

Abstract
Methods to construct 2’-deoxy-2’-fluoro-nucleosides have undergone limited improvement in the last twenty years in spite of substantially increased value of these compounds as pharmaceuticals and as tools for studying biological processes. We herein describe a consolidated approach to synthesize precursors to these commercially and scientifically valuable compounds via diastereocontrolled fluorination of the readily available precursor 2-deoxy-d-ribonolactone. With employment of appropriate sterically bulky silyl protecting groups at 3 and 5 positions, controlled electrophilic fluorination of the Li-ribonolactone enolate by N-fluorodibenezenesulfonamide yielded the corresponding 2-deoxy-2-fluoro-arabino-lactone in high isolated yield (72 %). The protected 2-deoxy-2, 2-difluoro-ribonolactone was obtained similarly in high yield from a second round of electrophilic fluorination (2 steps, 51% from protected ribonolactone starting material). Accomplishment of the difficult ribo-fluorination of the lactone was achieved by the directive effects of a diastereoselectively installed α-trimethylsilyl group. Electrophilic fluorination of a protected 2-deoxy-2-trimethylsilyl-arabino-lactone via enolate generation provided the protected 2-deoxy-2-fluoro-ribo-lactone as the exclusive fluorinated product. The reaction also yielded the starting material, the desilylated protected 2-deoxy-ribonolactone, which was recycled to provide a 38% chemical yield of the fluorinated product (versus initial protected ribonolactone) after consecutive silylation and fluorination cycles. Using our fluorinated sugar precursors we prepared the 2’-fluoro-arabino-, 2’-fluoro-ribo- and 2’,2’-difluoro-nicotinamide adenine dinucleotides (NAD+) of potential biological interest. These syntheses provide the most consolidated and efficient methods for production of sugar precursors of 2’-deoxy-2’-fluoronucleosides and have the advantage of utilizing an air-stable electrophilic fluorinating agent. The fluorinated NAD+s are anticipated to be useful for studying a variety of cellular metabolic and signaling processes.
Introduction
The efficient introduction of fluorine atoms into bioactive organic molecules has attracted considerable attention in recent years owing to the unique properties of the fluorine substituent.1-4 The selective replacement of hydrogen or oxygen with fluorine can change a compound’s biological activity, metabolic stability, chemical stability, lipophilicity, acidity and dipole properties with modest change in steric bulk.2-4 A broad class of particularly relevant compounds are the fluorinated carbohydrates1, 5, 6 and nucleosides.1, 3, 4 Selective fluorination in the sugar moiety of glycosides or nucleosides has proven useful to numerous investigations of enzyme mechanism where either sugars or nucleosides are substrates.6-14 Some of these compounds are potent drugs. For example, gemcitabine (Gemzar, Eli Lilly, 2’-deoxy-2’,2’-difluorocytidine) is used clinically to treat numerous cancers including ovarian,15 pancreatic16 and breast17 cancers, with sales in excess of 1.6 billion dollars per year.18 Of the mono-substituted 2’-fluoro-nucleosides, Clofarabine (Clorar, Genzyme, 2-Chloro-2’-deoxy-2’-fluoro-arabino-adenosine), has been approved for pediatric patients with relapsed or refractory acute lymphocytic leukemia19 and annual sales now exceed 100 million dollars.20
Our laboratory is interested in general and flexible methods to construct 2’-fluoro-nucleosides, nucleotides and dinucleotides, particularly derivatives of nicotinamide riboside. In previous studies we demonstrated that 2’-deoxy-2’-fluoro-arabino-nicotinamide-mononucleotide is a potent mechanism-based inhibitor (apparent Ki = 61 nM) of the signaling enzyme cell developmental protein 38 (CD38).10, 12 The 2’-fluoro-NAD+s and related compounds are likely to be valuable for the study of sirtuin enzyme mechanism,11, 21 and for studying the chemical properties of poly-ADP-ribosylpolymerases.22 Fluorinated NAD+s could be useful for identifying ADP-ribosyltransfer sites on proteins as well. For example, we previously demonstrated that the 2’-deoxy-2’-fluoro-arabino-furanosyl modification is suitably robust for MS/MS approaches used to characterize amino acid post-translational modifications.10 ADP-ribosylation sites are poorly surveyed within the proteome and yet these modifications are of heightened interest as they are implicated in important biological effects.22-24 The synthesis of 2’-deoxy-2’-fluoro-arabino-NAD+ was previously described,25 but syntheses of 2’-deoxy-2’-fluoro-ribo-NAD+ and 2’-deoxy-2’,2’-difluoro-NAD+ or their nucleoside precursors have not appeared in the literature. The difluoro derivatives in particular could be quite interesting based upon their anticipated chemical stability and altered electronic properties.
Methods to synthesize the 2-deoxy-2-fluoro-d-furanose precursors to 2’-fluorinated nucleosides have not experienced substantial recent innovation,1, 3, 26 despite the fact that interest and demand for these compounds have only increased in recent years. The synthetic methods currently available vary in efficiency and all depend on routes relying on different initial precursors to the respective final products. For example, the best method to make a protected 2-deoxy-2-fluoro-arabino-furanose requires only 2 steps (Scheme 1, Part I) from a protected ribose (58% overall yield).25, 27, 28, 29 On the other hand, 2-deoxy-2-fluoro-ribofuranose has no concise or efficient synthesis30 and still requires 6 steps from arabinose (Scheme 1 Part II; 4% overall yield).31 Alternatively, it can be obtained via a 10-step non-diastereoselective method in 11% overall yield.32-34 An efficient but non-diastereoselective route is established for the synthesis of protected 2-deoxy-2,2-difluoro ribonofuranose in 5 steps (Scheme 1 Part III; crude yield 25%) via coupling of ethyl-bromodifluoroacetate and isopropylidene glyceraldehyde. The route is not diastereoselective and depends upon crystallization of the preferred isomer.35 This procedure was developed by Eli Lilly for commercial synthesis of gemcitabine. 2-Deoxy-2,2-difluororibofuranose can also be obtained stereoselectively from glucose or mannose36, but that method involves 8 steps and very low overall yield (<15%). We wondered if it might be possible to improve and perhaps consolidate synthetic approaches to these desirable sugars by employing electrophilic fluorine as a means to modify protected 2-deoxy-ribonolactone, a starting material that can be obtained readily, abundantly and cheaply.
Scheme 1.
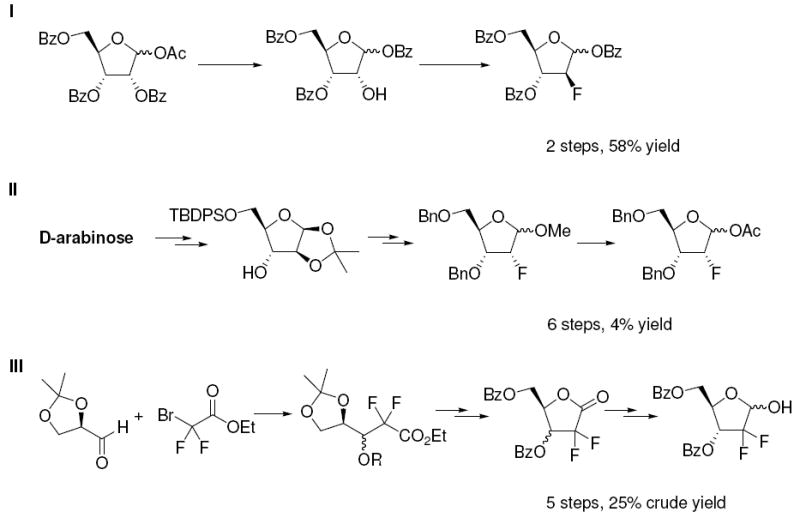
Existing Methods for the Syntheses of 2-Deoxy-2-fluoro-arabino/ribofuranoses
The strategy we considered is visually described in Scheme 2. We contemplated a general solution to the introduction of fluorine by controlling the reactivity of a 2-deoxy-ribonolactone enolate with an electrophilic fluorinating reagent. It is well known that lactone enolate can be functionalized in this manner, although stereocontrol is an issue and the studied systems predominantly are not subject to elimination.1 To be successful, we needed to solve 1) the competing β-elimination of the enolate species and 2) to rigorously control stereochemistry for the electrophilic substitution. We herein describe a successful solution to these problems with significant attenuation of the β-elimination as the undesired side reaction by utilization of bulky silyl protecting groups. Furthermore we establish a novel approach to the ribo-isomer using an α-silyl group. These methods allow us to synthesize the corresponding 2-deoxy-2-fluoro-arabino, 2-deoxy-2-fluoro-ribo- and 2-deoxy-2,2-difluorolactones and furanoses in diastereoselective manner with markedly improved synthetic efficiency. We also report the successful synthesis of the corresponding 2’-fluoro-NAD+s. Importantly, the successful development of a consolidated and flexible synthetic methodology to obtain all three types of 2-deoxy-2-fluoro-(ribo or arabino)-furanoses is anticipated to improve access to a large family of medicinally important 2’-fluoro-substituted nucleosides.
Scheme 2.
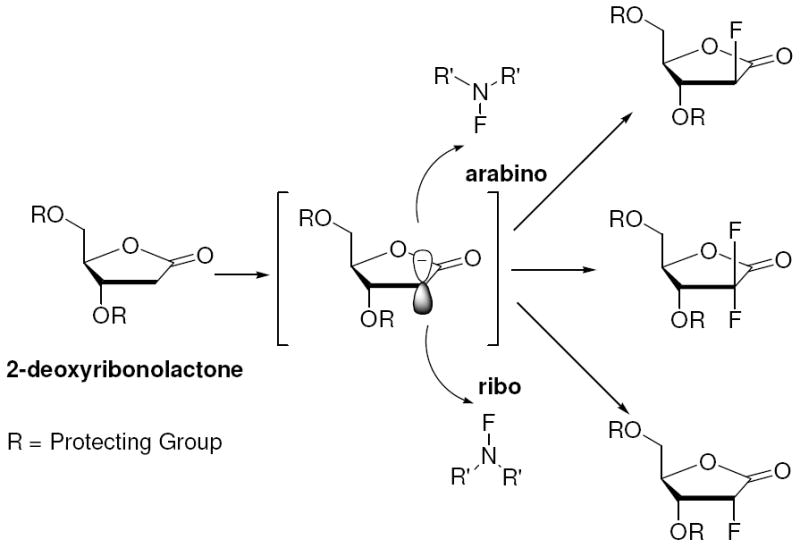
General Strategy for Diastereoselective Electrophilic Fluorination of Protected 2-Deoxyribonolactone
Results and Discussion
Diastereoselective electrophilic fluorination of protected γ-lactone was encouraged by existing precedents wherein β-elimination is not a problem. For example, Liotta and co-workers reported fluorination of 2,3-dideoxy-lactone 1 with N-fluorodibenezenesulfonimide (NFSi) to furnish the 2-fluorolactone 2 in 50-70% yield (Scheme 3).37 However, very limited success has been achieved when the β-position is substituted with oxygen. In one of the very few examples, Dehoux et al. applied these conditions to compound 3, and obtained the fluorinated compound 4 in only 16% yield (Scheme 3).38 Although the authors did not specifically address the reason for the poor outcome, we suspected competing elimination was likely the cause, since the alkoxide anion is significantly less basic than the enolate (Δ pKa ≈ 10) providing a strong thermodynamic driving force for elimination. We began our study by using the para-chlorobenzoyl-protected deoxyribonolactone 5, which was reacted under Liotta’s procedure (NFSi, LiHMDS in THF at -78°C), and furnished only the α, β-unsaturated γ-lactone 6 in 65% yield (Scheme 4). Since the driving force for elimination in our case was so much greater than in the Dehoux case, we were not surprised by this result.
