fig. 1D; Table 1) (1405.1 ± 4.9 cells per mm2 up from 988.5 ± 104.7 cells per mm2 at 3 weeks, n = 5, p = 0.0159) but did not significantly increase in dy/dy brainstem (Fig. 1D; Table 1) (1054.4 ± 47.6 compared with 1084.6 ± 104.1 at 3 weeks, n = 5, p = 0.9369). Again, CC1(+) cell density did not significantly drop in dy/dy brainstem between 6 weeks and 3 weeks, suggestive of a slow or stalled differentiation rather than a loss of cells. In addition to the brain structures reported in Figure 1, several different cortical regions were evaluated (lateral septum and anterior commissure) as well as total cerebellar white matter, spinal cord, and optic nerve (see Table 1). In each region, CC1(+) cell density was either similar or lower in dy/dy animals compared with wild-type littermates, at both 3 weeks and 6 weeks. In all cases, CC1(+) density increased in wild-type regions between 3 weeks and 6 weeks, whereas in some regions, e.g., corpus callosum, dy/dy CC1(+) density caught up to wild-type density by 6 weeks, but in some regions, e.g., cerebellum, it did not. These differences suggest regional variation in the role of laminin in modulating timely development of appropriate oligodendrocyte numbers.
We next evaluated levels of myelin basic protein (MBP) in dy/dy brains versus wild-type littermates (Fig. 1E,F). In agreement with a previous study by Chun et al. (2003), less MBP protein was observed in dy/dy cerebral cortices compared with wild-type cerebral cortices (Fig. 1F) (69.8 ± 8.3% of protein in wild-type littermate cerebral cortices, n = 3, p = 0.049). We also monitored the expression of 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP) and found reduced CNP levels in dy/dy cerebral cortices compared with wild-type littermates (73.3 ± 10.6%, n = 3, p = 0.047) (data not shown). In addition, the overall organization of white matter tracts was visualized using MBP immunoreactivity, with dy/dy MBP(+) tracts appearing grossly normal but often appearing more sparse compared with wild-type counterparts (Fig. 1E).
Correlation between delayed oligodendrocyte differentiation and myelination abnormalities in dy/dy brains
Given that delayed oligodendrocyte differentiation showed a differential time course in dy/dy cerebella compared with that in cerebral cortices, we next sought to determine whether the degree of myelination in the dy/dy cerebella correlated with its differentiation delay. Overall, the gross organization of white matter appeared normal in dy/dy cerebellum, as reflected by MBP and neurofilament immunoreactivity (Fig. 2A). However, dy/dy cerebella showed a small, but significant, reduction in MBP levels relative to that found in wild-type littermates (Fig. 2B) (84.1 ± 0.12%, n = 4, p = 0.025). It should also be noted that at 3 weeks of age dy/dy cerebellar white matter regions showed low-level laminin-α2 immunoreactivity, particularly associated with blood vessels (Fig. 2C). This dy/dy laminin-α2 immunoreactivity, however, was substantially less than in wild-type littermates, and by 6 weeks, dy/dy cerebella did not show any residual laminin-α2 reactivity that was detectable above that in negative controls (Fig. 2C). Next, we assessed myelin ultrastructure using transmission electron microscopy and found that dy/dy cerebellar axons, relative to their wild-type littermates, exhibited thinner myelin at 6 weeks of age; representative micrographs are shown in Figure 2D. Individual cerebellar g-ratios were plotted as a function of axon diameter (Fig. 2E); a significant trend (p < 0.001) was observed such that the median g-ratio was larger in dy/dy cerebella (0.874) compared with that in wild-type cerebella (0.832). Cerebellar g-ratios were also grouped by axon diameter to reveal that dy/dy cerebellar axons of all sizes showed significantly thinner myelin (Fig. 2F). Thus, in the cerebellum, delayed oligodendrocyte maturation correlated with dysmyelination; i.e., thinner myelin.
Figure 2.
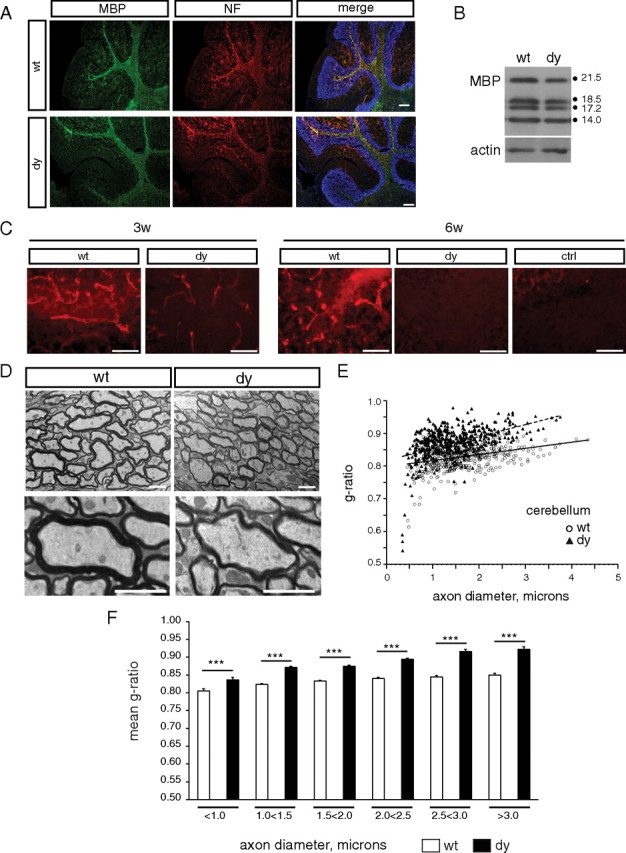
Myelin abnormalities in dy/dy cerebella. A, Immunocytochemistry to visualize MBP (green) and neurofilament (NF; red) in wild-type (wt) and dy/dy (dy) 3-week-old cerebella. Sections are counterstained with DAPI (blue) to visualize nuclei. Scale bar, 200 μm. B, Lysates from 3-week-old cerebella were evaluated by Western blot to detect MBP relative to actin as a loading control. MBP isoforms 21.5, 18.5, 17.2 and 14.0 kDa are indicated. C, Laminin α2 subunit immunoreactivity in wild-type and dy/dy cerebellar white matter at 3 weeks (3w) and 6 weeks (6w). Low-intensity laminin-α2 immunoreactivity was observed in 3-week-old dy/dy cerebella but this residual immunoreactivity was absent at 6 weeks. Scale bars, 50 μm. D, EM analysis of the cerebellum revealed thinner myelin. Representative electron micrographs of central cerebellar sagittal sections from 6-week-old mice; scale bars of 2 μm are shown in both low- and high-magnification images. E, Cerebellar g-ratios were plotted as a function of axon diameter for wild type (open circles) and dy/dy (black triangles); increased median g-ratios were observed overall in dy/dy cerebella (0.832 in wild-type vs 0.874 in dy/dy cerebella, p < 0.001). F, Cerebellar g-ratios grouped by axon diameter revealed significantly thinner myelin in dy/dy cerebellar axons of all sizes. Mean g-ratio ± SEM are shown (***p < 0.001).
