

Abstract
Glutamate (Glu) is associated with excitotoxic cell damage. Memantine modulates the glutamate induced excitotoxicity in Alzheimer’s disease (AD). No information is available as to the influence of memantine on in vivo brain glutamate levels.
Hippocampal Glu levels were measured in cognitively impaired and normal individuals (n=10) before and after six months of memantine treatment, using three dimensional high spatial resolution (0.5 cm3 voxels) proton magnetic resonance spectroscopy at 3 Tesla. These measurements were also repeated in a non-treated cognitively normal group (n=6).
Treatment with memantine decreased Glu/Cr (creatine) ratio in the left hippocampal region. Memantine reduced hippocampal glutamate levels, which may be consistent with its anti-excitotoxic property.
Keywords: Alzheimer’s disease, memantine, glutamate, hippocampus, magnetic resonance spectroscopy, excitotoxicity
Introduction
Glutamate (Glu) excitotoxicity through N-methyl-D-aspartic acid receptor (NMDAr) is a feature of Alzheimer’s disease (AD) (Lipton2004). Glutamate is the main excitatory neurotransmitter in hippocampal circuits (Ottersen1991) and the hippocampus, crucial for memory performance, is an early affected target in the course of AD. Memantine, an NMDAr antagonist, is an approved treatment of moderate to severe AD. The mechanism of memantine is based on the modulation of NMDAr stimulation by glutamate. Recent reports suggest that it is also effective in mild to moderate stages (Bakchine and Loft2007).
Proton magnetic resonance spectroscopy (1H-MRS) enables a non-invasive sampling of biochemical composition of the brain. Studies of hippocampal region show a reduction in N-acetylaspartate (NAA) levels (Schuff et al.1997;Block et al.2002) in AD patients as compared with normal controls. Some authors suggested that the decrease in NAA was a disease, but not age-related phenomenon, as they observed that NAA levels declined significantly with age in the frontal cortex, but not in the hippocampus (Chen et al.2000). Also reduction in cortical glutamate were reported in a few single voxel 1H-MRS AD studies (Antuono et al.2001;Hattori et al.2002), but the effects of memantine on in vivo hippocampal glutamate levels has not been examined.
Methods
Subjects
The subjects were recruited at the Center for Brain Health of the New York University (NYU) School of Medicine. All gave their written informed consent to NYU IRB (Institutional Review Board) approved protocols and received full clinical assessments (general medical, neurological, psychiatric, and laboratory examinations), a blood work-up and an MRI (to exclude possible confounding comorbidities affecting cognition). The assessment included a Mini Mental State Examination (MMSE) (Folstein1983) and a Global Deterioration Scale (GDS) (Reisberg et al.1993). A semi-structured interview based on the Brief Cognitive Rating Scale (BCRS) (Reisberg et al.1983) was used to query the informant for the subject’s recent memory performance and global functioning. A diagnosis of mild cognitive impairment (MCI) corresponded to GDS rating of 3 (Reisberg et al.1993). A diagnosis of AD was based on DSM IV criteria (American Psychiatric Association1994) and severity assessed using the GDS (subjects scored GDS=4 or 5 (Reisberg et al.1993)).
The study included two treated groups: normal elderly (NE-T, n=3) and cognitively impaired elderly (IE-T, n=7, 4 MCI and 3 AD patients) and two non-treated groups: normal elderly (NE-NT, n=3) and normal young (NY-NT, n=3). For all groups, the MRS examination was performed at baseline and after 6 months: in the treated groups this was before and after treatment. Memantine treatment began with 5 mg daily that gradually increased to 10 mg twice a day, over four weeks. For the next five months a dose of 20mg/ day was maintained.