Scheme 3.
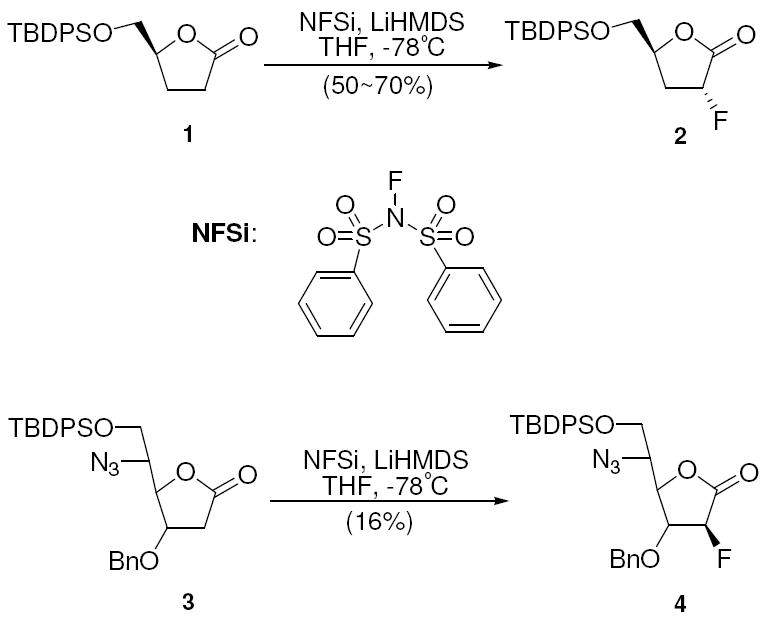
Electrophilic Fluorination of 2,3-Dideoxy-lactone 1 and 2-Deoxy-lactone 3
Scheme 4.
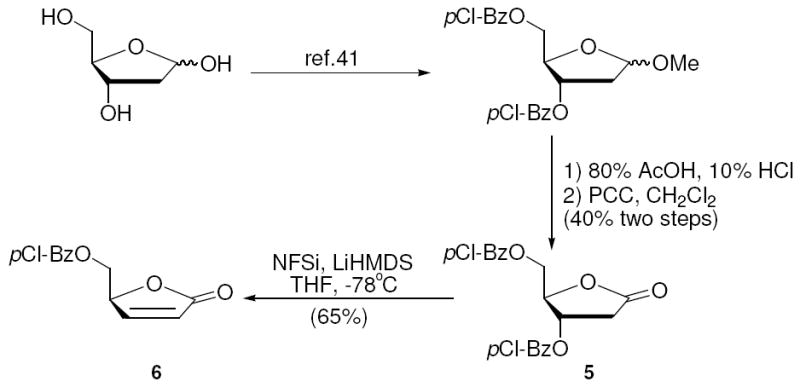
Attempted Fluorination of Lactone 5
We speculated that the elimination process could be diminished by moving to silyl protecting groups, which form leaving groups less basic than alkoxides (alcohol pKa = 16 versus silanol pKa =1139, 40), but can be potentially bulkier and more readily installed. We hypothesized that a sufficiently bulky group might sterically demand a 3-endo-ring pucker of the deoxy-ribonolactone ring, which would minimize steric interactions of the bulky group. Such a ring conformation would organize the 3-oxygen into a pseudo-equatorial position, thus diminishing the tendency of the silanoate to undergo elimination upon enolate formation.
Protection of deoxy-ribonolactone with t-butyldimethylsilyl (TBDMS) groups provided lactone 7. When 7 and NFSi were dissolved in THF and cooled to −78 °C, slow addition of LiHMDS resulted in the formation of 8 and 9 (Scheme 5). 2-Deoxy-2-fluoro-arabino-lactone 8 was obtained in 58% yield after silica gel chromatography, supplying a more than 3 fold improvement in yield for electrophilic fluorination versus the Dehoux result, encouraging our strategy. Favorably, the reaction yielded only the arabino-isomer, which can be attributed to the sterically bulky TBDMS group preventing a syn approach of the bulky fluorinating agent to the enolate. Although promising, the bulky TBDMS did not completely inhibit β-elimination as evident by the formation of 9. To increase silyl bulk we turned to the triisopropylsilyl (TIPS) group and synthesized the TIPS protected lactone 10. Lactone 10 was reacted as before and β-elimination was diminished enabling diastereoselective fluorination to give only the arabino isomer 11 in 72% isolated yield (Scheme 6). Stereochemistry was confirmed by NOE measurements (see experimental). Subsequently, 11 was reduced with DIBAL-H to obtain the TIPS protected 2-deoxy-2-fluoro-arabino-furanose 12 in 91% yield.
Scheme 5.
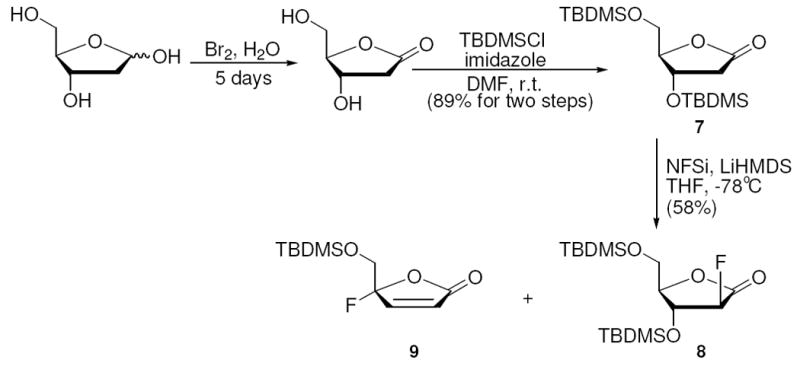
Synthesis of TBDMS-protected 2-Deoxy-2-fluoro-arabino-lactone
Scheme 6.
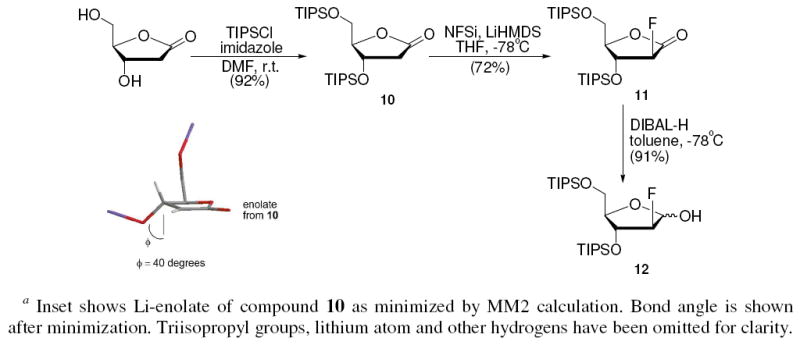
Synthesis of Silyl-protected 2-Deoxy-2-fluoro-arabino-furanosea
These results demonstrate that β-elimination in electrophilic fluorination of 2-deoxy-ribonolactone can be successfully mitigated by silyl steric bulk at the susceptible β-position. An MM2 minimization of the lithium enolate provided evidence that the steric bulk of the TIPS group organizes the 3-siloxy group into a geometry that is 40 degrees from the ideal angle for elimination (inset Scheme 6). The effect of the TIPS on preventing the elimination is impressive, as approximately 14 orders of magnitude of anion stability (Δ pKa) favor elimination of the silanoate from the enolate. Finally, in 2 steps from a protected 2-deoxy-ribonolactone, diastereoselective production of a 2-deoxy-2-fluoro-arabino-furanoside was achieved in 66% yield, surpassing the yield of the previous methodology (58% yield in 2 steps)25, 28, 29 and avoiding the use of the caustic substance diethylaminosulfur trifluoride (DAST).
We considered the monofluoro substituted lactone 11 to be a possible precursor to the corresponding 2-deoxy-2,2-difluororibonolactone. We felt it was only required that we repeat the fluorination procedure to achieve this valuable compound. Favorably, the second fluorination under the same reaction conditions provided the desired 2-deoxy-2,2-difluoro-ribonolactone 13 in 71% yield (Scheme 7). Reduction with DIBAL-H provided lactol 14 in 3 steps with 47% yield from 10. This straightforward method surpassed the non-diastereoselective method of Chou (5 steps, 25% yield, Scheme 1) for selectivity, brevity and yield.
Scheme 7.
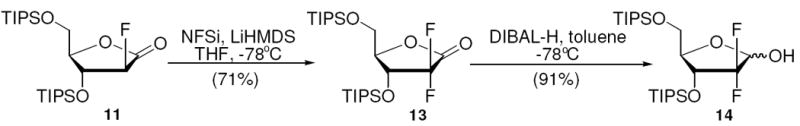
Synthesis of Silyl-protected 2-Deoxy-2,2-difluoro-ribofuranose
The absence from the literature of a convenient synthesis of 2-deoxy-2-fluoro-ribofuranose30 prompted us to investigate the possibility of converting the 2-deoxy-ribonolactone to the 2-deoxy-2-fluoro-ribo isomer. We supposed that a bulky protecting group at the 3 position would enforce steric arrangements in enolate reactions with electrophiles. To force stereochemistry of the fluoro-electrophile into the ribo-configuration we thought of using a removable directing group introduced at the α position. One attempt along these lines is shown in Scheme 8, and is based on a previous strategy of Kirk.41 2-Deoxy-2-bromo-lactone 15 was prepared by direct bromination of 7 (Scheme 8). Fluorination of 15 occurred stereoselectively, providing 16, of which the stereochemistry was assigned based upon coupling constants. Somewhat surprisingly, debromination of 16 with tributyltin hydride gave only the undesired arabino-fluoro compound 8 as the major product with no ribo-isomer formed (Scheme 8).
Scheme 8.
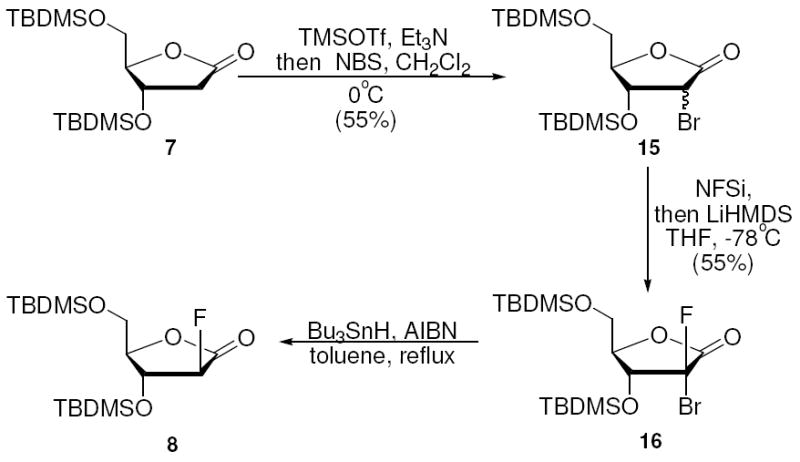
Debromination Approach to 2-Deoxy-2-fluoro-arabinolactone
This failure led us to consider an α-silyl lactone as a means for diastereocontrolled formation of the 2-deoxy-2-fluoro-ribo-lactone. Purified α-silyl ketones have been used as substrates in electrophilic fluorinations,42, 43 mostly for regio-controlled fluorination away from the silyl-group, although fluorination at the silylated-carbon suggested the potential workability of our strategy.43 We imagined that fluorination of an α-silyl lactone would generate the desired ribo-fluoro configuration, as a consequence of the tendancy of the bulky α-silyl and 3 position protecting group to move into a trans disposition in the enolate, thus permitting electrophilic fluorination syn to the 3-substitutent at the α-carbon. Subsequent proteo-desilylation with retention of stereochemistry was envisioned to provide the desired compound.