We next evaluated whether the optic nerve, previously reported to show myelin abnormalities (Chun et al., 2003), also showed delayed oligodendrocyte maturation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). CC1 immunoreactivity revealed fewer mature oligodendrocytes per area in dy/dy optic nerves relative to those in wild-type littermates (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material; Table 1). We also confirmed that dy/dy optic nerves at 6 weeks of age were dysmyelinated relative to wild-type littermates using transmission electron microscopy (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). In particular, significantly increased g-ratios were observed in a subset of axons larger than 1.0 μm in diameter (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). We next analyzed a variety of brain regions using transmission electron microscopy to determine whether, in general, delays or deficits in oligodendrocyte maturation corresponded with abnormal myelination (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We observed significant increases in g-ratio, indicative of thinner myelin, in cerebellum (Fig. 2), optic nerve (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and a subset of small caliber axons in the brainstem (supplemental Fig. 2E, available at www.jneurosci.org as supplemental material). In contrast, however, the anterior commissure and corpus callosum showed no significant change in g-ratio but showed increased percentages of unmyelinated axons (supplemental Fig. 2A–D, available at www.jneurosci.org as supplemental material). We noted that, overall, the degree of regional abnormalities in oligodendrocyte maturation at 6 weeks in general correlated with the degree of myelin abnormalities at 6 weeks (Figs. 1, 2; Table 1; supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material).
Delayed differentiation of oligodendrocyte progenitors in dy/dy brains
An observed decrease in mature oligodendrocytes at 3 weeks could result from a decreased availability of oligodendrocyte progenitors in future white matter target regions. Alternatively, oligodendrocyte progenitors may be available in target regions, but may differentiate poorly or on a slower time scale. A third possibility is that progenitors differentiate in a timely manner and in appropriate numbers, but that these newly matured oligodendrocytes are then selectively eliminated via cell death. To distinguish between these possibilities, we first investigated whether dy/dy brains contained an adequate supply of oligodendrocyte progenitors in the correct locations by examining the numbers of PDGFRα(+) cells per area in the same eight regions that were analyzed for the presence of mature oligodendrocytes (Table 2). At 3 weeks, we found that levels of oligodendrocyte progenitors differed significantly between dy/dy brains and their wild-type littermates (Fig. 3). In 3-week-old corpus callosum, we found a significant increase in the number of PDGFRα(+) cells per mm2 in dy/dy animals compared with wild-type littermates (Fig. 3A; Table 2) (1098.7 ± 107.0 vs 763.4 ± 65.5, respectively, n = 3, p = 0.0193). By 6 weeks, however, the density of PDGFRα(+) cells in the corpus callosum of both wild-type and dy/dy animals had decreased, although dy/dy corpus callosum still had an increased density of PDGFRα(+) oligodendrocyte progenitors relative to wild-type littermates (Fig. 3A; Table 2) (917.4 ± 78.9 cells per mm2 in dy/dy vs 413.5 ± 59.1 in wild type, n = 3, p = 0.0212).
Table 2.
PDGFRα(+) cells per square millimeter
| Region | 3 weeks |
6 weeks |
3 wk vs 6 wk |
||||||
|---|---|---|---|---|---|---|---|---|---|
| wt | dy | p value: wt vs dy | wt | dy | p value: wt vs dy | p value: wt | p value: dy | ||
| 1 | Corpus callosum | 763.4 ± 65.5 | 1098.7 ± 107.0 | 0.0193* | 413.5 ± 59.1 | 917.4 ± 78.9 | 0.0212* | 0.0540, n.s. | 0.0265* |
| 2 | Lateral septum | 735.6 ± 107.4 | 982.4 ± 109.2 | 0.3624, n.s. | 449.2 ± 127.6 | 547.1 ± 62.5 | 0.3690, n.s. | 0.3074, n.s. | 0.0124* |
| 3 | Anterior commissure | 868.3 ± 80.0 | 1307.6 ± 73.5 | 0.0993, n.s. | 501.1 ± 125.9 | 726.7 ± 42.1 | 0.1630, n.s. | 0.2112, n.s. | 0.0229* |
| 4 | Cortex, all | 790.9 ± 50.9 | 1136.4 ± 73.6 | 0.0045** | 439.4 ± 19.7 | 823.1 ± 39.7 | 0.0137* | 0.0294* | 0.0166* |
| 5 | Cerebellum, central | 255.7 ± 18.0 | 622.6 ± 129.6 | 0.1108, n.s. | 364.2 ± 12.4 | 534.3 ± 40.0 | 0.0367* | 0.0134* | 0.5860, n.s. |
| 6 | Cerebellum, all | 333.6 ± 28.7 | 636.3 ± 100.9 | 0.1317, n.s. | 407.6 ± 19.8 | 593.5 ± 21.6 | 0.0340* | 0.1478, n.s. | 0.7600, n.s. |
| 7 | Brain stem | 259.7 ± 13.6 | 259.4 ± 34.1 | 0.9953, n.s. | 475.7 ± 1.4 | 499.2 ± 95.4 | 0.8305, n.s. | 0.0032** | 0.1239, n.s. |
| 8 | Spinal cord | 478.1 ± 165.4 | 706.8 ± 169.1 | 0.0104* | |||||
| 9 | Optic nerve | 745.8 ± 22.0 | 1256.9 ± 83.8 | 0.0766, n.s. | |||||
wt, Wild type; dy, dy/dy; wk, week; n.s., not significant; *p < 0.05;
**p < 0.01.
Figure 3.
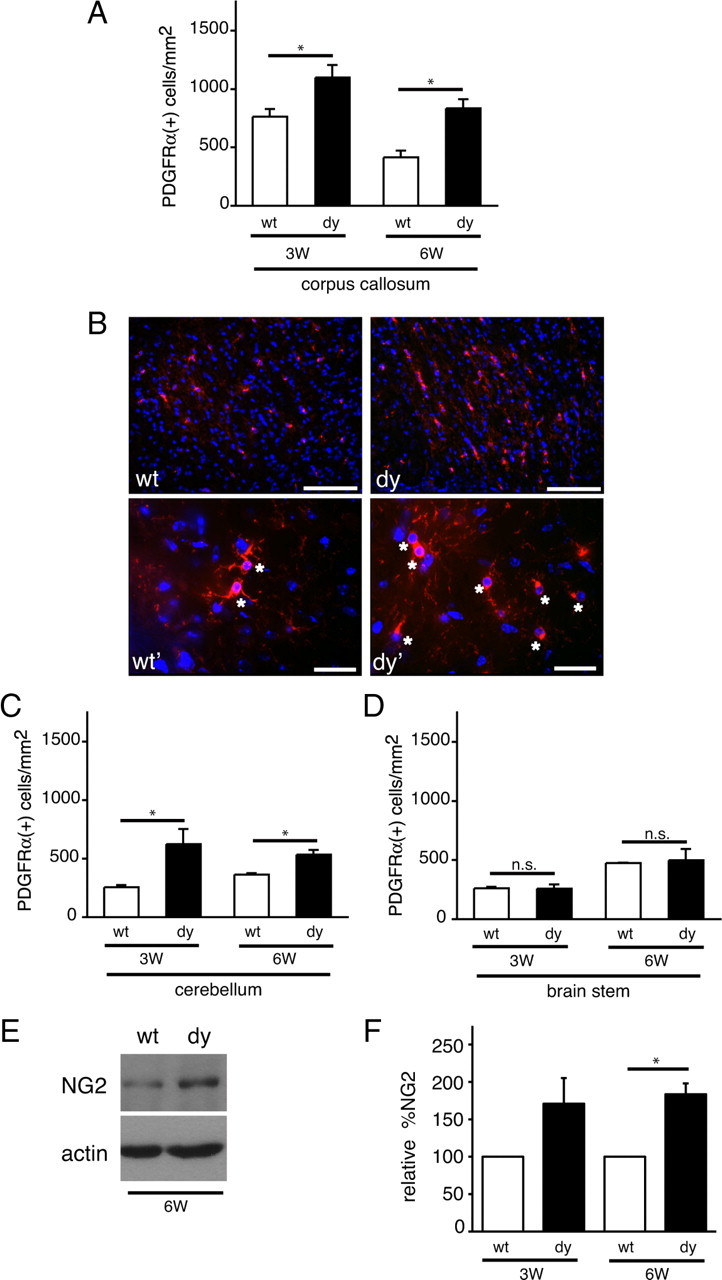
Oligodendrocyte progenitors accumulate in dy/dy brains. A, Oligodendrocyte progenitor cells per square millimeter in the corpus callosum were measured in wild-type (wt; white bars) and dy/dy (dy; black bars) littermates at 3 weeks (3W) and 6 weeks (6W) of age using PDGFRα immunocytochemistry. Graphs are mean (±SEM) counts obtained from 7 different areas of the corpus callosum (n = 3; *p < 0.05). B, Top panels, Representative images of PDGFRα(+) oligodendrocyte progenitors (red) in the corpus callosum of 3-week-old wild-type and dy/dy brains. Nuclei are visualized using DAPI (blue). Scale bar, 100 μm. Bottom panels, Representative images of PDGFRα(+) oligodendrocyte progenitors (red; indicated by *) in the corpus callosum of 3-week-old wild-type (wt') and dy/dy (dy') brains obtained at higher magnification. Scale bar, 25 μm. C, Oligodendrocyte progenitor cells per mm2 in the proximal cerebellar white matter were measured in wild-type (white bars) and dy/dy (black bars) littermates at 3 weeks and 6 weeks of age. Graphs are mean (±SEM) counts obtained from 6 different areas of the proximal cerebellar white matter (n = 3; *p < 0.05). D, Oligodendrocyte progenitor cells per mm2 in the brainstem were measured in wild-type (white bars) and dy/dy (black bars) littermates at 3 weeks and 6 weeks of age. Graphs are mean (±SEM) counts obtained from 3 different areas of the brainstem (n = 3; n.s, not significant). E, Lysates from 6-week-old cerebella were evaluated by Western blot to detect NG2 protein relative to actin as a loading control. A representative blot from a wild-type and dy/dy littermate is shown. F, Relative densitometry of NG2 protein levels at 3 weeks and 6 weeks in dy/dy cerebella compared with paired littermate wild-type cerebella (n = 4, *p < 0.05).
