

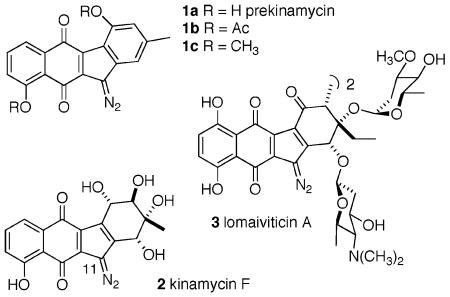
The diazoparaquinone family of antibiotics, exemplified by the naturally occurring species prekinamycin (1a),1 kinamycin F (2),2 and lomaiviticin A (3),3 has had a long and storied history, featuring two major structural revisions (N-cyanocarbazole → diazofluorene for the kinamycins,4 and diazobenzo[b]fluorene → diazobenz[a]-fluorene for isoprekinamycin (not shown)5). Following these corrections, two mechanism-of-action hypotheses have been proposed. Jebaratnam, using diazofluorene as a model system, suggested that oxidative activation of the diazo function might lead to DNA damaging reactive oxygen species via a putative C(11) radical.6 Dmitrienko, employing the isoprekinamycin structure as a probe molecule, favored an alternative mechanism whereby enhanced diazonium character of the N2 group as a consequence of an internal hydrogen bond network provided a site for nucleophilic attack by DNA amino groups, possibly leading to DNA damaging C(11) radicals as well.7 A disclosure by He et al. on the structure and biological activity of lomaiviticin A (3) attributed its profound cytotoxicity (IC50's of 0.7–6.0 nM for a variety of tumor cell lines)3 to double-stranded DNA cleavage under reducing conditions.
Neither Jebaratnam nor Dmitrienko utilized a diazoparaquinone-containing species for their studies, and an alternative mechanism-of-action that directly incorporates both this specific functionality and the reductive activation observation of He can be envisioned (Scheme 1). This proposal does not stray far from orthodox and precedented mechanisms of known paraquinoid antitumor agents, such as mitomycin and the anthracyclins.8 The observation that lomaiviticin A operates through bioreductive activation is suggestive of one-electron addition to the quinone of generic diazoparaquinone 4 to give a reactive semiquinone 5 following protonation (not obligatory). The radical 5 so generated can be represented by the resonance form 6. This pivotal radical (or radical anion) 5/6 can furnish the second key intermediate of this hypothesis, a C(11) radical 7, provided that C(11) is pyramidalized sufficiently to permit adequate overlap between the enol (enolate) orbitals and σ*C–N for rapid loss of N2. If this sequence occurs in proximity to DNA, then H-atom abstraction seems plausible by analogy with the similar reaction of a related indenyl monoradical derived from neocarzinostatin and p-hydroxythiophenol,9 and the hydroxymethylacylfulvene-containing intermediate 8 along with a DNA radical will be formed. At this point in the mechanistic speculation, there are two distinct avenues by which these two species, 8 and DNA•, might react further to lead to the observed result with lomaiviticin A. Generation of a DNA-based radical (C(4′), C(5′), or C(1′)) itself may be sufficient to cause strand cleavage, as per the putative O2-mediated mechanisms-of-action of other radical-generating DNA damaging agents, such as bleomycin and the enediynes.10 In addition, a rebound addition of DNA• into the enone of 8 might also contribute to the formation of DNA lesions at low O2 concentrations.11 In addition to the potential for radical-based DNA damage, the quinone methide moiety within 8 may interact further with DNA to lead to strand scission products by analogy with the documented DNA alkylating chemistry of the structurally related hydroxymethylacylfulvenes.12 The dimeric nature of lomaiviticin A is suggestive of double reaction to afford the observed double strand cleavage.
Scheme 1.

A Proposal for the Mechanism-of-Action of the Diazoparaquinone Antibiotics
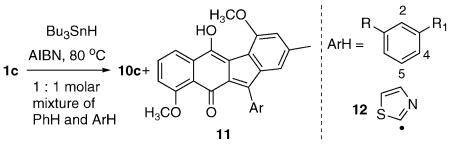
A test of this hypothesis was planned using readily available prekinamycin (1a)13 and Bu3SnH/AIBN in PhH (80 °C) as a surrogate for the unidentified e−/H+ or H• transfer agent available in the cellular milieu, with the anticipation that species of type 8 (or nucleophile-trapped adducts thereof) might be formed (Scheme 2).14 In fact, treatment of this substrate under standard radical-generating conditions provided a good yield of a single characterizable product, the C(11) benzene adduct 10a, following aqueous workup and SiO2 chromatography. Presumably, loss of any attached tin residue and air oxidation preceded isolation of 10a. Control experiments in which ambient light was included or omitted led to similar yields of 10a, whereas omission of either the tin reagent, AIBN, or heat returned starting material. Similar chemistry was observed with the derived diacetate 1b and the dimethyl ether 1c,13 and correlations as indicated (Scheme 2) confirmed that the identical C(11) adducts were formed in each case. Subsequent studies were conducted with the dimethyl ether 1c as a concession to ease of chromatographic isolation and characterization.