α-Silyl lactone 17 was generated diastereoselectively (the arabino-configuration was determined by NOE correlation between H2 and H4) from 7 in 72% yield (Scheme 9). Interestingly, treatment of 17 with NFSi afforded the desired ribo-fluoro product 18 in 33% yield with 2-deoxyribonolactone 7 in 61% yield. The formation of 2-fluoro-arabino-lactone 8 was not observed. The recovered compound 7 was resubmitted to silylation-fluorination conditions to give 18 in 38% combined yield after one recycle (Scheme 9). Subsequent reduction of 18 to lactol 19 was effected in 95% yield. The three-step silylation, fluorination and reduction sequence afforded the 2-deoxy-2-fluororibofuranose in 22% yield. This method is by far the most efficient chemical synthesis of 2-deoxy-2-fluoro-ribofuranose ever reported.
Scheme 9.
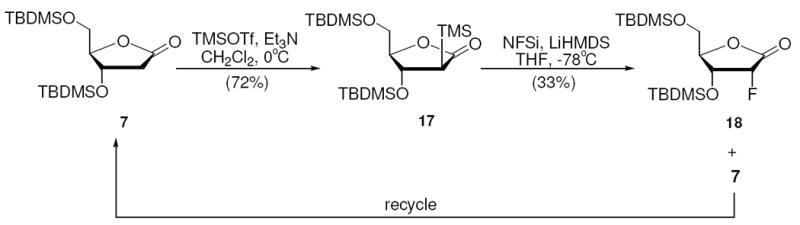
Synthesis of 2-Deoxy-2-fluoro-ribolactone
The result confirmed our expectation that the α-trimethylsilyl (TMS) substituent can control the stereochemistry of the fluorine substituent. The coincident desilylation observed in the process, although desirable and stereoselective, raises the question of the origin of the selectivity. Is it determined at the enolate reaction to the fluorine, or in the desilylation process, or both? At this point, we do not have a clear answer. Additionally, we have limited insight into the cause of the desilylation (without fluorination) that limits yield. We speculate that the reaction conditions stimulate a significant percentage of the C-silyl starting material to isomerize to the O-silyl ketene acetal, before it can reacts with NFSi, and when quenched the O-silyl derivative decomposes to 7. Low temperature interconversions of C-silylated esters and O-silylated ketene acetals have been reported.44 Further investigations into this reaction are ongoing in our laboratory.
At this point, with all possible 2-deoxy-2-fluoro-furanoses in hands, we were prepared to couple nicotinamide to sugars to obtain the corresponding 2’-deoxy-2’-fluoro-nicotinamide nucleosides and 2’-deoxy-2’,2’-difluoronucleosides, preferably with stereocontrol to yield the desired β-isomers. Our main concern was that the silyl groups restrict the levels of acidity we could employ in the coupling reaction.
The only reported synthesis of 1-(2’-deoxy-2’-fluoro-arabinofuranosyl)-nicotinamide was from 1,2:5,6-di-O-isopropylidene-D-allofuranose (8 steps in 24% yield).25 We here describe the synthesis of the 1-(2’-deoxy-2’-fluoro-arabinofuranosyl)-nicotinamide in 5 steps, 38% isolated yield from protected 2-deoxyribonolactone. 12 was activated with methanesulfonylchloride and triethylamine, which formed only the α-chloro sugar 20 (Scheme 10). 20 was coupled with nicotinamide in acetonitrile and dichloromethane mixed solvent with stoichiometric amount of AgSbF6. Crude product 21 was a mixture of both isomers with β being the major isomer. After deprotection with fluoride, β and α-isomers were separated by preparative HPLC (β : α = 3.5). The 1H NMR spectrum obtained for 22 (β) agreed with literature data.25 Yield of 22 (β) from 12 was 62% and required only one purification step.
Scheme 10.
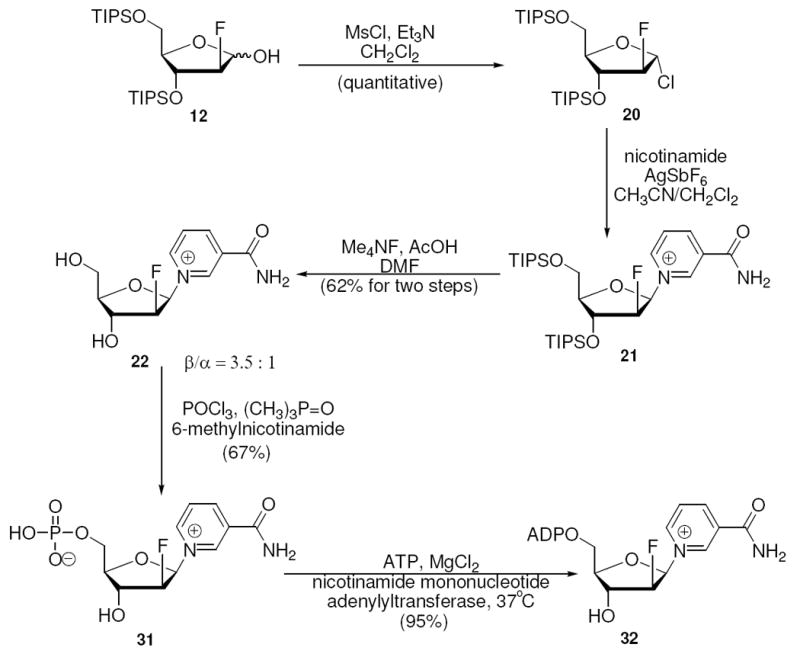
Synthesis of 2’-Deoxy-2’-fluoro-arabino-NAD+
A description of the synthesis of 1-(2’-deoxy-2’-fluoro-ribofuranosyl)-nicotinamide has not appeared in the chemical literature although the compound’s stability to hydrolysis has been described.45 We herein describe the synthesis of this compound in 6 steps and 15% isolated yield from 2-deoxy-ribonolactone. The furanose 19 was converted to the chloro sugar 23 by treatment with methanesulfonylchloride and triethylamine, the reaction occurred quantitatively but produced both α- and β-isomers (Scheme 11). Coupling of the chloro-sugar to nicotinamide with AgSbF6 provided a mixture of nicotinamide adducts 24, followed by deprotection and HPLC purification to provide the desired nicotinamide substituted 2’-deoxy-2’-fluoro-ribo-nucleoside 25 in 45% yield from the lactol 19. The corresponding α isomer was isolated in 46% yield. Stereochemistries were assigned by NOEs (see experimental).
Scheme 11.
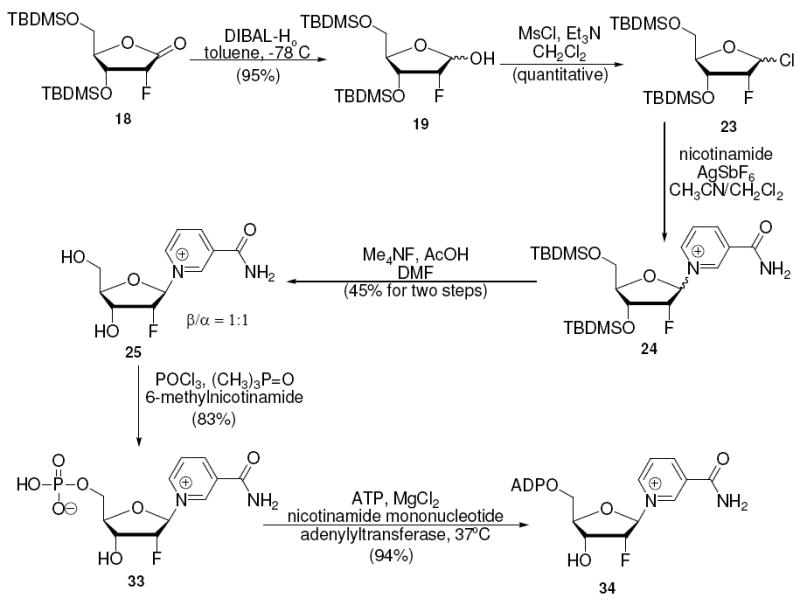
Synthesis of 2’-Deoxy-2’-fluoro-ribo-NAD+
The synthesis of the 1-(2’-deoxy-2’,2’-difluoro-ribofuranosyl)-nicotinamide has never been reported. The completion of the synthesis of the β-isomer in 6 steps with 18% isolated yield and 23% for the α-isomer are reported here. We prepared the 1-mesylate 26 similarly to a reported method.35 It appears that the additional fluorine at the 2-position deactivates the mesylate from nucleophilic chlorination which occurs for the arabino- and ribo-fluoro analogues (Schemes 10 and 11). Reaction with nicotinamide yielded nucleoside 27 in both α and β-configuration. Subsequent deprotection and HPLC purification provided β-28 in 38% yield and the α-anomer in 50% yield. Poor stereochemical control from the mesylate is known, with few preferable alternatives.35 NOEs between the H1’ and H4’ in the β-nucleoside and NOEs between the H1’ and H3’ and nicotinamide H2 and H4’ for the α isomer confirmed stereochemistries.
The complete syntheses of the nicotinamide substituted mononucleotides and dinucleotides were straightforward and only the syntheses of the difluoro-nucleotide and difluoro-dinucleotide are described. The monofluoro derivatives were prepared by similar methods and the preparation of these compounds is described in the experimental and in Scheme 10 and 11. 1-(2-deoxy-2,2-difluororibosyl)-nicotinamide 28 (β-isomer) was phosphorylated with POCl3 in trimethylphosphate in the presence of 6-methylnicotinamide, a hindered weak base which controls acidity, to yield 2’-deoxy-2’,2’-difluoro-nicotinamide mononucleotide (2’-deoxy-2’,2’-difluoro-NMN) 29 in 78% isolated yield (Scheme 12). Although previously unstudied, we found that the 2-fluoro-NMN compounds could be adenylated enzymatically with yeast nicotinamide mononucleotide adenylyltransferase46 (NMNAT-1, see supporting information). In this case, reaction with ATP furnished 2’-deoxy-2’,2’-difluoro-NAD+ 30 in 90% yield versus ATP, which was limiting, with recovery of unreacted 29. These steps complete the first reported syntheses of 2’,2’-difluoro-NMN and 2’,2’-difluoro-NAD+. The dinucleotide was completed in 8 steps with 14% overall yield from 2-deoxyribonolactone 10. Similarly with these methods 2’-fluoro-arabino-NMN and NAD+ 31, 32 (7 steps 22.4% yield from 10) were synthesized as well as 2’-fluoro-ribo-NMN and NAD+ 33, 34 (8 steps 12% yield from 7).
Scheme 12.