To investigate whether increased numbers of PDGFRα(+) oligodendrocyte progenitors occurred regionally versus globally, we evaluated progenitor numbers in cerebellum, brainstem, spinal cord, and in a variety of additional brain regions in which reduced numbers of mature oligodendrocytes had been observed (Fig. 3; Table 2). We first noted that the central white matter of the cerebellum had a similar pattern of elevated numbers of PDGFRα(+) cells, such that dy/dy cerebella at 3 weeks showed significantly higher numbers of PDGFRα(+) cells per mm2 compared with wild-type littermates (Fig. 3C) (622.6 ± 129.6 vs 255.7 ± 18.0, respectively, n = 3, p = 0.0486). By 6 weeks, however, PDGFRα(+) cell density in wild-type cerebella remained unchanged compared with 3 week levels, whereas PDGFRα(+) cell density in dy/dy cerebella remained significantly elevated (Fig. 3C; Table 2) (534 ± 40.0 cells per mm2 vs 364.2 ± 12.4, n = 3, p = 0.0367). In dy/dy brainstem, however, we did not observe any change in PDGFRα(+) cell density (Fig. 3D; Table 2) (259.4 ± 34.1 cells per mm2 in dy/dy vs 259.7 ± 13.6 in wild type, n = 3, p = 0.9953), which may reflect the fact that the brainstem is one of the earliest regions to myelinate, and, thus, oligodendrocyte development may have normalized by 3 weeks.
In addition to elevated numbers of oligodendrocyte progenitors, dy/dy brain lysates were observed to have elevated levels of NG2 (Figs. 3E,F) (183.5 ± 28.9%, n = 4, p = 0.022, at 6 weeks), a chondroitin sulfate proteoglycan expressed in oligodendrocyte progenitors but not in mature oligodendrocytes (Stallcup, 1981; Nishiyama et al., 1996). NG2 immunostaining confirmed that dy/dy cerebral cortices had a higher degree of NG2 immunoreactivity in the cerebellum and in the corpus callosum compared with wild-type littermates (data not shown). Overall, the decreased expression of maturation markers (i.e., CC1 and MBP) accompanied by accumulation of progenitor markers (i.e., PDGFRα and NG2) indicates that the timing of oligodendrocyte maturation could be altered in laminin-α2-deficient brains.
The Src family kinase Fyn is dysregulated in dy/dy brains
The Src family kinase (SFK) Fyn is required for normal myelination such that mice lacking Fyn, as well as mice expressing a catalytically inactive Fyn, have myelination defects (Umemori et al., 1994; Osterhout et al., 1999; Sperber et al., 2001). Fyn activity is tightly regulated by phosphorylation at several highly conserved tyrosines: an autophosphorylation site at tyrosine 418, and an inhibitory site at the C-terminal tyrosine 529. Because Fyn has been shown to be activated downstream of laminin in cultured oligodendrocytes (Colognato et al., 2004), we examined Fyn in laminin-deficient brains. Western blot analysis of dy/dy cortical lysates, however, revealed no significant differences in levels of Fyn protein compared with wild-type littermates (Fig. 4A,B) (114.7 ± 1.7%, n = 3). We next evaluated the regulation of Fyn using antibodies against the SFK phosphorylation sites at Y418 (catalytic) and Y529 (regulatory). Oligodendrocytes express two SFKs, Fyn and Lyn, but Lyn protein and Lyn activity is virtually undetectable in myelin by 2 weeks of age (Krämer et al., 1999). By 3 weeks of age, Fyn and its kinase activity are elevated and correlate with the period of active myelination (Krämer et al., 1999; Lu et al., 2005). Using Western blots, we observed that levels of phosphorylated Y529 (relative to total Fyn levels) were increased by almost twofold in 3-week-old dy/dy cortices compared with wild-type cortices (Fig. 4A,B) (190.9 ± 36.5%, n = 3, p = 0.0022) and remained elevated at 6 weeks of age, albeit with increased variability (174.9 ± 102.6%, n = 6, p = 0.24) (data not shown). The increased levels of phosphorylated Y529 indicated that Fyn was dysregulated in laminin-deficient mice.
Figure 4.

Fyn is dysregulated in dy/dy brains. A, Lysates from 3-week-old cerebral cortices (CTX) were evaluated by Western blot to detect Fyn, phosphorylated tyrosine 529 (pY529), Csk, Cbp, and actin. Representative blots from 3-week-old wild-type (wt) and dy/dy (dy) littermates are shown. B, Densitometric analysis of Fyn, phospho-Fyn, and Fyn regulatory proteins Csk and Cbp: mean Fyn protein, mean phospho-Y529 immunoreactivity normalized to total Fyn protein, mean Csk protein, and mean Cbp protein are shown (n = 3; **p < 0.01; *p < 0.05). Fyn, Csk, and Cbp protein levels were normalized to actin protein levels. C, Lysates from 3-week-old cerebella (CB) were evaluated by Western blot to detect Fyn, phosphorylated tyrosine 529 (pY529), Csk, Cbp, and actin. Representative blots from 3-week-old wild-type and dy/dy littermates are shown. D, Densitometric analysis of Fyn, phospho-Fyn, and Fyn regulatory proteins Csk and Cbp: mean Fyn protein, mean phospho-Y529 immunoreactivity normalized to total Fyn protein, mean Csk protein, and mean Cbp protein are shown (n = 3; *p < 0.05). Fyn, Csk, and Cbp protein levels were normalized to actin protein levels.