MRS imaging and data post-processing
All MRI and 1H-MRS were performed in a 3 Tesla whole-body scanner (Trio, Siemens AG, Erlangen, Germany) using a TEM3000 transmit-receive head coil (MRInstruments, Minneapolis, MN). Subject was lying in the supine position with legs bent 45 degrees. The foam wedges placed on both sides of the head immobilized it in the coil. Each subject’s head was positioned using the imager’s standard orthogonal laser beams. Contiguous sagittal, and coronal T1-weighted spin-echo (TE/TR=7.3/600 ms) were obtained at 240×240 mm2 field-of-view (FOV) 512×512 matrix and 5 mm slice thickness. A paraxial MRI angulated along the anterior-posterior hippocampal axis followed, as shown in Figure 1. These MRI were used to manually image-guide a 9 cm left-right (LR) ×7cm anterior-posterior (AP) ×2cm inferior-sperior (IS) =126 cm3 volume-of-interest (VOI) paralleling the hippocampal axis to cover both hippocampi (Figure 1). Our custom three dimensional chemical shift imaging (3D CSI) based shimming produced consistent 19±4 Hz full-width at half maximum water from this VOI (Hu et al.1995).
Figure 1.
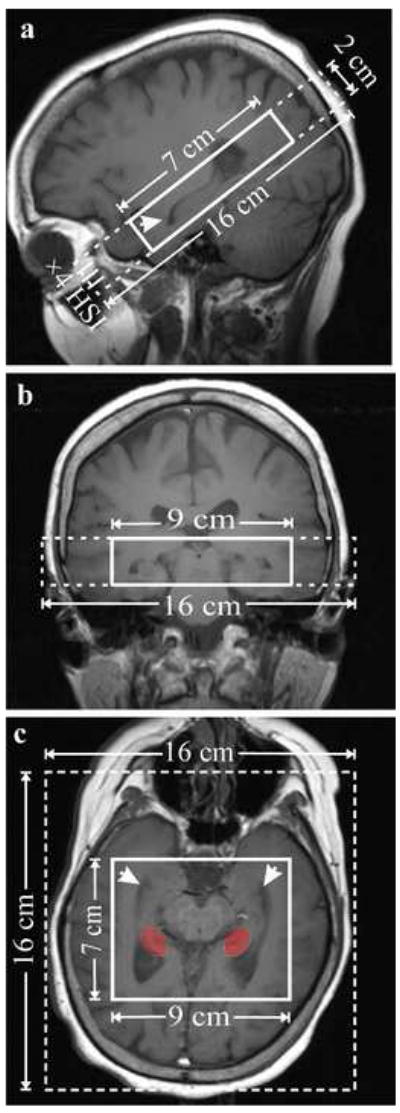
Sagittal (a) and coronal (b) and paraxial (c) T1-weighted images, along the hippocampus axis, depicting the position and angulation of the 16AP×16LR×2IS cm3 FOV (dashed line) and 7AP×9LR×2IS cm3 VOI (solid line) centered on the hippocampi (arrows). The red indicates the region of the hippocampus from where the spectra were sampled.
The VOI was selectively excited using TR/TE = 1600/ 39 ms point resolved spectroscopy (PRESS) with WET (water suppression enhanced through T1 effects), as shown in Figure 2 (Bottomley1987;Ogg et al.1994). No outer-volume-suppresion was applied. The PRESS partitioned the VOI using Hadamard encoding along a 2 cm FOV into four slices along its short - IS direction, each of which was subdivided with uniform sampling 16×16 2D-CSI in the LR×AP planes as shown in Figures 1 and 2 (Goelman et al.2006b). At a FOV of 16×16 cm2 this lead to a localization grid comprising nominal 1.0×1.0×0.5=0.5 cm3 voxels. The actual voxel size (full width at half maximum of the point spread function) for this uniform 2D phase encoding is 1.12×1.12×0.5=0.63 cm3 (Mareci and Brooker1991;Brooker et al.1987). (Note that in the third, Hadamard direction the nominal equals the actual voxel size (Goelman et al.2006a)). For consistency reasons, however, in the ensuing discussion we will refer to the nominal voxel size. The MR signal was digitized for 512 ms [determined by the 150 – 230 ms T2 values of cerebral metabolites (Zaaraoui et al.2007)] with 1024 complex points for a 2 Hz/point spectral resolution.
Figure 2.