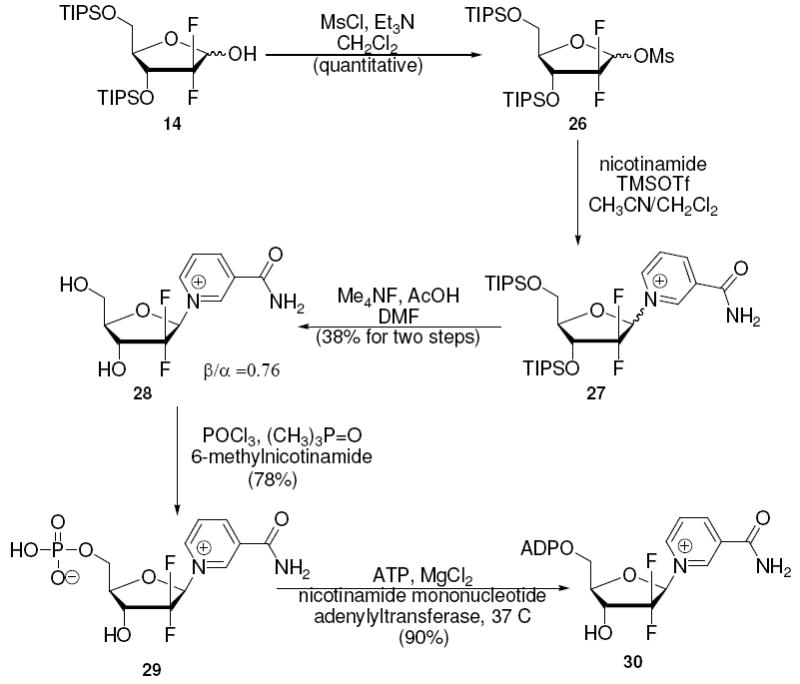
Synthesis of 2’-Deoxy-2’,2’-difluoro-NAD+
Conclusions
We have developed general methods of achieving diastereoselective electrophilic fluorination of 2-deoxy-ribonolactone to produce each of the corresponding α-fluoro substituted isomers (Scheme 13). Fluorinated furanoses, appropriate for nucleoside synthesis are made in increased yield and efficiency versus previously reported methods with the advantage of avoiding use of DAST. In addition, consolidation and synthetic brevity are achieved with complete control of diastereoselectivity of fluorination. In the case of 2-deoxy-2,2-difluoro-ribofuranose, the yield almost doubles the previously reported yield (47% versus 25%) while decreasing the number of required synthetic steps from 5 to 3 (Scheme 13). Moreover the synthesis here is diastereochemically pure. With respect to the ribo-fluoro derivatives, we found that we could control stereochemistry into the difficult ribo-configuration by utilization of an α-silyl group. The corresponding ribofuranose was made in 3 steps (with one recycle) slashing half the steps from the previous shortest synthesis, while increasing yield by 32% (from 4% to 36%, Scheme 13).
Scheme 13.
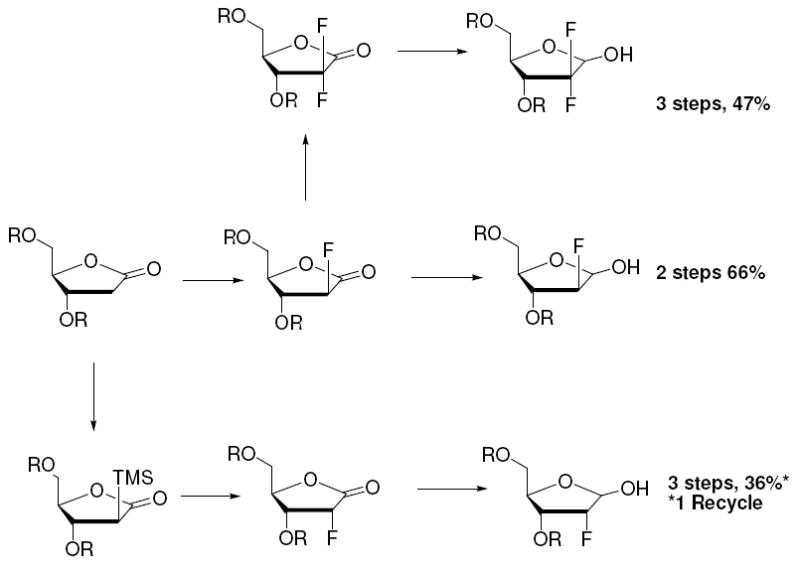
Syntheses of Protected 2-Deoxy-2-fluoro-furanose from 2-Deoxy-ribonolactone
Central to the success of our strategy was the ability of bulky silyl protecting groups at 3 and 5 positions of the lactone to attenuate the tendancy of the lactone enolate to undergo elimination prior to reacting with the electrophilic fluorinating agent. The silyl groups could have forced late stage manipulations of the protecting groups for synthesis of nucleosides, but we found that the silyl-protected lactols were easily activated to chloro sugars (in arabino and ribo), and in the arabino case, only the α-isomer was generated, allowing preferential synthesis of the β-nicotinamide substituted nucleoside. The gem-difluoro sugar was activated to mesylate.35 We anticipate that the methods reported here will improve and simplify synthetic access to a variety of 2’-fluorosubstituted nucleosides, particularly the 2’-deoxy-2’-fluoro-ribofuranoside derivatives. In addition, we are exploring other types of diastereocontrolled electrophilic modification of 2-deoxy-ribonolactones and are investigating introduction of chlorine, nitrogen and other heteroatoms. We expect to report the chemical and biochemical properties of the 2’-fluorinated NAD+ derivatives in future publications.
Experimental Section
2-deoxy-3,5-di-O-(p-chlorobenzoyl)-D-ribonolactone (5)
A solution of methyl-2-deoxy-3,5-di-O-(p-chlorobenzoyl)-D-ribofuranoside (500 mg, 1.18 mmol) in 20 mL of 80% acetic acid aqueous solution and 2 mL of 10% aqueous HCl was heated to reflux for an hour and then cooled to room temperature. Water was added, organic phase was washed with water, saturated NaHCO3 solution, brine and dried over anhydrous Na2SO4. Solvent was removed under vacuo and crude product was dissolved in 10 mL of CH2Cl2, to this solution was added PCC (294 mg, 1.36 mmol). Reaction was stirred at room temperature and monitored by TLC, once it was completed, PCC was filtered off, filtrate was concentrated and purified by column chromatography (hexanes:ethyl acetate 4:1) to afford 5 (190 mg, 0.46 mmol, 40% yield for two steps) as white solid. mp = 89-90 °C. 1H NMR (CDCl3, 400 MHz), δ ppm: .2.81 (dd, J= 2.1, 18.8 Hz, 1H), 3.12 (dd, J= 7.5, 18.9 Hz, 1H), 4.60 (dd, J= 3.7, 12.3 Hz, 1H), 4.68 (dd, J= 3.8, 12.3 Hz, 1H), 4.92 (m, 1H), 5.58 (dt, J= 1.8, 7.5 Hz, 1H), 7.42 (m, 4H), 7.93 (m, 4H). 13C NMR (125 MHz, CDCl3), δ ppm: 34.9, 63.9, 71.9, 82.2, 127.0, 127.4, 129.09, 129.11, 131.0, 131.2, 140.3, 140.6, 164.98, 165.02, 173.5. HRMS (ESI): calcd. for C19H14Cl2O6: 408.0167; Found: 408.017.
(S)-4-hydroxymethyl-2-buten-4-olide (6)
To a flame-dried round-bottom flask were added 5 (155 mg, 0.38 mmol) and NFSi (120 mg, 0.38mmol) in 5 mL of anhydrous THF. The solution was cooled to -78°C and LiHMDS in THF solution (0.456 mL, 0.456 mmol) was added dropwise. Reaction mixture was allowed to stir at -78°C for additional hour and then quenched by saturated NH4Cl solution. Organic layer was washed by saturated NaHCO3 solution, water, brine and dried over anhydrous Na2SO4. Column chromatography (hexanes: ethyl acetate 4:1~2:1) gave 6 (62 mg, 0.24 mmol, 65%) as white solid. mp = 115-116 °C. 1H NMR (CDCl3, 400 MHz), δ ppm: 4.55 (dd, J= 4.8, 12.0 Hz, 1H), 4.61 (dd, J= 3.7, 12.1 Hz, 1H), 5.34 (m, 1H), 6.22 (dd, J= 2.1, 5.8 Hz, 1H), 7.40 (d, J= 8.6 Hz, 2H), 7.48 (dd, J=1.5, 5.7 Hz, 1H), 7.91 (d, J= 8.6 Hz, 2H). 13C NMR (125 MHz, CDCl3), δ ppm: 63.1, 76.9, 80.8, 123.6, 127.5, 128.91, 128.96, 131.1, 131.6, 140.2, 152.1, 165.2, 172.1. HRMS (ESI): calcd. for C12H9ClO4: 252.0189; Found: 252.0188.
2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-D-ribonolactone (7)
To a solution of 2-deoxy-D-ribose (1.0 g, 7.45 mmol) in 6 mL of water was added Br2 (2 mL). The flask was sealed and the content was stirred at room temperature for 5 days. The resulting mixture was neutralized by adding silver carbonate until the pH was 7. The mixture was filtered and the filtrate was concentrated under reduced pressure to yield 2-deoxyribonolactone as a yellow oil. Without further purification, the crude product was dissolved in 20 mL of anhydrous DMF, and imidazole (2.53 g, 37.3 mmol) and t-butyldimethylsilyl chloride (4.5 g, 29.8 mmol) were added. The resulting solution was stirred at room temperature for 24 h, and quenched by addition of water. Water layer was extracted by ethyl acetate (3×10 mL), organic layers were combined, washed with brine and dried over anhydrous Na2SO4. Crude product was concentrated in vacuo. Flash chromatography (hexanes:ethyl acetate 20:1) afforded 7 (3.2 g, 8.9 mmol, 89% yield after two steps) as white solid. mp = 72-73 °C.1H NMR (CDCl3, 400 MHz), δ ppm: 0.038 (s, 3H), 0.051 (s, 3H), 0.062 (s, 6H), 0.085 (s, 18H), 2.36 (dd, J= 2.6, 17.7 Hz, 1H), 2.79 (dd, J= 6.7, 17.7 Hz, 1H), 3.73 (dd, J= 2.5, 11.5 Hz, 1H), 3.78 (dd, J= 3.4, 11.5 Hz, 1H), 4.30 (dd, J= 2.5, 5.2 Hz, 1H), 4.48 (dt, J= 2.3, 6.6 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.7, -5.5, -4.9, -4.8, 17.9, 18.2, 25.7, 25.8, 39.0, 62.5, 69.6, 88.1, 175.8. HRMS (ESI): calcd. for C17H36O4Si2: 360.2152; Found: 360.2155.
2-deoxy-2-fluoro-3,5-di-O-(t-butyldimethylsilyl)-D-ribonolactone (8)
To a flame-dried 100 mL round-bottom flask were added 7 (1.8 g, 5 mmol) and NFSi (2.36 g, 7.5 mmol) in 20 mL of anhydrous THF. The solution was cooled to -78°C and 6.5 mL (6.5 mmol) of a 1 M solution of LiHMDS in THF was added dropwise over a period of 10 mins. This was allowed to stir at -78°C for an additional hour and was quenched by saturated NH4Cl. The mixture was allowed to warm to room temperature, water layer was extracted by ethyl acetate (3×10 mL), organic layers were combined, washed with saturated NaHCO3, water, brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Crude product was purified by flash chromatography (hexanes:ethyl acetate 20:1) to afford both 8 (1.1 g, 29 mmol, 58%) and 9 (0.5 g, 1.9 mmol, 38%) also as white solid. 8 mp = 49-50°C. 1H NMR (CDCl3, 400 MHz), δ ppm: 0.060 (s, 3H), 0.065 (s, 3H), 0.107 (s, 3H), 0.128 (s, 3H), 0.87 (s, 9H), 0.89 (s, 9H), 3.76 (dd, J= 2.5, 12.4 Hz, 1H), 3.95 (dt, J= 2.1, 12.4 Hz, 1H), 4.10 (dt, J= 2.0, 7.7 Hz, 1H), 4.70 (dt, J= 7.8, 18.9 Hz, 1H), 5.09 (dd, J= 8.0, 51.7 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: -5.5, -5.4, -5.2, -4.8, 17.9, 18.2, 25.5, 25.7, 59.4, 71.3, 71.5, 80.6, 80.7, 91.3, 93.3, 168.5, 168.7. HRMS (ESI): calcd. for C17H35FO4Si2: 378.2058; Found: 378.206.