Similar to dy/dy cortices, the cerebella of dy/dy mice expressed normal levels of Fyn (Fig. 4C,D) (103.3 ± 8.7% at 3 weeks, n = 3; 95.5 ± 35.2% at 6 weeks) (data not shown). Levels of phosphorylated Y529 (relative to total Fyn), however, were higher in dy/dy cerebella than in wild-type cerebella (Fig. 4C,D) (147.3 ± 17.1%, n = 3, p = 0.026), although the degree of elevation was not as strong as in the cortex (190.9 ± 36.5% in the cortex, vs 147.3 ± 17.1% in the cerebellum). In contrast to cerebral cortices, however, the levels of phosphorylated Y529 in laminin-deficient cerebella returned to normal at 6 weeks (111.1 ± 34.6%, p = 0.56) (data not shown). Together, these results indicate that Fyn is dysregulated in the brains of laminin-deficient mice; however, the cortex and cerebellum exhibited regional variation in terms of how much Fyn dysregulation occurred. It should be noted, however, that no significant changes in Fyn protein, Fyn phosphorylation, or Csk protein were observed between the cerebellum and cerebral cortex of wild-type mice (n = 3) (data not shown). The observed Fyn dysregulation in laminin-deficient mice indicates that similar signaling abnormalities may contribute to the CNS pathology seen in MDC1A.
dy/dy brains contain elevated levels of Csk and Cbp, negative regulators of Fyn
Based on our observations that Fyn was regulated inappropriately in laminin-deficient brains, we next examined the expression of known negative regulators of Fyn and other Src family kinases. The protein Csk, shown previously to be expressed in oligodendrocytes, has been shown to be the principal kinase to regulate Fyn via phosphorylation of the inhibitory Y529 site (Takeuchi et al., 1993; Colognato et al., 2004). And, in the presence of laminin-2, cultured oligodendrocytes were previously found to have decreased levels of phosphorylated Fyn Y529, suggesting that Csk activity can be affected by laminin (Colognato et al., 2004). In conjunction with elevated levels of Y529 phosphorylation, dy/dy cortices and cerebella at 3 weeks of age showed increased Csk protein compared with wild-type littermates (Fig. 4A,B, Csk in dy/dy cortices, 180.7 ± 30.3%, n = 3, p = 0.027; Fig. 4D, 154.6 ± 31.5%, p = 0.134 in cerebella; Csk normalized to actin levels in each lysate). By 6 weeks, Csk levels decreased in both wild-type and dy/dy cortices, compared with their respective levels at 3 weeks (data not shown).
Cbp is required for normal Csk function. This transmembrane adaptor protein is found in lipid raft microdomains and has been shown to recruit Csk to the membrane where myristoylated molecules such as Fyn are located (Kawabuchi et al., 2000; Takeuchi et al., 2000). We found that Cbp levels in the cortices of 3-week-old dy/dy mice were significantly elevated compared with wild-type levels (Fig. 4A,B) (147.8 ± 34.9%, n = 4, p = 0.029). Cbp levels in dy/dy cerebella were also elevated relative to Cbp in wild-type cerebella (Fig. 4D) (163.8 ± 68.5%, n = 3, p = 0.18). By 6 weeks, Cbp levels in dy/dy cortices and cerebella remained somewhat elevated, but only by ∼28% of wild-type levels (128.7 ± 35.9%, n = 3, p = 0.31 in cortices; and 128.6 ± 54.5%, n = 3, p = 0.65 in cerebella). Csk is known to bind to a phosphorylated tyrosine 317 on Cbp; this binding both activates Csk and increases Csk affinity for Fyn (Matsuoka et al., 2004). In dy/dy cortices, we observed levels of Cbp Y317 phosphorylation (relative to total Cbp) that were similar to wild-type levels at all time points and regions investigated (data not shown). Together, the elevated levels of both Csk and Cbp in laminin-deficient dy/dy cerebral cortices suggest that these molecules may contribute to Fyn dysregulation.
Laminin promotes oligodendrocyte progenitor cell differentiation
While we observed changes in Fyn regulation in vivo, it remained unclear whether these changes were occurring in oligodendrocyte progenitors or oligodendrocytes failing to make appropriate laminin contact. To determine whether the loss of laminin altered Fyn phosphorylation in oligodendrocyte progenitor cells, we placed isolated rat OPCs in contact with laminin-2 substrates and monitored Fyn Y529 phosphorylation (Fig. 5). We found that on laminin substrates Fyn phosphorylation at Y529 was decreased substantially (Fig. 5A,B) (37.44 ± 5.8% relative to phosphorylation levels on PDL substrates, p = 0.0045). This laminin-mediated suppression of Fyn phosphorylation in cultured rodent OPCs suggested that the abnormally high degree of phosphorylation of Fyn Y529 in dy/dy brains may be due to loss of appropriate laminin contact.
Figure 5.

Laminin promotes the differentiation of oligodendrocyte progenitors using a Fyn-dependent mechanism. A, Oligodendrocyte progenitor cells added to PDL or laminin-2 (Lm) substrates for 2 h were lysed and evaluated by Western blot to detect phospho-Y529 relative to total Fyn. B, Graph depicts mean pY529 phosphorylation, relative to total Fyn levels, on laminin-2 substrates (Lm) relative to PDL substrates (**p < 0.01, n = 3). C, Oligodendrocyte progenitor cells were added to PDL or laminin-2 (Lm) substrates for 24 h in minimal medium, in the presence of 1 μm PP2 or equivalent volume of DMSO vehicle as a control (ctrl). CNP immunoreactivity (green) was performed to visualize newly formed oligodendrocytes, in conjunction with DAPI to view total nuclei (blue). Scale bar, 50 μm. D, Graph depicts the mean percentage of cells that show CNP immunoreactivity per field, in 3 independent experiments (n = 3; *p < 0.05 for comparisons between PDL and Lm under control conditions, and, between control and PP2 conditions on Lm).
The decreased numbers of mature oligodendrocytes and increased numbers of oligodendrocyte progenitors that were together observed in laminin-deficient brains indicated that laminin contact with oligodendrocyte progenitors might be able to promote or change the timing of differentiation into newly formed oligodendrocytes. To determine whether laminin contact could promote oligodendrocyte progenitor cell differentiation we placed oligodendrocyte progenitors on laminin versus PDL substrates for 24 h and evaluated the percentage of cells that expressed CNP, a protein expressed by newly formed oligodendrocytes but not oligodendrocyte progenitors (Fig. 5C,D). We found that oligodendrocyte progenitors grown on laminin showed an ∼2-fold increase in the percentage of cells expressing CNP after 24 h (Fig. 5D) (n = 3, p = 0.0128). In contrast, cells treated with PP2, a Src family kinase inhibitor, did not show any laminin-mediated enhancement of oligodendrocyte progenitor differentiation (Fig. 5D) (n = 3, p = 0.9801). Together, these results suggest that laminin promotes the transition from oligodendrocyte progenitor cell to newly formed oligodendrocyte and this laminin-mediated effect depends on Src family kinase activity. After 4 d on laminin or PDL substrates, however, we did not observe any substrate-mediated difference in the percentage of CNP(+) cells (data not shown), suggesting that laminin may accelerate differentiation but is not required for it per se. This finding would be consistent with in vivo observations in which laminin-deficient animals at 3 weeks show delays in differentiation that are partially or fully remedied by 6 weeks, depending on the region (Figs. 1–3; Tables 1–2).