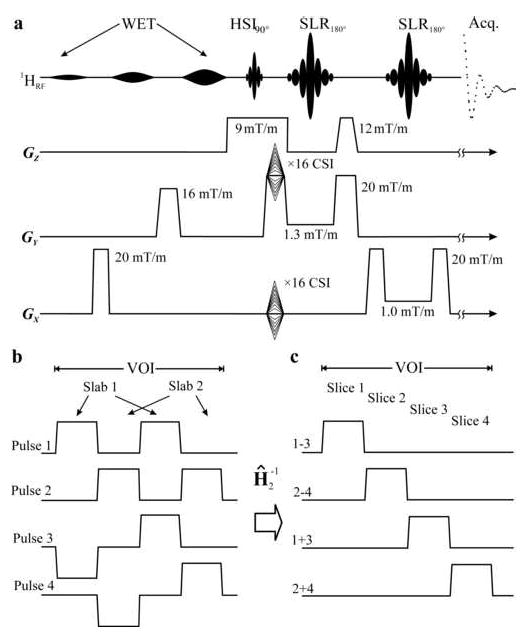
Top, a: Schematic description of the TE=39ms PRESS with WET water suppression. Localization along X and Y (LR×AP) was ×16 CSI. The ×4 partitions along Z (IS) were achieved by sequentially encoding two slabs each TR=1600 ms, with 2nd order HSI, as shown in 2b, with the 5.12 ms PRESS 90° pulse. The two refocusing 180°s were 12.8 ms long numerically optimized Shinnar-LeRoux pulses under the selective-gradient strengths indicated. Bottom, left, b: The spatial excitation profiles of the two 2nd order HSI slabs in the VOI. The goal was to reduce the B1 demands and the inter-slice “bleed” (Goelman et al.2006b;Goelman et al.2006a). Bottom, right, c: The four slice profiles obtained by the two inverse 2nd order Hadamard transforms, Ĥ2−1, indicated as i±j with i and j indicating the pulse numbers in 2b.
Reducing the chemical shift displacement artifact for more accurate localization at the higher B0 requires stronger selective gradients and commensurate larger radio-frequency (RF) pulse bandwidths (larger, B1). To retain the VOI size, yet minimize the chemical shift displacement, the novel multi-slab approach of Fig. 2b, was used (Goelman et al.2006b). Exploiting the property that Hadamard slices do not have to be contiguous, we segmented the VOI into two slabs, only half of which was excited at each acquisition, as shown in Fig. 2b. Consequently, only half the B1 needed to cover the whole VOI was required, allowing use of the strong, 9 mT/m, HSI slice-select gradient shown in Fig. 2a. Since the slabs excited different parts of the VOI and each required under 800 ms to acquire, both were obtained sequentially during each TR=1600 ms, halving the 16×16×4 MRSI matrix acquisition time to 13.7 min. (27.5 min. for the two averages applied) at a near-ideal 100% receiver duty cycle due to this “multiplexing in space and time” scheme (Goelman et al.2006b). The four slices were reconstructed by two 2nd order inverse Hadamard transforms as shown in Fig. 2c.
The 1H-MRS was post-processed offline using in-house software. Residual water signals were removed in the time domain using the method of (Marion et al.1989), and the time data apodized with a 4Hz Lorentzian, zero-filled from 1024 to 2048 points in the time domain and from a spatial grid of 16×16 to 32×32 in the LR×AP planes, yielding interpolated 0.125 cm3 voxels (no spatial filters were applied). Although zero-filling does not add any information content to the raw data, our rationale for choosing this strategy is that it can increase the effective resolution by providing overlapping voxels, thereby reducing partial volume artifacts (Du et al.1994;Bernstein et al.2001). This was followed by Fourier transforms along the LR, AP and time directions, and Hadamard reconstruction along the IS direction (Goelman et al.2006b). Automatic frequency and zero-order phase corrections were made in each voxel in reference to its NAA peak. Relative metabolites levels were estimated from their peak areas using parametric spectral modeling and least-squares optimization of Soher et al., as shown in Figure 3b (Soher et al.1998). Eight model functions (aspartate, choline, Cr, Glu, glutamine, myo-inositol, NAA and taurine) were used to fit our data. The software also accounted for metabolite line-widths differences amongst voxels (although it assigns a single linewidth to all metabolites within a given voxel) that could arise from magnetic field inhomogeneities due to variable susceptibility gradients around the hippocampus.