4-(t-butyldimethylsiloxy)methyl-4-fluoro-2-buten-4-olide (9)
mp = 85-87 °C. 1H NMR (CDCl3, 500 MHz), δ ppm: 0.045 (s, 3H), 0.057 (s, 3H), 0.85 (s, 9H), 3.86 (dd, J= 11.3, 15.9 Hz, 1H), 4.04 (dd, J= 7.8, 11.3 Hz, 1H), 6.25 (d, J= 5.7 Hz, 1H), 7.33 (d, J= 5.7 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.62, -5.58, 18.1, 25.6, 63.5, 63.8, 114.4, 116.3, 125.01, 125.05, 149.7, 149.8, 168.39, 168.41. HRMS (ESI): calcd. for C11H19FO3Si: 246.1087; Found: 246.1085.
2-deoxy-3,5-di-O-(triisopropylsilyl)-D-ribonolactone (10)
2-Deoxy-D-ribonolactone (1.1 g, 8.3 mmol) was dissolved in 10 mL of anhydrous DMF, to this solution were added imidazole (3.4 g, 50 mmol) and triisopropylsilyl chloride (6.4 g, 33 mmol) were added. The resulting solution was stirred at room temperature for 24 h, and quenched by addition of water. Water layer was extracted by ethyl acetate (3 × 10 mL), organic layers were combined, washed with brine and dried over anhydrous Na2SO4. Crude product was concentrated in vacuo. Column chromatography (hexanes:ethyl acetate 25:1~20:1) provided 10 (3.4 g, 7.7 mmol, 92%) as colorless oil. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.05 (stack, 42H), 2.41 (dd, J= 1.9, 17.6Hz, 1H), 2.86 (dd, J= 6.6, 17.6 Hz, 1H), 3.83 (dd, J= 2.6, 9.4 Hz, 1H), 3.91 (dd, J= 3.0, 11.4 Hz, 1H), 4.39 (s, 1H), 4.65 (d, J= 6.5, 1H).13C NMR (125 MHz, CDCl3), δ ppm: 11.9, 12.1, 18.0, 39.7, 63.4, 70.1, 88.9, 176.1. HRMS (ESI): calcd. for C23H48O4Si2: 444.3091; Found: 444.3094. MM2 calculations on lithium enolate of 10 were performed using CHEM3D version 10.0 Minimized energy to minimum RMS gradient of 0.100
2-deoxy-2-fluoro-3,5-di-O-(triisopropylsilyl)-D-ribonolactone (11)
Compound 11 was obtained according to the fluorination procedure to synthesize 8, using 10 (2 g, 4.5 mmol) as the starting material. Column chromatography (hexanes:ethyl acetate 30:1) provided 11 (1.5 g, 3.25 mmol, 72%) as a white solid. mp = 38-39 °C. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.07 (stack, 42H), 3.91 (dd, J= 2.4, 12.1 Hz, 1H), 4.08 (dt, J= 2.1, 12.1 Hz, 1H), 4.16 (dt, J= 2.1, 7.0 Hz, 1H), 4.92 (dt, J= 7.2, 18.8 Hz, 1H), 5.10 (dd, J= 7.4, 51.3 Hz, 1H).13C NMR (100 MHz, CDCl3), δ ppm: 11.9, 12.1, 17.7, 17.80, 17.84, 17.86, 60.3, 71.6, 71.8, 81.8, 81.9, 91.7, 93.7, 168.6, 168.8. NOE identified between H2 and H4 in NOESY. HRMS (ESI): calcd. for C23H47FO4Si2: 462.2997; Found: 462.2993.
2-deoxy-2-fluoro-3,5-di-O-(triisopropylsilyl)-D-arabino-furanose (12)
11 (200 mg, 0.43 mmol) was dissolved in 1.5 mL of anhydrous toluene and cooled to -78°C. To this solution was added 3.02 mL of DIBAL-H in THF solution (3.02 mmol). The reaction mixture was held at -78°C at all time. Two hours later, the mixture was quenched by methanol at -20°C and additional cold methanol was added. The mixture was then allowed to warm slowly to room temperature and was washed with 0.1 M HCl. Aqueous later was extracted with ether, the combined organic layer was washed with saturated NaHCO3, water, brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Column chromatography (hexanes:ethyl acetate 15:1) afforded 12 (181 mg, 0.39 mmol, 91%) as colorless oil. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.07 (stack, 54H), 3.46 (d, J= 10.9 Hz, 1H), 3.60 (m, 1.27H), 3.78 (m, 1.57H), 3.95 (q, J= 3.7 Hz, 0.28H), 4.32 (m, 1H), 4.49 (dd, J= 0.9, 12.6 Hz, 1H), 4.63 (t, J= 4.0 Hz, 0.14H), 4.67 (t, J= 4.0 Hz, 0.14 H), 4.81 (t, J= 4.1 Hz, 0.28H), 4.83 (dd, J= 0.8, 50.2 Hz, 1H), 5.31 (m, 0.3H), 5.35 (t, J= 10.1 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: 12.0, 12.2, 18.0, 63.2, 75.4, 75.5, 75.6, 87.6, 87.7, 95.9, 97.5, 97.7, 98.8, 98.9, 100.8, 101.0. HRMS (ESI): calcd. for C23H49FO4Si2: 464.3153; Found: 464.3162.
2-deoxy-2,2-difluoro-3,5-di-O-(triisopropylsilyl)-D-ribonolactone (13)
Compound 13 was obtained according to the fluorination procedure to synthesize 8, using 11 (92 mg, 0.2 mmol) as the starting material. Column chromatograph (hexanes:ethyl acetate 40:1) provided 13 (68 mg, 0.14 mmol, 71%) as pale yellow oil. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.08 (stack, 42H), 3.96 (dd, J= 2.4, 12.0 Hz, 1H), 4.08 (dt, J= 2.6, 12.1 Hz, 1H), 4.31 (m, 1H), 4.76 (dt, J= 6.0, 11.2 Hz, 1H). 13C NMR (100 MHz, CDCl3), δ ppm: 11.4, 11.45, 11.8, 12.1, 17.2, 17.3, 17.36, 17.43, 59.6, 68.3, 68.5, 68.6, 68.7, 82.5, 82.6, 109.9, 112.4, 115.0, 163.3, 163.6, 164.0. HRMS (ESI): calcd. for C23H46F2O4Si2: 480.2903; Found: 480.2901.
2-deoxy-2,2-difluoro-3,5-di-O-(triisopropylsilyl)-D-ribofuranose (14)
Compound 14 was obtained according to the reduction procedure to synthesize 12, using 13 (160 mg, 0.33 mmol) as the starting material. Column chromatography (hexanes:ethyl acetate 10:1) provided 14 (146 mg, 0.3 mmol, 91%) as colorless oil. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.04 (stack, 78H), 3.48 (d, J= 11.3 Hz, 1H), 3.67 (m, 1.8H), 3.81 (m, 1.8H), 3.88 (dt, J= 2.1, 11.2 Hz, 1H), 4.02 (m, 0.8H), 4.24 (m ,1H), 4.39 (dt, J= 2.0, 10.7 Hz, 1H), 4.67 (m, 0.8H), 5.02 (dd, J= 5.1, 9.8 Hz, 0.8H), 5.11 (dd, J= 6.1, 11.3 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: 11.87, 11.90, 12.1, 12.3, 17.72, 17.73, 17.78, 17.83, 17.86, 17.89, 62.2, 62.34, 62.36, 69.6, 69.7, 69.8, 70.0, 71.9, 72.0, 72.1, 72.3, 83.95, 84.02, 85.3, 95.3, 95.5, 95.6, 95.8, 96.0, 96.2, 96.3, 96.5, 119.5, 120.1, 121.5, 121.6, 122.1, 123.6, 124.2. HRMS (ESI): calcd. for C23H48F2O4Si2: 482.3059; Found: 482.3054.
2-deoxy-2-bromo-3,5-di-O-(t-butyldimethylsilyl)-D-ribono, arabino-lactones (15)
To a solution of 7 (180 mg, 0.5 mmol) and triethylamine (303 mg, 3 mmol) in 6 mL of CH2Cl2 at 0°C was added TMSOTf (333 mg, 1.5 mmol), and the solution was stirred at this temperature for 30 mins. A solution of NBS (134 mg, 0.75 mmol) in 1.5 mL of CH2Cl2 was added, and the stirring was continued for 1 h at 0°C. Reaction mixture was poured into saturated NaHCO3 solution, extracted with CH2Cl2 (3×5 mL). Combined organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Flash chromatography (hexanes: ethyl acetate 35:1) afforded a mixture of two isomers of 15 (120 mg, 0.27 mmol, 55%, 1:1.4, arabino/ribono) as pure pale yellow liquid. A small amount of this mixture was separated to obtain the pure compounds. Stereochemistry was determined by an observed NOE between the 2 and 4 protons of the arabino-isomer. Arabino-15 : 1H NMR (CDCl3, 500 MHz), δ ppm: 0.041 (s, 3H), 0.053 (s, 3H), 0.10 (s, 3H), 0.13 (s, 3H), 0.85 (s, 9H), 0.90 (s, 9H), 3.76 (dd, J= 2.0, 12.2 Hz, 1H), 3.93 (dd, J= 2.2, 12.2 Hz, 1H), 4.31 (m, 1H), 4.39 (t, J= 5.0 Hz, 1H), 4.47 (d, J= 5.7 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.6, -5.5, -5.0, -4.8, 18.1, 18.2, 25.6, 25.8, 46.1, 60.3, 68.8, 85.0, 170.9. HRMS (ESI): calcd. for C17H35BrO4Si2: 438.1257; Found: 438.1261. Ribono-15 : 1H NMR (CDCl3, 500 MHz), δ ppm: 0.058 (s, 3H), 0.062 (s, 3H), 0.116 (s, 3H), 0.172 (s, 3H), 0.87 (s, 9H), 0.88 (s, 9H), 3.79 (dd, J= 3.0, 12.0 Hz, 1H), 3.92 (dd, J= 3.3, 12.0 Hz, 1H), 4.21 (m, 1H), 4.37 (d, J= 6.9 Hz, 1H), 4.67 (dd, J= 6.1, 6.6 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.5, -5.4, -5.0, -4.1, 17.8, 18.2, 25.6, 25.7, 46.1, 60.2, 75.6, 85.3, 170.1. HRMS (ESI): calcd. for C17H35BrO4Si2: 438.1257; Found: 438.1258.
(2-R and 2-S)-2-deoxy-2-bromo-2-fluoro-3,5-di-O-(t-butyldimethylsilyl)-D-ribonolactone (16)
Compound 16 was obtained according to the fluorination procedure to synthesize 8, using 15 (400 mg, 0.91 mmol) as the starting material. Column chromatography (hexanes:ethyl acetate 30:1) provided 16 (230 mg, 0.5 mmol, 55%) as pale yellow oil. 1H NMR (CDCl3, 500 MHz), δ ppm: 0.058 (s, 3H), 0.065 (s, 3H), 0.13 (s, 3H), 0.17 (s, 3H), 0.86 (s, 9H), 0.93 (s, 9H), 3.77 (dd, J= 1.9, 12.7 Hz, 1H), 4.00 (m, 2H), 4.53 (dd, J= 8.0, 15.4 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.5, -5.4, -5.2, -4.6, 18.0, 18.2, 25.5, 25.7, 58.3, 72.0, 72.2, 80.6, 80.7, 98.2, 100.5, 165.6, 165.8. HRMS (ESI): calcd. for C17H34BrFO4Si2: 456.1163; Found: 456.1178.