Scheme 2.

Preliminary Phenylation of Prekinamycin and Derivatives
The clean formation of the benzene adducts 10a–c under these relative mild and neutral conditions raises the key question, what is the reactive intermediate that precedes C(11)/PhH bond formation? Evidence that bears on this issue was gathered by examining the relative rates of aromatic solvent incorporation (versus benzene) for the substrate 1c in 1:1 molar mixtures of benzene with a variety of electron-rich or electron-deficient solvents, eq 1 and Table 1. The observed relative rates with 1c would be difficult to reconcile with an electrophilic aromatic substitution mechanism from a orthoquinonemethide-type electrophile as in 8, given the acceleration (relative to benzene) of the electron-deficient solvents, chlorobenzene (entry b), cyanobenzene (entry c), and 1,3-dicyanobenzene (entry g). Direct addition of the putative sp2 radical of 7 to the aromatic solvent cannot be dismissed so readily, however, in light of the precedent provided by aromatic radical substitution data for a related radical 1215 and also for Ph• (not shown).15 That is, a general accelerating effect of most substituents relative to H attends the radical aromatic substitution reactions of 12 and Ph•, as is observed with 1c. An analysis of the o:m:p ratios provides further evidence consistent with the radical addition mechanism. Thus, additions that place radical density on the substituent-bearing carbon are generally favored, whereas it is difficult to rationalize the observed regiochemistry of addition with the electron-deficient entries if electrophilic chemistry were operational. The reoxidation of the presumably first-formed cyclohexadienyl radicals, even under reducing tin hydride conditions has been described,16 although the precise source of the oxidant in the 1c case has not been identified.
Table 1.
Relative Rates of Aromatic Solvent Addition for 1c and the Reference sp2 Radical 12
| ArH | relative ratea,b | o:m:p | relative rate (12)a,d | o:m:p | ||
|---|---|---|---|---|---|---|
| R | R1 | |||||
| a | CH3 | H | 2.2 ± 0.1 | 62:23:15 | 2.1 | 69:23:17 |
| b | Cl | H | 1.5 ± 0.2 | 48:32:20 | 0.9 | 49:33:19 |
| c | CN | H | 2.1 ± 0.3 | 43:25:32 | 2.0 | 56:18:26 |
| d | OCH3 | H | 3.1 ± 0.7 | 72:16:12 | 2.2 | 63:14:23 |
| e | CH3 | CH3 | 4.0 ± 0.9 | 50:50:0c | ||
| f | OCH3 | OCH3 | 4.2 ± 1.5 | 31:69:0c | ||
| g | CN | CN | 6.1 ± 0.5e | 24:76:0c | ||
Rate relative to benzene.
Quintuple measurements with standard deviation.
Ratio of 2:4:5 adducts.
Data from ref 15.
Extrapolated from the results of 1:9 and 1:13 ratios of 1,3-dicyanobenzene to benzene solvent due to solubility constraints.
The intermediacy of a putative sp2 radical akin to 7 in the chemistry of 1c suggests that running this reductive sequence in the presence of ever-increasing concentrations of tin hydride might diminish the overall yield of aromatic trapping products [10c + 11]. This prediction was born out for the benzene/benzonitrile pair (Figure 1), with scarcely any change in the observed trapping ratios.
Figure 1.
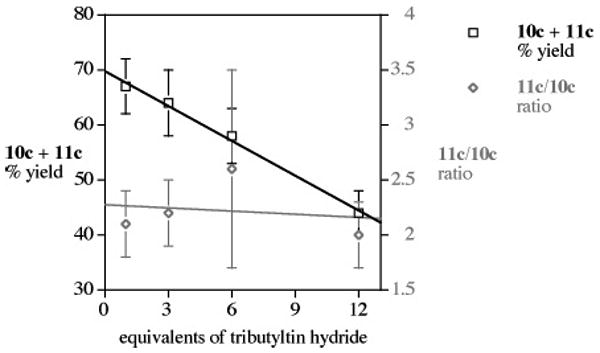
Yield and ratio of aromatic trapping products 10c/11c upon increasing [Bu3SnH].