Figure 3.
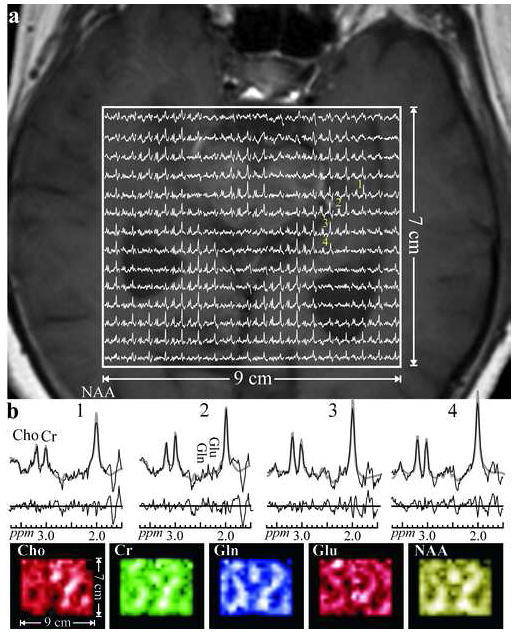
a. Real part of the 18×14 (LR×AP) axial matrix of 1H spectra in the VOI of Fig. 1c (Solid frame) superimposed on the MRI of this slice for anatomical reference (right side of the figure corresponds to the left side of the head). Spectra representing (0.5 cm)3=0.125 cm3 interpolated voxels share the same 1.6 - 3.7 ppm and intensity scales. The linewidths in this slice were 12.1±1.4 Hz (mean ± standard deviation).
b: Top: Voxels marked “1-4” from the left hippocampus in the matrix above; raw spectra (solid thin lines) overlaid with the metabolite fitted function (gray thick curves) with the residual (raw spectrum – fit) plotted below. Spectra and residuals are on the same horizontal and PPM scales. Note the SNR, spectral resolution and quality of the fit (reflected from the noise-only residual, from these small voxels acquired in 27 minutes.
b: Bottom: Axial metabolic maps of the N-acetylaspartate (NAA), glutamate (Glu), glutamine (Gln), creatine (Cr), and choline (Cho) obtained from the fit of the above matrix. Note the excellent correspondence between the metabolic images and the morphology of the image above, reflecting the overall quality of the spectra, spatial resolution and localization. The metabolite maps presented are formed from the quantified amplitude of each metabolite.
In every case three hippocampal regions were established: anterior, middle and posterior. The regions were equally spaced along the anterior-posterior axis of the hippocampus (from the most anterior to the most posterior margin of the hippocampus on the axial projections). Depending on the subject’s anatomy and a slice, anterior hippocampal margin was demarcated by the border between amygdala and the hippocampus or was visible posterior to the temporal bone at the level of the temporal horn. Posterior margin was demarcated by the fimbria. Voxels from the posterior part of each hippocampus were interactively selected, for two main reasons: First, the hippocampus is not a homogenous structure (Vermathen et al.2000), thus one standard region was selected. Second, the posterior yields the highest quality spectra. Glutamate and NAA are presented as ratio to Cr in order to correct for variations in susceptibility, coil loading, partial volume and changes in the receiver gain over time. For subjects, the ratio is the average value derived from all voxels in the posterior region.
Statistical analysis
U Mann-Whitney and χ2 tests were used to compare the demographic and metabolites data between subject groups. The rates of change in metabolites over time were expressed as
The within-group comparisons between baseline and follow-up metabolite ratios were done with Wilcoxon test.