Debromination of 16 yielding 8
16 (60 mg, 0.13 mmol), tributyltinhydride (83 mg, 0.28 mmol) and AIBN (3 mg, 0.018 mmol) were dissolved in 1 mL of toluene and stirred at 90°C for 24 h. Solvent was evaporated and residue was dissolved in acetonitrile, washed with hexanes to remove organotin compounds. Solvent was again concentrated in vacuo, and an 1H NMR spectrum identified it as compound 8 described previously.
2-deoxy-2-trimethylsilyl-3,5-di-O-(t-butyldimethylsilyl)-D-arabinolactone (17)
To a solution of 7 (1 g, 2.78 mmol), and triethylamine (1.68 g, 16.68 mmol) in 28 mL of CH2Cl2 at 0°C was added TMSOTf (1.85 g, 8.34 mmol) dropwise. The solution was stirred at this temperature for another 2 h and then quenched with saturated NH4Cl. The mixture was allowed to warm to room temperature, water layer was extracted by CH2Cl2 (3×10 mL), organic layers were combined, washed with saturated NaHCO3, water, brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Crude product was purified by flash chromatography (hexanes:ethyl acetate 25:1) to afford 17 (850 mg, 1.97 mmol, 71%) as a white solid. mp = 70-71°C. 1H NMR (CDCl3, 400 MHz), δ ppm: 0.067 (s, 6H), 0.074 (s, 6H), 0.19 (s, 9H), 0.85 (s, 9H), 0.89 (s, 9H), 2.17 (d, J= 2.5 Hz), 3.50 (dd, J= 7.7, 10.7 Hz, 1H), 3.76 (dd, J= 7.1, 12.2 Hz, 1H), 4.27 (m, 1H), 4.45 (t, J= 2.2 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.4, -5.3, -4.7, - 4.2, -1.7, -0.8, 17.7, 18.4, 25.6, 25.8, 25.90, 25.94, 42.2, 62.2, 71.8, 87.4, 177.7. HRMS (ESI): calcd. for C20H44O4Si3: 432.2547; Found: 432.2553.
2-deoxy-2-fluoro-3,5-di-O-(t-butyldimethylsilyl)-D-ribonolactone (18)
Compound 18 was obtained according to the fluorination procedure to synthesize 8, using 17 (780 mg, 1.81 mmol) as the starting material. Column chromatograph (hexanes:ethyl acetate 30:1~10:1) provided both 18 (226 mg, 0.60 mmol, 33%) and 7 (400 mg,1.11 mmol, 61%) as white solid. mp = 75-77°C. 1H NMR (CDCl3, 400 MHz), δ ppm: 0.046 (s, 3H), 0.061 (s, 3H), 0.087 (s, 6H), 0.86 (s, 9H), 0.87 (s, 9H), 3.77 (dd, J= 1.7, 11.9 Hz, 1H), 3.84 (dd, J= 2.6, 12.0 Hz, 2H), 4.37 (d, J= 2.1 Hz, 1H), 4.43 (d, J= 5.2 Hz, 1H), 5.21 (dd, J= 5.3, 50 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: -5.7, -5.6, -5.3, -4.9,-4.8, 18.2, 18.3, 25.6, 25.8, 62.3, 70.3, 70.4, 71.8, 84.2, 85.8, 86.4, 88.1, 171.1. HRMS (ESI): calcd. for C17H35FO4Si2: 378.2058; Found: 378.2059.
2-deoxy-2-fluoro-3,5-di-O-(t-butyldimethylsilyl)-D-ribofuranose (19)
Compound 19 was obtained according to the reduction procedure to synthesize 12, using 18 (100 mg, 0.22 mmol) as the starting material. Column chromatography (hexanes:ethyl acetate 15:1) provided 19 (95 mg, 0.205 mmol, 95%) as colorless oil. 1H NMR (CDCl3, 500 MHz), δ ppm: 1.02 (stack, 96 H), 3.28 (d, J= 7.3 Hz, 2.2H), 3.68 (dd, J= 3.7, 11 Hz, 1H), 3.76 (dd, J= 2.7, 11 Hz, 1H), 3.80 (dd, J= 1.8, 11 Hz, 2.2H), 3.92 (dd, J= 2.2, 11 Hz, 2.2H), 4.09 (dt, J= 1.9, 6.7 Hz, 2.2H), 4.14 (dd, J= 1.2, 12.3 Hz, 1H), 4.23 (s, 1H), 4.49 (dd, J= 1.7, 2.8 Hz, 1H), 4.59 (dd, J= 3.7, 53.4 Hz, 2.2H), 4.71 (ddd, J= 3.8, 10.4, 23.5 Hz, 2.2H), 4.85 (dt, J= 4.4, 51.7 Hz, 1H), 5.24 (dd, J= 4.2, 8 Hz, 1H), 5.27 (t, J= 6.1 Hz, 2.2H). 13C NMR (125 MHz, CDCl3), δ ppm: 11.83, 11.85, 11.9, 12.2, 17.71, 17.72, 17.81, 17.85, 17.87, 17.88, 61.8, 63.5, 69.8, 69.9, 71.9, 72.0, 84.0, 85.67, 85.70, 87.6, 89.2, 93.5, 95.0, 95.7, 95.9, 99.1, 99.3. HRMS (ESI): calcd. for C17H37FO4Si2: 380.2214; Found: 380.2212.
1-chloro-2-deoxy-2-fluoro-3,5-di-O-(triisopropylsilyl)-D-arabinofuranose (20)
12 (40 mg, 0.086 mmol) was dissolved in 0.5 mL of CH2Cl2 and triethylamine (12.2 mg, 0.12 mmol). To this solution was added at 0°C methanesulfonyl chloride (11.5 mg, 0.1 mmol). After 3 h of stirring under argon at room temperature, the mixture was evaporated in vacuo, and the residue was taken up in ethyl acetate. The solution was washed with saturated NaHCO3, followed by 1 M HCl, water and brine. Solvent was concentrated under reduced pressure to afford 20 (40 mg, 0.086 mmol, almost quantitatively) as yellow liquid. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.08 (stack, 42H), 3.89 (dd, J= 3.7, 11.7 Hz, 1H), 3.96 (dd, J= 2.9, 11.7 Hz, 1H), 4.30 (dd, J= 3.4, 8.3 Hz, 1H), 4.58 (dd, J= 5.1, 14.8 Hz, 1H), 5.12 (d, J= 51.7Hz, 1H), 6.15 (d, J= 12.5 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: 11.9, 12.0, 17.80, 17.84, 17.9, 61.4, 75.1, 75.4, 88.61, 88.64, 95.3, 95.6, 103.8, 105.3. HRMS (ESI): calcd. for C23H48ClFO3Si2: 482.2815; Found: 482.2822.
1-(2’-deoxy-2’-fluoro-3’,5’-di-O-(triisopropylsilyl)arabinofuranosyl)-nicotinamide (21)
20 (25 mg, 0.052 mmol) and nicotinamide (15 mg, 0.12 mmol) were dissolved in 1 mL of CH2Cl2. To this solution at 0°C was added a ice-cold solution of nicotinamide (15 mg, 0.12 mmol) and AgSbF6 (36 mg, 0.104 mmol) in 1.5 mL of acetonitrile. Reaction mixture was kept at room temperature overnight. Solvent was evaporated under reduce pressure and the residue was redissolved in methanol and pass through a short pad of celite. Concentrated crude product (which contained a mixture of α and β isomers) was examined by NMR and used for the next step without further purification.
1-(2’-deoxy-2’-fluoro-arabinofuranosyl)-nicotinamide (22)
To a solution of 21 (25 mg, 0.044 mmol) in 1 mL of DMF were added acetic acid (10.6 mg, 0.176 mmol) and tetramethylammonnium fluoride (16.4 mg, 0.176 mmol). The reaction was stirred at room temperature overnight, then was concentrated and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compounds were eluted at a flow rate of 2 mL/min) to afford 22 (β-isomer, tR = 8 min, 7 mg, 0.027 mmol, 62%) and α-isomer (tR = 6.7 min, 2 mg, 0.008 mmol, 18%). β-isomer 1H NMR (D2O, 400 MHz), δ ppm: 3.88 (dd, J= 4.8, 13.0 Hz, 1H), 4.0 (dd, J= 1.9, 12.9 Hz, 1H), 4.28 (dd, J= 4.7, 8.3 Hz, 1H), 4.51 (dt, J= 5.5, 17.6 Hz, 1H), 5.5 (dt, J= 4.6, 51.3 Hz, 1H), 6.68 (dd, J= 4.7, 9.8 Hz, 1H), 8.23, (t, J= 6.6 Hz, 1H), 8.95 (d, J= 8.1 Hz, 1H), 9.18 (d, J= 6.2 Hz, 1H), 9.57 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 59.4, 71.1, 71.3, 85.07, 85.11, 93.9, 94.0, 94.1, 95.6, 128.1, 133.8, 141.5, 143.8, 146.1, 165.7. HRMS (ESI): calcd. for C11H13FN2O4: 256.0859; Found: 256.0865.
(1R,2R)-1-chloro-2-deoxy-2-fluoro-3,5-di-O-(t-butyldimethylsilyl)-D-ribofuranose (23)
Compound 23 was obtained according to the chlorination procedure to synthesize 20, using 19 (10 mg, 0.021 mmol) as the starting material to afford 23 (10 mg, 0.021 mmol, almost quantitatively) as yellow liquid. 1H NMR (CDCl3, 500 MHz), δ ppm: 1.02 (stack, 51 H), 3.86 (stack, 3.4H), 4.02 (dd, J= 1.8, 11.8 Hz, 0.7H), 4.10 (m, 0.7H), 4.31 (d, J= 2.1 Hz, 1H), 4.55 (quintet, J= 3 Hz, 1H), 4.82 (dt, J= 4.7, 49 Hz, 1H), 4.95 (dd, J= 3.3, 45 Hz, 0.7H), 6.06 (d, J= 11.1 Hz, 0.7H), 6.21 (d, J= 4.1 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: 11.8, 11.9, 12.1, 12.2, 17.77, 17.81, 17.84, 17.86, 17.95, 17.96, 31.5, 52.5, 61.6, 62.3, 68.6, 68.8, 85.8, 87.9, 88.7, 89.6, 92.8, 93.0, 93.4, 93.7, 95.6, 97.2. HRMS (ESI): calcd. for C17H36ClFO3Si2: 398.1876; Found: 398.1880.
1-(2’-deoxy-2’-fluoro-3’,5’-di-O-(t-butyldimethylsilyl)-D-ribofuranosyl)-nicotinamide (24)
23 (11 mg, 0.026 mmol) and nicotinamide (8 mg, 0.065 mmol) were dissolved in 1 mL of CH2Cl2. To this solution at 0°C was added a ice-cold solution of nicotinamide (8 mg, 0.065 mmol) and AgSbF6 (8.9 mg, 0.026 mmol) in 1.5 mL of acetonitrile. Reaction mixture was kept at room temperature overnight. Solvent was evaporated under reduce pressure and the residue was redissolved in methanol and pass through a short pad of celite. Concentrated crude product was examined by NMR and used for the next step without further purification.