Dysregulation of p27 occurs in the dy/dy cerebellum
Because the timing of oligodendrocyte differentiation is altered in laminin-deficient brains, we examined the expression of the cyclin-dependent kinase inhibitor, p27, a protein known to exert temporal control over oligodendrocyte development. p27 is known to be upregulated progressively as OPCs undergo cell division, and p27 protein levels peak with the onset of active myelination (for review, see Nguyen et al., 2006). There were small decreases in p27 levels in both the dy/dy cerebral cortex (85.2 ± 22.3%, n = 3, p = 0.38) and dy/dy cerebellum (93.7 ± 32.5%, n = 3, p = 0.93) compared with their wild-type littermates at 3 weeks (data not shown). By 6 weeks, however, p27 levels were substantially higher in both brain regions of dy/dy mice relative to that in wild-type littermates (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material) (298.4 ± 223.1%, n = 3, p = 0.12 in the cortex; 224.0 ± 127.2%, n = 3, p = 0.047 in the cerebellum). Interestingly, we also observed increased levels of the RNA binding protein, QKI, in the cerebella of dy/dy brains (183.0 ± 73.2% of wild-type levels, n = 4, p = 0.035, data not shown). QKI is a downstream target of Fyn and has been shown to stabilize mRNAs critical to the myelination process including p27 and MBP transcripts (Larocque and Richard, 2005; Lu et al., 2005). Finally, we monitored oligodendrocyte progenitor p27 protein levels at 2 and 24 h after exposure to laminin (supplemental Fig. 3C,D, available at www.jneurosci.org as supplemental material). At 2 h postattachment, no differences were observed in p27 levels between laminin and PDL substrates. We found that by 24 h, however, cells grown on laminin showed lower levels of p27 (67.3 ± 8.5%) than cells grown on PDL (supplemental Fig. 3D, available at www.jneurosci.org as supplemental material) (n = 3, p = 0.0442).
Oligodendrocyte survival is unchanged in adult laminin-deficient mice
Cultured oligodendrocytes have been shown to die more readily in the presence of antibodies that block the laminin receptor α6β1 integrin (Frost et al., 1999; Colognato et al., 2002), and newly formed oligodendrocytes show increased cell death in the developing brainstem of α6-integrin knock-out mice (Colognato et al., 2002). These findings prompted us to ask whether in laminin-deficient brains, the decrease in mature oligodendrocyte numbers could be the result of increased cell death. To determine whether white matter tracts showed normal levels of death we performed TUNEL on wild-type and dy/dy littermate brains at 3 weeks and 6 weeks (Fig. 6; Table 3). We evaluated 9 separate fields within the corpus callosum of each animal and found no difference in TUNEL(+) cells per area between wild-type and dy/dy animals (Fig. 6A; Table 3) (p = 0.6100, n = 4 at 3 weeks; p = 0.8863, n = 4 at 6 weeks). To determine whether mature oligodendrocytes were dying selectively in dy/dy brains, we performed TUNEL in conjunction with CC1, an antibody that labels the cell bodies of mature oligodendrocytes. We found very few dying CC1(+) cells within the corpus callosum (<1% in all animals evaluated), and we did not find any change in TUNEL in the CC1-positive cell population of dy/dy brains at either 3 or 6 weeks (Fig. 6B,C; Table 3) (n = 3; p = 0.6991 for 3 weeks; p = 0.7336 for 6 weeks). We also determined the percentage of TUNEL(+) in the PDGFRα(+) population and in this case observed virtually no overlap between TUNEL and PDGFRα(+) cells (Table 3; 0.3 ± 0.3% in wild type and none in dy/dy, n = 3).
Figure 6.

Oligodendrocyte death in dy/dy adult brains. A, The mean number of TUNEL-positive cells per area (mm2) is shown for wild-type (wt) and dy/dy (dy) corpus callosum at 3 weeks and 6 weeks (n = 4). B, The percentage of TUNEL-positive cells within the CC1(+) cell population is shown for wild-type and dy/dy corpus callosum at 3 weeks and 6 weeks (n = 3). C, CC1 immunocytochemistry (green) to visualize mature oligodendrocyte cell bodies was performed in combination with TUNEL to detect dying cells (red). Representative fields are shown from 3-week-old wild-type and dy/dy corpus callosum. Arrows depict TUNEL-positive cells; scale bar denotes 100 μm. n.s., Not significant.
Table 3.
TUNEL
| 3 weeks |
6 weeks |
|||||
|---|---|---|---|---|---|---|
| wt | dy | p value | wt | dy | p value | |
| TUNEL(+) cells per mm2 | ||||||
| Corpus callosum | 54.2 ± 4.9 | 48.4 ± 13.4 | 0.6100 (n = 4) | 33.2 ± 7.0 | 31.2 ± 18.3 | 0.8863 (n = 4) |
| Cerebellar white matter | 60.1 ± 16.0 | 56.2 ± 12.5 | 0.6221 (n = 4) | 39.7 ± 8.6 | 26.5 ± 5.7 | 0.1437 (n = 3) |
| Brainstem | 42.2 ± 12.5 | 44.2 ± 8.9 | 0.8528 (n = 4) | 38.9 ± 9.4 | 41.4 ± 9.0 | 0.6904 (n = 4) |
| %TUNEL(+) of CC1(+) | ||||||
| Corpus callosum | 0.3 ± 0.2 | 0.5 ± 0.4 | 0.6991 (n = 3) | 0.4 ± 0.2 | 0.6 ± 0.3 | 0.7336 (n = 3) |
| Cerebellar white matter | 0.3 ± 0.3 | 1.5 ± 0.8 | 0.1835 (n = 3) | 0.6 ± 0.6 | 0.3 ± 0.3 | 0.7664 (n = 3) |
| Brainstem | 0.3 ± 0.3 | 0.0 ± 0.0 | N.A. (n = 3) | 0.0 ± 0.0 | 0.8 ± 0.5 | N.A. (n = 3) |
| %TUNEL(+) of PDGFRα(+) | ||||||
| Corpus callosum | 0.3 ± 0.3 | 0.0 ± 0.0 | N.A. (n = 3) | 0.2 ± 0.2 | 0.0 ± 0.0 | N.A. (n = 3) |
| Cerebellar white matter | 0.6 ± 0.6 | 1.4 ± 0.2 | 0.2586 (n = 3) | 0.3 ± 0.3 | 0.6 ± 0.4 | 0.5647 (n = 3) |
| Brainstem | 1.3 ± 0.7 | 1.6 ± 0.8 | 0.8396 (n = 3) | 0.0 ± 0.0 | 0.6 ± 0.6 | N.A. (n = 3) |
wt, Wild type; dy, dy/dy; N.A., not applicable.