 |
(1) |
This latter point is significant in that it argues against the intervention of a competing mechanism (i.e., 8 combining with the aromatic solvent) upon formation of 11. The remainder of the reaction mixture provided little characterizable material, but typically about 5–15% of the formal dimer of radical 7 was isolated from many trials. The observed drop-off in overall yield of [10c + 11c] (Figure 1) only makes sense in the context of the Scheme 1 mechanism if the aryl solvent trapping rate is not too dissimilar from the rate of sp2 radical/H–SnBu3 reaction. A rate constant for this latter process with Ph• has been estimated to be ∼109 M−1 s−1 (80 °C),17 and given the concentration differences ([aromatic solvents] ≈ 25–150 × [Bu3SnH]), then the rate constant for sp2 radical addition (e.g., 7) to the aromatic solvent would be on the order of 107 M−1 s−1, a value not too far removed, given the approximations involved, from the measured rate constant for phenyl radical addition to chlorobenzene (∼106 M−1 s−1, 25 °C).18
Supplementary Material
Acknowledgments
Support from the General Medical Sciences Institute of the National Institutes of Health (GM 37681) is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for 10a–e and 11a–g. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gould SJ, Chen J, Cone MC, Gore MP, Melville CR, Tamayo N. J Org Chem. 1996;61:5720–5721. [Google Scholar]
- 2.Gould SJ. Chem Rev. 1997;97:2499–2509. doi: 10.1021/cr9600215. and references therein. [DOI] [PubMed] [Google Scholar]
- 3.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 4.(a) Gould SJ, Tamayo N, Melville CR, Cone MC. J Am Chem Soc. 1994;116:2007–2208. [Google Scholar]; (b) Mithani S, Weeratunga G, Taylor NJ, Dmitrienko GI. J Am Chem Soc. 1994;116:2209–2210. [Google Scholar]
- 5.Proteau PJ, Li Y, Chen J, Williamson RJ, Gould SJ, Laufer RS, Dmitrienko GI. J Am Chem Soc. 2000;122:8325–8326. [Google Scholar]
- 6.Arya DP, Jebaratnam DJ. J Org Chem. 1995;60:3268–3269. [Google Scholar]
- 7.Laufer RS, Dmitrienko GI. J Am Chem Soc. 2002;124:1854–1855. doi: 10.1021/ja0167809. [DOI] [PubMed] [Google Scholar]
- 8.Moore HW. Science. 1977;197:527–532. doi: 10.1126/science.877572. [DOI] [PubMed] [Google Scholar]
- 9.Sugiyama H, Fujiwara T, Saito I. Tetrahedron Lett. 1994;35:8825–8828. [Google Scholar]
- 10.Murphy JA, Griffiths J. Nat Prod Rep. 1993:551–564. doi: 10.1039/np9931000551. [DOI] [PubMed] [Google Scholar]
- 11.Xu Yj, Xi Z, Zhen Ys, Goldberg IH. Biochemistry. 1997;36:14975–14984. doi: 10.1021/bi972101o. [DOI] [PubMed] [Google Scholar]
- 12.(a) Woynarowski JM, Napier C, Koester SK, Chen SF, Troyer D, Chapman W, MacDonald JR. Biochem Pharmacol. 1997;54:1181–1193. doi: 10.1016/s0006-2952(97)00321-3. [DOI] [PubMed] [Google Scholar]; (b) Herzig MCS, Arnett B, MacDonald JR, Woynarowski JM. Biochem Pharmacol. 1999;58:217–225. doi: 10.1016/s0006-2952(99)00085-4. [DOI] [PubMed] [Google Scholar]; (c) McMorris TC. Bioorg Med Chem. 1999;1:881–886. doi: 10.1016/s0968-0896(99)00016-4. [DOI] [PubMed] [Google Scholar]
- 13.Hauser FM, Zhou M. J Org Chem. 1996;61:5722. [Google Scholar]
- 14.The reactions of Bu3SnH with diazocarbonyls have scarcely been explored. A relevant example is provided by Kim: Kim S, Cho JR. Bull Korean Chem Soc. 1993;14:664–665.
- 15.Vernin G, Jauffred R, Ricard C, Dou HJM, Metzger J. J Chem Soc, Perkin Trans. 1972;2:1145–1150. [Google Scholar]
- 16.Beckwith ALJ, Bowry VW, Bowman WR, Mann E, Parr J, Storey JMD. Angew Chem, Int Ed. 2004;43:95–98. doi: 10.1002/anie.200352419. and references therein. [DOI] [PubMed] [Google Scholar]
- 17.Garden SJ, Avila DV, Beckwith ALJ, Bowry VW, Ingold KU, Lusztyk J. J Org Chem. 1996;61:805–809. doi: 10.1021/jo951245a. [DOI] [PubMed] [Google Scholar]
- 18.Scaiano JC, Stewart LC. J Am Chem Soc. 1983;105:3609–3614. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.