1-(2-deoxy-2-fluoro-D-ribofuranosyl)-nicotinamide (25)
Compound 25 was obtained according to the deprotection procedure to synthesize 22, using 24 (12.6 mg, 0.026 mmol) as the starting material. Crude product was purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compounds were eluted at a flow rate of 2 mL/min) to afford 25 (β-isomer, tR = 14.5 min, 3 mg, 0.012 mmol, 45%) and α-isomer (tR = 10.6 min, 3.1 mg, 0.012 mmol, 46%). β-isomer: 1H NMR (D2O, 600 MHz), δ ppm: 3.75 (dd, J= 2.4, 13.2 Hz, 1H), 3.97 (dd, J= 2.4, 13.8 Hz, 1H), 4.31 (m, 1H), 4.35 (m, 1H), 5.20 (dd, J= 4.2, 49.2 Hz, 1H), 6.47 (d, J= 14.4 Hz, 1H), 8.10 (t, J= 7.8 Hz, 1H), 8.80 (dd, J= 1.2, 7.8 Hz, 1H), 9.16 (d, J= 6.6 Hz, 1H), 9.54 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 55.15, 55.18, 55.22, 58.8, 67.3, 67.4, 85.8, 94.2, 95.7, 97.2, 97.5, 128.5, 134.1, 140.8, 142.9, 145.9, 165.2. NOESY: NOE correlation between sugar H3’ and nicotinamide H2. HRMS (ESI): calcd. for C11H13FN2O4: 256.0859; Found: 256.0863. α-isomer: 1H NMR (D2O, 500 MHz), δ ppm: 3.82 (dd, J= 4.5, 10.5 Hz, 1H), 4.0 (dd, J= 2.0, 10.5 Hz, 1H), 4.61 (m, 1H), 4.81 (m, 1H), 5.65 (dt, J= 4.5, 43.5 Hz, 1H), 6.79 (dd, J= 3.5, 8.5 Hz, 1H), 8.29, (dd, J= 5.5, 7.0 Hz, 1H), 9.01 (d, J= 6.5 Hz, 1H), 9.17 (d, J= 5.5 Hz, 1H), 9.40 (s, 1H). NOESY: NOE correlations between sugar H4’ and nicotinamide H2 sugar H4’ and nicotinamide H4 sugar H1’ and H3’.
1-methylsulfonyl-2-deoxy-2,2-difluoro-3,5-di-O-(triisopropylsilyl)-D-ribofuranose (26)
14 (146 mg, 0.3 mmol) was dissolved in 1.1 mL of CH2Cl2 and triethylamine (42 mg, 0.42 mmol). To this solution was added at 0°C methanesulfonyl chloride (41 mg, 0.35 mmol). After 3 h of stirring under argon at room temperature, the mixture was evaporated in vacuo, and the residue was taken up in ethyl acetate. The solution was washed with saturated NaHCO3, followed by 1 M HCl, water and brine. Solvent was concentrated under reduced pressure to afford 26 (165 mg, 0.3 mmol, almost quantitatively) as yellow liquid. 1H NMR (CDCl3, 400 MHz), δ ppm: 1.07 (stack, 74H), 3.07 (s, 1.9H), 3.08 (s, 3H), 3.83 (dd, J= 3.8, 11.4 Hz, 0.64H), 3.89 (m, 2H), 4.00 (m, 1.6H), 4.26 (dd, J= 4.0, 8.1 Hz, 1H), 4.47 (dd, J= 4.7, 16.5 Hz, 1H), 4.59 (m, 0.64H), 5.83 (d, J= 7.0 Hz, 0.64H), 5.92 (d, J= 6.8 Hz, 1H). 13C NMR (125 MHz, CDCl3), δ ppm: 11.84, 11.85, 12.1, 12.2, 17.67, 17.73, 17.76, 17.79, 17.81, 17.87, 17.91, 40.0, 40.2, 46.3, 61.5, 61.8, 69.0, 69.2, 69.4, 70.98, 71.12, 71.2, 71.4, 84.7, 84.8, 88.0, 99.4, 99.6, 99.9, 100.1, 100.3, 100.5. HRMS (ESI): calcd. for C24H50F2O6SSi2: 560.2835; Found: 560.284.
1-(2’-deoxy-2’,2’-difluoro-3’,5’-di-O-(triisopropylsilyl)-ribofuranosyl)-nicotinamide (27)
26 (420 mg, 0.75 mmol) and nicotinamide (732 mg, 6 mmol) were dissolved in 20 mL of CH3CN/CH2Cl2 (1:1). To this solution was added TMSOTf (167 mg, 0.75 mmol) under argon, the reaction mixture was kept refluxing overnight. Solvent was evaporated in vacuo, concentrated crude product was examined by NMR and used for the next step without further purification.
1-(2’-deoxy-2’,2’-difluoro-ribofuranosyl)-nicotinamide (28)
Compound 28 was obtained according to the deprotection procedure to synthesize 22, using 27 (440 mg, 0.75 mmol) as the starting material. Crude product was purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compounds were eluted at a flow rate of 2 mL/min) to afford 28 (β-isomer, tR = 17.8 min, 78 mg, 0.28 mmol, 38%) and the α-isomer (tR = 14.8 min, 103 mg, 0.37 mmol, 50%). β-isomer: 1H NMR (D2O, 600 MHz), δ ppm: 3.83 (d, J= 11.4 Hz, 1H), 4.01 (d, J= 12.6 Hz, 1H), 4.24 (d, J= 7.8 Hz, 1H), 4.46 (dd, J= 10.8, 20.4 Hz, 1H), 6.56 (d, J= 8.4 Hz, 1H), 8.20 (t, J= 6.6 Hz, 1H), 8.92 (d, J= 7.8 Hz, 1H), 9.20 (d, J= 6.6 Hz, 1H), 9.61 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 58.4, 67.3, 67.5, 67.6, 83.2, 83.3, 93.3, 93.4, 93.5, 93.6, 119.4, 121.7, 124.0, 128.6, 134.3, 141.4, 143.6, 146.9, 165.4. HRMS (ESI): calcd. for C11H12F2N2O4: 274.0765; Found: 274.0771. α-isomer: 1H NMR (D2O, 500 MHz), δ ppm: 3.78 (dd, J= 4.5, 11.0 Hz, 1H), 3.91 (dd, J= 1.5, 11 Hz, 1H), 4.59 (m, 1H), 6.75 (t, J= 5.0 Hz, 1H), 8.24, (dd, J= 5.0, 6.5 Hz, 1H), 8.98 (d, J= 6.5 Hz, 1H), 9.11 (d, J= 4.5 Hz, 1H), 9.34 (s, 1H).
2’-deoxy-2’,2’-difluoro-ribo-nicotinamide mononucleotide (29)
To a flame-dried round-bottom flask were added 28 (5 mg, 0.018 mmol), 6-methylnicotinamide (12.4 mg, 0.091 mmol) and 0.5 mL of trimethyl phosphate. At 0°C, 13.9 mg (0.091 mmol) of phosphorous oxychloride was added to the reaction mixture. This solution was stirred at 0°C for another 2 hours. Ice was added to quenched the reaction, pH was adjusted to 7 by adding NaOH solution and phosphate buffer. Crude product was concentrated and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compound was eluted at a flow rate of 2 mL/min) to afford 29 (tR = 6.2 min, 5 mg, 0.014 mmol, 78%). 1H NMR (D2O, 500 MHz), δ ppm: 4.02 (dd, J= 3.2, 13.4 Hz, 1H), 4.19 (dt, J= 2.4, 13.4 Hz, 1H), 4.43 (d, J= 8.4 Hz, 1H), 4.65 (stack, 2H), 6.75 (dd, J= 2.3, 8.9 Hz, 1H), 8.38 (t, J= 6.5 Hz, 1H), 9.10 (d, J= 8.2 Hz, 1H), 9.36 (d, J= 5.8 Hz, 1H), 9.78 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 59.7, 69.4, 69.5, 69.6, 69.8, 86.80, 86.84, 94.0, 94.2, 94.4, 94.5, 115.9, 118.4, 120.9, 123.4, 128.5, 134.1, 140.9, 143.2, 146.8, 165.4. HRMS (ESI): calcd. for C11H14F2N2O7P: 355.0501; Found: 355.0503.
2’-deoxy-2’,2’-difluoro-NAD+ (30)
A single reaction (50 μL) containing 6 mM of 29, 2 mM of ATP, 10 mM of MgCl2, 1 μL of pyrophosphatase (1 unit), 5 μL of NMNAT-1 (13.5 μM) and 50 mM phosphate buffer (pH~7.4) was incubated at 37°C for 1 hour. The reaction was terminated by addition of 3 μL of 10% TFA and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compound was eluted at a flow rate of 2 mL/min, tR = 12.7 min, 90 % versus ATP with recovery of unreacted 30). 1H NMR (500 MHz, D2O), δ ppm: 4.28 (stack, 2H), 4.36 (m, 1H), 4.42 (s, 1H), 4.55 (stack, 3H), 4.73 (stack, 2H), 6.16 (d, J= 5.6Hz, 1H), 6.69 (d, J= 9.3 Hz, 1H), 8.38 (dd, J= 6.5, 7.9 Hz, 1H), 8.41 (s, 1H), 8.60 (s, 1H), 9.04 (d, J= 8.2 Hz, 1H), 9.38 (d, J= 6.3 Hz, 1H), 9.54 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 59.7, 65.18, 65.22, 69.3, 69.49, 69.55, 69.7, 70.3, 74.3, 83.9, 84.0, 86.76, 86.81, 94.0, 94.2, 94.3, 94.5, 118.4, 119.7, 121.8, 123.8, 128.4, 133.9, 140.2, 140.8, 143.1, 146.7, 148.9, 151.6, 154.7, 165.2. HRMS (ESI): calcd. for C21H26F2N7O13P2: 684.1026; Found: 684.1019.
2’-deoxy-2’-fluoro-arabino-nicotinamide mononucleotide (31)
Compound 31 was obtained according to the phosphorylation procedure to synthesize 29, using 22 (7 mg, 0.027 mmol) as the starting material. Crude product was concentrated and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compound was eluted at a flow rate of 2 mL/min) to afford 31 (tR = 5.4 min, 6 mg, 0.018 mmol, 67%). 1H NMR (D2O, 400 MHz), δ ppm: 4.04 (m, 1H), 4.19 (m, 1H), 4.34 (m, 1H), 4.56 (dt, J= 5.0, 17.8 Hz, 1H), 5.51 (dt, J= 4.6, 51.4 Hz, 1H), 6.67 (dd, J= 4.8, 8.8 Hz, 1H), 8.24 (t, J= 7.1 Hz, 1H), 8.92 (d, J= 7.9 Hz, 1H), 9.31 (d, J= 6.1 Hz, 1H), 9.39 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 55.17, 55.20, 55.23, 60.60, 60.62, 72.6, 72.8, 90.17, 90.19, 98.5, 98.8, 99.2, 100.7, 128.3, 133.9, 140.3, 142.6, 145.8, 165.6. MS (M+): calculated: 337.06; found: 337.45.