Furthermore, we evaluated overall TUNEL(+) cells per area, the percentage of TUNEL(+) cells within the CC1(+) population, and the percentage of TUNEL(+) cells within the PDGFRα(+) population in two additional brain regions: the cerebellar white matter and the brainstem (Table 3). In the cerebellar white matter, a minimum of 10 fields were evaluated per animal (n = 3), and in the brainstem, a minimum of 3 different fields were evaluated per animal (n = 3). Overall, very little death was observed, with no significant differences between wild type and dy/dy at either 3 weeks or 6 weeks (Table 3). dy/dy animals showed increased death within the CC1(+) cell population of cerebellar white matter at 3 weeks; however, this increase was not statistically significant (Table 3; 0.3 ± 0.3% in wild type vs 1.5 ± 0.8% in dy/dy, n = 3, p = 0.1835). Likewise, dy/dy animals had increased death within the PDGFRα(+) cell population of cerebellar white matter at 3 weeks; however, this increase was not statistically significant (Table 3; 0.6 ± 0.6% in wild type vs 1.4 ± 0.2% in dy/dy, n = 3, p = 0.2586). These findings indicate that, at least at 3 weeks of age and beyond, preferential death of cells in the oligodendrocyte lineage is unlikely to play a major role in altering the numbers of oligodendrocytes and oligodendrocyte progenitors. It remains a possible, however, that oligodendrocyte death at earlier time points could occur and contribute to the decrease in oligodendrocyte numbers observed at 3 weeks.
Discussion
In the CNS of laminin-α2-deficient dy/dy mice, oligodendrocyte progenitors accumulate due to delayed differentiation. By 6 weeks, however, oligodendrocyte development begins to catch up across multiple regions of the dy/dy brain and spinal cord. In several regions, however, elevated numbers of oligodendrocyte progenitors persist in conjunction with fewer mature oligodendrocytes and abnormal myelination. These findings indicate that laminin-α2 deficiency stalls or delays oligodendrocyte lineage progression, and, while widespread, the degree of developmental delay varies regionally. Fyn-signaling abnormalities were, furthermore, identified in dy/dy brains, which may, at least in part, account for perturbed oligodendrocyte lineage progression (Model, Fig. 7). In particular, Fyn was inappropriately phosphorylated at its inhibitory tyrosine; this dysregulation was accompanied by an increase in Csk and Cbp, oligodendrocyte proteins that suppress Fyn activity. These findings provide new insight into the molecular mechanisms by which α2-containing laminins regulate oligodendrocyte progenitor maturation.
Figure 7.

Laminin and oligodendrocyte progenitor development. Oligodendrocyte progenitor differentiation is inappropriately delayed in the brains of laminin-deficient mice. Regulatory molecules that can suppress Fyn activity, Csk and Cbp, are elevated in laminin-deficient brains. A normal role of laminin is, therefore, to suppress Csk and Cbp, and thus promote Fyn activation and oligodendrocyte maturation. In addition, laminin may activate Fyn independent of Csk/Cbp suppression (Colognato et al., 2004). It remains unknown if laminin is able to promote oligodendrocyte differentiation through Fyn-independent mechanisms (dashed line).
The dy/dy mouse model for muscular dystrophy is the result of laminin-α2 deficiency. While the mutation has not been identified, it has been linked to the LAMA2 locus (Sunada et al., 1994). Chun et al. (2003) reported that 5-week-old dy/dy mice have CNS myelination abnormalities that include less MBP protein in the forebrain, and fewer mature oligodendrocytes and myelinated axons in the corpus callosum and optic nerve. In the current study, we observed that in 3-week-old mice a similar decrease in mature oligodendrocytes exists in the corpus callosum, as well as in many other brain regions. By 6 weeks, however, the density of CC1(+) cells had risen in most brain regions but still remained lower than in wild-type littermates. In addition to decreased numbers of oligodendrocytes, we found that dy/dy brains, in almost all regions examined, showed significant increases in the number of oligodendrocyte progenitors. The presence of excess progenitors suggested that the observed reduction in oligodendrocytes is neither due to a failure of oligodendrocyte progenitors to be generated from germinal zones, nor due to a failure of progenitors to arrive at white matter tracts. Instead, oligodendrocyte differentiation may be stalled or delayed such that progenitors accumulate at their target destinations.
Laminin-deficient brains showed delayed differentiation in almost all regions evaluated. The timing of this delay, however, varied regionally. In the cerebellum, for instance, oligodendrocyte density was relatively normal at 3 weeks but lagged behind at 6 weeks both in terms of oligodendrocyte density (Fig. 1; Table 1) and thinner myelin, as evidenced by increased g-ratios (Fig. 2). Conversely, delays in oligodendrocyte differentiation in the corpus callosum, while pronounced at 3 weeks, were no longer significant at 6 weeks and corresponded with relatively normal myelin thickness, albeit with an ∼37% increase in unmyelinated axons (Fig. 1; Table 1; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In almost every region, therefore, an inverse relationship between increased numbers of oligodendrocyte progenitors and decreased numbers of mature oligodendrocytes was observed (Tables 1, 2). Some exceptions to this correlation were found; in particular, the brainstem contained decreased numbers of mature oligodendrocytes without a corresponding increase in the numbers of oligodendrocyte progenitors (Tables 1, 2) or significant changes in myelination (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). It is possible, therefore, that early myelinating regions such as the brainstem either are not influenced by laminin or have compensatory changes that occur by 3 weeks of age.
Laminin-deficient MDC1A patients also show regional differences in brain abnormalities. In one study, white matter signals were abnormal to varying degrees in the cerebral cortex of all patients, whereas the cerebellum and the brainstem showed T2 abnormalities in only ∼20% of patients (Leite et al., 2005). These findings indicate that laminin's influence on CNS development may be different in different brain regions, or, alternatively, that development in different brain regions may be differentially modulated by other factors that in turn influence their response to laminins. Another possibility is that differential compensation by additional laminin heterotrimers may take place in different brain regions; e.g., the loss of α2-laminins is compensated for in the cerebellum but not the cortex. This is observed in the peripheral nervous system, where α2-laminin deficiencies cause myelination abnormalities; these animals show upregulated laminin-α4 expression, but only in selected regions (Patton et al., 1997; Yang et al., 2005). In the current study, we found that residual laminin-α2 immunoreactivity was detectable in the cerebellum and brainstem of 3-week-old dy/dy brains but was undetectable by 6 weeks (Fig. 2; supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material). These findings suggest that variable degrees of laminin-α2 expression could also account for the different degrees of oligodendrocyte differentiation delay observed regionally in dy/dy brains.
In addition to a differentiation delay, another possible explanation to account for fewer mature oligodendrocytes is increased cell death. Laminin substrates are known to promote the survival of newly formed oligodendrocytes in vitro (Frost et al., 1999; Colognato et al., 2002). And, loss of the laminin receptor α6β1 integrin causes increased death in premyelinating axon tracts in both the brainstem and the cerebellum (Colognato et al., 2002; Benninger et al., 2006). At 3 weeks, however, dy/dy mice did not show increased death in either oligodendrocytes or oligodendrocyte progenitors (Fig. 6; Table 3). A small increase in both the percentage of TUNEL-positive-CC1(+) cells and TUNEL-positive-PDGFRα(+) cells was observed in laminin-deficient cerebellar white matter (Table 3), but these increases were not statistically significant. There is therefore currently no evidence to support a role for death in the diminished numbers of mature oligodendrocytes at 3 weeks. Decreased survival of oligodendrocytes at earlier time points, however, cannot be ruled out. Unfortunately, dy/dy mice cannot be identified at earlier ages (see Materials and Methods). Additional studies using laminin gene knock-out mice will therefore be useful to investigate the consequence of laminin loss on cell survival earlier in the developing brain.
What are the potential signaling mechanisms by which laminins regulate the transition from oligodendrocyte progenitor to myelinating oligodendrocyte? Several signaling molecules are influenced by laminins in oligodendrocyte cultures, including ILK, Fyn, FAK, MAPK, and PI3K (Colognato et al., 2002, 2004; Baron et al., 2003; Chun et al., 2003; Hoshina et al., 2007). In the current study, we examined whether Fyn was regulated by laminins in vivo and found that phosphorylation of the Fyn negative regulatory tyrosine was enhanced in dy/dy brains (Fig. 4). And, we found that oligodendrocyte progenitor cell attachment to laminin caused a decrease in phosphorylation of this same negative regulatory tyrosine (Fig. 5). Together, these findings suggest that laminin regulates the transition from oligodendrocyte progenitor to oligodendrocyte, at least in part, by modulating Fyn regulation. Fyn activity peaks during active myelination, and Fyn knock-out mice have less forebrain myelination; together these findings suggest that changes in Fyn regulation could alter the timing of oligodendrocyte development (Umemori et al., 1994; Sperber et al., 2001). Fyn-null mice also show changes in the ratio of exon2-containing MBP isoforms such that the 21.5 kDa isoform is decreased relative to other isoforms (Lu et al., 2005). In laminin-deficient mice, the ratio of the 21.5 kDa MBP isoform was also decreased, such that the percentage of 21.5 kDa MBP protein (relative to total MBP protein) drops by ∼50% (6.2 ± 7.1% total MBP protein in the wild type vs 3.1 ± 0.8% in dy/dy cerebral cortex at 3 weeks). The 21.5 kDa isoform is highly associated with active myelination; therefore, a trend toward a decrease in the 21.5 kDa MBP isoform is consistent with the hypothesis that laminin deficiency leads to a delayed developmental progression. Due to high variability, however, these trends were not statistically significant (n = 3, p = 0.4916).
In T-cells, Fyn activity is tightly regulated by Csk, the C-terminal Src Kinase (Palacios and Weiss, 2004). Csk in turn is regulated by its ability to bind to the transmembrane adapter molecule Cbp. Csk is the principal kinase that phosphorylates Fyn and other SFKs at SFK negative regulatory tyrosines and has thus been shown to suppress the ability of Fyn to phosphorylate downstream targets. Csk expression is normally highest in the developing embryonic brain, when Fyn activity is low (Inomata et al., 1994). During myelination, Csk levels decrease substantially compared with those in the embryonic brain. Here, we show that elevated levels of Csk and Cbp found in dy/dy brains correlate with increased phosphorylation of the Fyn negative regulatory tyrosine, providing a potential molecular basis for Fyn dysregulation in laminin-deficient brains. We cannot rule out, however, that increased levels of Csk and Cbp are also found in non-oligodendrocyte lineage cells.
Evidence from the current study indicates that the inappropriate accumulation of oligodendrocyte progenitors could reflect a dysregulation of the signaling mediators that control the timing of oligodendrocyte development. The timing of cell cycle exit regulates myelination onset, although the precise mechanisms that dictate when and where oligodendrocytes initiate the final phases of myelination remain to be elucidated. Several cyclin-dependent-kinase inhibitors, such as p27 and p57, however, are known to regulate the withdrawal of oligodendrocyte progenitors from the cell cycle (Nguyen et al., 2006). The levels of p27 in particular have been shown to peak with the onset of myelination. Here, we report that (1) laminin contact with OPCs suppresses p27 accumulation, and (2) p27 is increased in laminin-deficient brains (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Overexpression of p27 has been shown to block both oligodendrocyte progenitor division and differentiation, and thus results in a stalled progenitor phenotype (Tang et al., 1999). Here, we noted that increased p27 coincided in some cases with the accumulation of OPCs, raising the possibility that laminin normally promotes OPC differentiation through p27. In support of this, integrin engagement by extracellular matrix has been shown to regulate p27 by activating signaling pathways that control p27 protein degradation (Fu et al., 2007).
p27 levels are also regulated by mRNA stability. The RNA binding protein, QKI, enhances the stability of p27 mRNA in developing oligodendrocytes (Larocque et al., 2005). QKI is in itself an essential molecule for myelination: quaking viable mice have a mutation that causes the loss of particular QKI isoforms important for MBP mRNA stability, causing these mice to have severe myelination defects (Larocque and Richard, 2005). We found that QKI levels in laminin-deficient brains were elevated (data not shown), providing a possible mechanism for the observed increase in p27. Fyn is known to phosphorylate QKI and regulate its ability to stabilize and traffic MBP mRNA (Lu et al., 2005), thus providing a potential link between Fyn dysregulation, QKI/p27 dysregulation, and delayed oligodendrocyte differentiation observed in laminin-deficient animals. In summary, the current study has for the first time shown that laminins promote the transition from oligodendrocyte progenitor cell to oligodendrocyte and do so, at least in part, by modulating Fyn signaling. These findings suggest that dysregulated Fyn signaling may contribute to pathology in MDC1A laminin deficiencies and offer new insight into how laminins regulate oligodendrocyte development.
Footnotes
This study was supported by a National Multiple Sclerosis Society Career Transition Fellowship, the National Institute of Neurological Disorders and Stroke (Grant 5R01NS054042), and the NY State Department of Health (C020929). We thank members of the Colognato laboratory for helpful discussions, Ms. Noreen Bukhari (Stony Brook University) for help with statistical analysis, Dr. Joel Levine (Stony Brook University) for the generous gift of NG2 antibody, and Dr. Jon Lindquist (Otto-von-Guericke University) for the generous gift of Cbp/PAG antibody. In addition, we are very grateful to Susan Van Horn of the Stony Brook Central Microscopy Imaging Center facility for TEM sample preparation and thank Ms. Van Horn and Dr. Wiebke Möbius (Goettingen University) for providing helpful advice on TEM.