2’-deoxy-2’-fluoro-arabino-NAD+ (32)
A single reaction (50 μL) containing 6 mM of 31, 2 mM of ATP, 10 mM of MgCl2, 1 μL of pyrophosphatase (1 unit), 5 μL of NMNAT-1 (13.5 μM) and 50 mM phosphate buffer (pH~7.4) was incubated at 37°C for 1 hour. The reaction was terminated by addition of 3 μL of 10% TFA and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compound was eluted at a flow rate of 2 mL/min, tR = 14.7 min, 95% versus ATP with recovery of unreacted 32). 1H NMR (D2O, 600 MHz), δ ppm: 4.41 (m, 1H), 4.46 (stack, 2H), 4.57 (stack, 3H), 4.67 (dd, J= 3.6, 5.4 Hz, 1H), 4.80 (dt, J= 4.8, 17.4 Hz, 1H), 4.90 (t, J= 6 Hz, 1H), 5.71 (dt, J= 4.8, 51 Hz, 1H), 6.20 (d, J= 6 Hz, 1H), 6.81, (dd, J= 4.8, 9.6 Hz, 1H), 8.37, (s, 1H), 8.40 (dd, J= 6.6, 7.8 Hz, 1H), 9.05, (d, J= 7.8 Hz, 1H), 9.38 (d, J= 6.6 Hz, 1H), 9.51 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 63.4, 65.4, 70.3, 70.8, 71.0, 74.1, 83.5, 83.8, 83.9, 86.8, 93.68, 93.73, 93.9, 95.3, 118.4, 128.3, 133.2, 140.0, 141.2, 143.4, 146.03, 148.9, 152.2, 165.1. MS (M+): calculated: 666.11; found: 666.60.
2’-deoxy-2’-fluoro-ribo-nicotinamide mononucleotide (33)
Compound 33 was obtained according to the phosphorylation procedure to synthesize 29, using 25 (3.5 mg, 0.014 mmol) as the starting material. Crude product was concentrated and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compound was eluted at a flow rate of 2 mL/min) to afford 33 (tR = 5.9 min, 3.8 mg, 0.011 mmol, 83%). 1H NMR (D2O, 600 MHz), δ ppm: 3.95 (d, J= 12.6 Hz, 1H), 4.16 (d, J= 13.2 Hz, 1H), 4.52 (stack, 2H), 5.40 (d, J= 50.4 Hz, 1H), 6.67 (d, J= 13.8 Hz, 1H), 8.31 (s, 1H), 9.00 (s, 1H), 9.35 (s, 1H), 9.74 (s, 1H). 13C NMR (150 MHz, D2O), δ ppm: 55.16, 55.20, 60.1, 60.6, 69.3, 69.4, 86.97, 86.98, 90.0, 91.5, 94.6, 94.8, 127.8, 133.4, 141.0, 143.4, 145.9, 165.7. HRMS (ESI): calcd. for C11H15FN2O7P: 337.0595; Found: 337.0599.
2’-deoxy-2’-fluoro-ribo-NAD+ (34)
A single reaction (50 μL) containing 3.8 mM of 33, 10 mM of ATP, 10 mM of MgCl2, 1 μL of pyrophosphatase (1 unit), 10 μL of NMNAT-1 (27 μM) and 50 mM phosphate buffer (pH~7.4) was incubated at 37°C for 1 hour. The reaction was terminated by addition of 3 μL of 10% TFA and purified by HPLC on a Waters RP-18 XBridge PrepShield 19 × 50mm column (solvent was 20 mM ammonium acetate, compound was eluted at a flow rate of 2 mL/min, tR = 18.6 min, 94% versus 33). 1H NMR (D2O, 600 MHz), δ ppm: 3.44 (dd, J= 7.2, 12 Hz, 1H), 3.54 (dd, J= 4.2, 11.4 Hz, 1H), 4.10 (m, 1H), 4.15 (dd, J= 4.8, 11.4 Hz, 2H), 4.27 (t, J= 2.4 Hz, 1H), 4.37 (dd, J= 1.8, 14.4 Hz, 1H), 4.40 (t, J= 3.6 Hz, 1H), 4.49 (stack, 2H), 5.30, (dt, J= 3.0, 51 Hz, 1H), 5.95, (d, J= 6.0 Hz, 1H), 6.41 (dd, J= 2.4, 13.8 Hz, 1H), 8.11, (s, 1H), 8.13 (dd, J= 6.6, 7.8 Hz, 1H), 8.74 (d, J= 8.4 Hz, 1H), 9.13 (d, J= 6.6 Hz, 1H), 9.32 (s, 1H). 13C NMR (125 MHz, D2O), δ ppm: 62.5, 63.45, 63.49, 65.36, 65.40, 67.7, 67.8, 70.4, 72.1, 73.9, 83.8, 83.9, 85.1, 85.2, 86.6, 94.1, 95.7, 97.2, 97.5, 102.4, 118.4, 128.7, 133.8, 139.2, 139.8, 140.3, 142.5, 145.99, 146.02, 149.0, 152.8, 155.4, 156.5, 165.0. HRMS (ESI): calcd. for C21H27FN7O13P2: 666.1121; Found: 666.1132.
Supplementary Material
Acknowledgments
We thank Ellison Medical Foundation for a New Scholar in Aging Award 2007 to Anthony A. Sauve. We also thank Cornell University for funding.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra for all new compounds described herein. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ma JA, Cahard D. Chem Rev. 2008;108:PR1–PR43. doi: 10.1021/cr800221v. [DOI] [PubMed] [Google Scholar]
- 2.Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 3.Kirk KL. Org Process Res Dev. 2008;12:305–321. [Google Scholar]
- 4.O’Hagan D. Chem Soc Rev. 2008;37(2):308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 5.Nicolaou KC, Mitchell HJ. Angew Chem Int Ed. 2001;40:1576–1624. [PubMed] [Google Scholar]
- 6.Williams SJ, Withers SG. Carbohydr Res. 2000;327:27–46. doi: 10.1016/s0008-6215(00)00041-0. [DOI] [PubMed] [Google Scholar]
- 7.Vocadlo DJ, Withers SG. Carbohydr Res. 2005;340:379–388. doi: 10.1016/j.carres.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 8.Vocadlo DJ, Davies GJ, Laine R, Withers SG. Nature. 2001;412:835–838. doi: 10.1038/35090602. [DOI] [PubMed] [Google Scholar]
- 9.Zechel DL, Withers SG. Curr Opin Chem Biol. 2001;5:643–649. doi: 10.1016/s1367-5931(01)00260-5. [DOI] [PubMed] [Google Scholar]
- 10.Sauve AA, Deng HT, Angeletti RH, Schramm VL. J Am Chem Soc. 2000;122:7855–7859. [Google Scholar]
- 11.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. J Biol Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Kriksunov IA, Jiang H, Graeff R, Lin H, Lee HC, Hao Q. Chem Biol. 2008;15:1068–1078. doi: 10.1016/j.chembiol.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter DJ, Merrill BM, Short SA. J Biol Chem. 1995;270:15551–15556. doi: 10.1074/jbc.270.26.15551. [DOI] [PubMed] [Google Scholar]
- 14.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. J Med Chem. 2005;48:5504–5508. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 15.Ferrandina G, Ludovisi M, Lorusso D, Pignata S, Breda E, Savarese A, Del Medico P, Scaltriti L, Katsaros D, Priolo D, Scambia G. J Clin Oncol. 2008;26:890–896. doi: 10.1200/JCO.2007.13.6606. [DOI] [PubMed] [Google Scholar]
- 16.Vulfovich M, Rocha-Lima C. Expert Rev Anticancer Ther. 2008;8:993–1002. doi: 10.1586/14737140.8.6.993. [DOI] [PubMed] [Google Scholar]
- 17.Silvestris N, Cinieri S, La Torre I, Pezzella G, Numico G, Orlando L, Lorusso V. Breast. 2008;17:220–226. doi: 10.1016/j.breast.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Eli Lilly. Eli Lilly 2007 Annual Report. 2007 [Google Scholar]
- 19.Bonate PL, Arthaud L, Cantrell WR, Jr, Stephenson K, Secrist JA, III, Weitman S. Nat Rev Drug Discov. 2006;5:855–863. doi: 10.1038/nrd2055. [DOI] [PubMed] [Google Scholar]
- 20.Genzyme. Genzyme 2007 Annual Report. 2007 [Google Scholar]
- 21.Sauve AA, Wolberger C, Schramm VL, Boeke JD. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 22.Hakme A, Wong HK, Dantzer F, Schreiber V. EMBO Rep. 2008;9:1094–1100. doi: 10.1038/embor.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith BC, Hallows WC, Denu JM. Chem Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 25.Sleath PR, Handlon AL, Oppenheimer NJ. J Org Chem. 1991;56:3608–3613. [Google Scholar]
- 26.Dax K, Albert M, Ortner J, Paul BJ. Carbohydr Res. 2000;327:47–86. doi: 10.1016/s0008-6215(00)00022-7. [DOI] [PubMed] [Google Scholar]
- 27.Reichman U, Watanabe KA, Fox JJ. Carbohydr Res. 1975;42:233–240. doi: 10.1016/s0008-6215(00)84265-2. [DOI] [PubMed] [Google Scholar]
- 28.Tewson TJ, Welch MJ. J Org Chem. 1978;43:1090–1092. [Google Scholar]
- 29.Watts JK, Damha MJ. Can J Chem. 2008;86:641–656. [Google Scholar]
- 30.Pankiewicz KW. Carbohydr Res. 2000;327:87–105. doi: 10.1016/s0008-6215(00)00089-6. [DOI] [PubMed] [Google Scholar]
- 31.Larsen CH, Ridgway BH, Shaw JT, Smith DM, Woerpel KA. J Am Chem Soc. 2005;127:10879–10884. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- 32.Mikhailopulo IA, Poopeiko NE, Sivets GG, Khripach NB. Nucleosides Nucleotides. 1995;14:383–384. [Google Scholar]
- 33.Wright JA, Taylor NF, Fox JJ. J Org Chem. 1969;34:2632–2636. doi: 10.1021/jo01261a031. [DOI] [PubMed] [Google Scholar]
- 34.Anderson CD, Goodman L, Baker BR. J Am Chem Soc. 1958;80:5247–5252. [Google Scholar]
- 35.Chou TS, Heath PC, Patterson LE, Poteet LM, Lakin RE, Hunt AH. Synthesis. 1992;6:565–570. [Google Scholar]
- 36.Fernandez R, Matheu MI, Echarri R, Castillon S. Tetrahedron. 1998;54:3523–3532. [Google Scholar]
- 37.McAtee JJ, Schinazi RF, Liotta DC. J Org Chem. 1998;63:2161–2167. [Google Scholar]
- 38.Dehoux C, Gorrichon L, Baltas M. Eur J Org Chem. 2001;6:1105–1113. [Google Scholar]
- 39.Tripp CP, Hair ML. Langmuir. 1995;11:149–155. [Google Scholar]
- 40.Damrauer R, Simon R, Krempp M. J Am Chem Soc. 1991;113:4431–4435. [Google Scholar]
- 41.Ge P, Kirk KL. J Org Chem. 1997;62:3340–3343. doi: 10.1021/jo962394b. [DOI] [PubMed] [Google Scholar]
- 42.Enders D, Potthoff M, Raabe G, Runsink J. Angew Chem Int Ed. 1997;36:2362–2364. [Google Scholar]
- 43.Enders D, Faure S, Potthoff M, Runsink J. Synthesis. 2001;15:2307–2319. [Google Scholar]
- 44.Welch JT, Plummer JS, Chou TS. J Org Chem. 1991;56:353–359. [Google Scholar]
- 45.Handlon AL, Xu C, Mullersteffner HM, Schuber F, Oppenheimer NJ. J Am Chem Soc. 1994;116:12087–12088. [Google Scholar]
- 46.Natalini P, Ruggieri S, Raffaelli N, Magni G. Biochemistry. 1986;25:3725–3729. doi: 10.1021/bi00360a037. [DOI] [PubMed] [Google Scholar]
- 47.Fox JJ, Hoffer M, Wempen I, Yung NC. J Am Chem Soc. 1961;83:4066–4070. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.