References
- Baron W, Decker L, Colognato H, ffrench-Constant C. Regulation of integrin growth factor interactions in oligodendrocytes by lipid raft microdomains. Curr Biol. 2003;13:151–155. doi: 10.1016/s0960-9822(02)01437-9. [DOI] [PubMed] [Google Scholar]
- Baron W, Colognato H, ffrench-Constant C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia. 2005;49:467–479. doi: 10.1002/glia.20132. [DOI] [PubMed] [Google Scholar]
- Benninger Y, Colognato H, Thurnherr T, Franklin RJ, Leone DP, Atanasoski S, Nave KA, Ffrench-Constant C, Suter U, Relvas JB. Beta1-integrin signaling mediates premyelinating oligodendrocyte survival but is not required for CNS myelination and remyelination. J Neurosci. 2006;26:7665–7673. doi: 10.1523/JNEUROSCI.0444-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery PC, ffrench-Constant C. Laminin-2/integrin interactions enhance myelin membrane formation by oligodendrocytes. Mol Cell Neurosci. 1999;14:199–212. doi: 10.1006/mcne.1999.0781. [DOI] [PubMed] [Google Scholar]
- Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T. Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J Cell Biol. 2003;163:397–408. doi: 10.1083/jcb.200304154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato H, Baron W, Avellana-Adalid V, Relvas JB, Baron-Van Evercooren A, Georges-Labouesse E, ffrench-Constant C. CNS integrins switch growth factor signalling to promote target-dependent survival. Nat Cell Biol. 2002;4:833–841. doi: 10.1038/ncb865. [DOI] [PubMed] [Google Scholar]
- Colognato H, Ramachandrappa S, Olsen IM, ffrench-Constant C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J Cell Biol. 2004;167:365–375. doi: 10.1083/jcb.200404076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato H, ffrench-Constant C, Feltri ML. Human diseases reveal novel roles for neural laminins. Trends Neurosci. 2005;28:480–486. doi: 10.1016/j.tins.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Colognato H, Galvin J, Wang Z, Relucio J, Nguyen T, Harrison D, Yurchenco PD, Ffrench-Constant C. Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development. 2007;134:1723–1736. doi: 10.1242/dev.02819. [DOI] [PubMed] [Google Scholar]
- Court FA, Wrabetz L, Feltri ML. Basal lamina: Schwann cells wrap to the rhythm of space-time. Curr Opin Neurobiol. 2006;16:501–507. doi: 10.1016/j.conb.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Frost EE, Buttery PC, Milner R, ffrench-Constant C. Integrins mediate a neuronal survival signal for oligodendrocytes. Curr Biol. 1999;9:1251–1254. doi: 10.1016/s0960-9822(99)80506-5. [DOI] [PubMed] [Google Scholar]
- Fu Y, Fang Z, Liang Y, Zhu X, Prins P, Li Z, Wang L, Sun L, Jin J, Yang Y, Zha X. Overexpression of integrin beta1 inhibits proliferation of hepatocellular carcinoma cell SMMC-7721 through preventing Skp2-dependent degradation of p27 via PI3K pathway. J Cell Biochem. 2007;102:704–718. doi: 10.1002/jcb.21323. [DOI] [PubMed] [Google Scholar]
- Hoshina N, Tezuka T, Yokoyama K, Kozuka-Hata H, Oyama M, Yamamoto T. Focal adhesion kinase regulates laminin-induced oligodendroglial process outgrowth. Genes Cells. 2007;12:1245–1254. doi: 10.1111/j.1365-2443.2007.01130.x. [DOI] [PubMed] [Google Scholar]
- Howe CL. Coated glass and vicryl microfibers as artificial axons. Cells Tissues Organs. 2006;183:180–194. doi: 10.1159/000096509. [DOI] [PubMed] [Google Scholar]
- Inomata M, Takayama Y, Kiyama H, Nada S, Okada M, Nakagawa H. Regulation of Src family kinases in the developing rat brain: correlation with their regulator kinase, Csk. J Biochem. 1994;116:386–392. doi: 10.1093/oxfordjournals.jbchem.a124536. [DOI] [PubMed] [Google Scholar]
- Jucker M, Tian M, Norton DD, Sherman C, Kusiak JW. Laminin alpha 2 is a component of brain capillary basement membrane: reduced expression in dystrophic dy mice. Neuroscience. 1996;71:1153–1161. doi: 10.1016/0306-4522(95)00496-3. [DOI] [PubMed] [Google Scholar]
- Kawabuchi M, Satomi Y, Takao T, Shimonishi Y, Nada S, Nagai K, Tarakhovsky A, Okada M. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 2000;404:999–1003. doi: 10.1038/35010121. [DOI] [PubMed] [Google Scholar]
- Krämer EM, Klein C, Koch T, Boytinck M, Trotter J. Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J Biol Chem. 1999;274:29042–29049. doi: 10.1074/jbc.274.41.29042. [DOI] [PubMed] [Google Scholar]
- Larocque D, Richard S. QUAKING KH domain proteins as regulators of glial cell fate and myelination. RNA Biol. 2005;2:37–40. doi: 10.4161/rna.2.2.1603. [DOI] [PubMed] [Google Scholar]
- Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. Protection of p27(Kip1) mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci. 2005;8:27–33. doi: 10.1038/nn1359. [DOI] [PubMed] [Google Scholar]
- Leite CC, Lucato LT, Martin MG, Ferreira LG, Resende MB, Carvalho MS, Marie SK, Jinkins JR, Reed UC. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol. 2005;35:572–579. doi: 10.1007/s00247-004-1398-y. [DOI] [PubMed] [Google Scholar]
- Lu Z, Ku L, Chen Y, Feng Y. Developmental abnormalities of myelin basic protein expression in fyn knock-out brain reveal a role of Fyn in posttranscriptional regulation. J Biol Chem. 2005;280:389–395. doi: 10.1074/jbc.M405973200. [DOI] [PubMed] [Google Scholar]
- Matsuoka H, Nada S, Okada M. Mechanism of Csk-mediated down-regulation of Src family tyrosine kinases in epidermal growth factor signaling. J Biol Chem. 2004;279:5975–5983. doi: 10.1074/jbc.M311278200. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Borgs L, Vandenbosch R, Mangin JM, Beukelaers P, Moonen G, Gallo V, Malgrange B, Belachew S. The Yin and Yang of cell cycle progression and differentiation in the oligodendroglial lineage. Ment Retard Dev Disabil Res Rev. 2006;12:85–96. doi: 10.1002/mrdd.20103. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Osterhout DJ, Wolven A, Wolf RM, Resh MD, Chao MV. Morphological differentiation of oligodendrocytes requires activation of Fyn tyrosine kinase. J Cell Biol. 1999;145:1209–1218. doi: 10.1083/jcb.145.6.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, Stein PL, Scherer SS, McMorris FA. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallcup WB. The NG2 antigen, a putative lineage marker: immunofluorescent localization in primary cultures of rat brain. Dev Biol. 1981;83:154–165. doi: 10.1016/s0012-1606(81)80018-8. [DOI] [PubMed] [Google Scholar]
- Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J Biol Chem. 1994;269:13729–13732. [PubMed] [Google Scholar]
- Takeuchi M, Kuramochi S, Fusaki N, Nada S, Kawamura-Tsuzuku J, Matsuda S, Semba K, Toyoshima K, Okada M, Yamamoto T. Functional and physical interaction of protein-tyrosine kinases Fyn and Csk in the T-cell signaling system. J Biol Chem. 1993;268:27413–27419. [PubMed] [Google Scholar]
- Takeuchi S, Takayama Y, Ogawa A, Tamura K, Okada M. Transmembrane phosphoprotein Cbp positively regulates the activity of the carboxyl-terminal Src kinase, Csk. J Biol Chem. 2000;275:29183–29186. doi: 10.1074/jbc.C000326200. [DOI] [PubMed] [Google Scholar]
- Tang XM, Beesley JS, Grinspan JB, Seth P, Kamholz J, Cambi F. Cell cycle arrest induced by ectopic expression of p27 is not sufficient to promote oligodendrocyte differentiation. J Cell Biochem. 1999;76:270–279. doi: 10.1002/(sici)1097-4644(20000201)76:2<270::aid-jcb10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Umemori H, Sato S, Yagi T, Aizawa S, Yamamoto T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature. 1994;367:572–576. doi: 10.1038/367572a0. [DOI] [PubMed] [Google Scholar]
- Yang D, Bierman J, Tarumi YS, Zhong YP, Rangwala R, Proctor TM, Miyagoe-Suzuki Y, Takeda S, Miner JH, Sherman LS, Gold BG, Patton BL. Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J Cell Biol. 2005;168:655–666. doi: 10.1083/jcb.200411158. [DOI] [PMC free article] [PubMed] [Google Scholar]