Results
1H-MRS
Sample spectra from a paraxial slice containing the bilateral hippocampi of a 72 year old healthy female elderly control is shown in Figure 3, superimposed on the MRI from the corresponding slice for anatomical reference. Metabolic maps from this slice are also presented to demonstrate the quality of localization, as reflected by the metabolic voids in the cerebrospinal fluid (CSF) spaces.
The SNRs for the metabolites, defined as their peak-height divided by twice the root-mean-square of the noise [see 4.3.14 in (Ernst et al.1987)], were (mean ± standard-deviation) NAA: 8.0±3.3, Cr: 5.3±1.9, Cho: 4.7±1.8 Glu: 6.4±3.2 and Gln: 3.3±0.3. Peak heights for this calculation were obtained by dividing their area by the line-width in that voxel, both estimated by the spectral modeling procedure, as shown in Fig. 3 (Soher et al.1998). The noise was estimated from a peak-free region ca. 3-4 ppm upfield from the NAA. SNRs under “3” were discarded as ”uncertain signal” for the metabolite in that voxel, e.g., in the ventricles. The linewidths determined from the spectral modeling procedure were 12.1±1.4 Hz. Recalling that this value also contains the 4 Hz line-broadening filter used in the post processing leads to an actual linewidth of 8.1±1.4 Hz, or a T2* of 39 ms.
Baseline
Cognitively normal elderly (NE-NT&NT-T, n=6) were slightly younger than impaired elderly subjects (IE-T n=7) (Z=-2.1, p<.05) and had lower BCRS ratings (Z=-2.9, p<.01) (Table 1). When whole treated and non-treated groups were compared significant differences were found only for age (Z=-2.6, p<.01) and BCRS (Z=-3.3, p<.01), with non-treated group being younger and having lower BCRS scores. Neither baseline Glu/Cr nor NAA/Cr ratios differed between normal elderly and cognitively impaired subjects, or between treated and non-treated subjects.
Table 1.
Study groups at baseline. Values are given as means (standard deviations).
| Group | NY-NT (n=3) | NE-NT&NE-T | NE-NT (n=3) | NE-T (n=3) | IE-T (n=7) | |
|---|---|---|---|---|---|---|
| Age | 29.9 (3.8) | 70.7 (1.9) | 70.3 (1.6) | 71.1 (2.4) | 74.1 (7.8) | |
| F/M | 2/1 | 4/2 | 2/1 | 2/1 | 3/4 | |
| Time to follow-up (months) | 6.7 (4.4) | 10.7 (2.9) | 12.7 (2.1) | 8.7 (2.3) | 6.5 (3.8) | |
| BCRS | 1.0 (.00) | 1.3 (.32) | 1.1 (.11) | 1.6 (.20) | 2.5 (.60) | |
| MMSE | 30 | 29.2(.75) | 29.0(1.0) | 29.3(0.6) | 26.9 (2.9) | |
| Glu/Cr | Left hippocampus | .56 (.23) | .91 (.54) | .74 (.42) | 1.10 (.69) | .97 (.56) |
| Right hippocampus | 1.19 (.85) | .82 (.36) | .95 (.47) | .68 (.22) | 1.30 (1.20) | |
| NAA/Cr | Left hippocampus | 1.65 (.26) | 1.57 (.58) | 1.59 (.65) | 1.56 (.65) | 1.75 (.62) |
| Right hippocampus | 1.82 (.09) | 1.57 (.70) | 1.85 (.99) | 1.29 (.12) | 1.64 (.77) | |
NY-NT: non-treated young controls
NE-NT: non-treated elderly controls
NE-T: cognitively normal elderly treated with memantine
IE-T: cognitively impaired elderly treated with memantine
The BCRS mean is derived only from first 5 items (concentration, recent memory, past memory, orientation and functional assessment staging) as they are on an ordinal scale, the remainder 6 items are on a categorical scale. The score of 1 for each of first five items corresponds to “no difficulties”.
Treatment effects
The rate of change for Glu/Cr in the left hippocampus was significantly reduced (Z=-2.8, p<.01) in the total treated group (NE-T&IE-T: -.04 (±.06) unit/month) as compared with the non-treated group (NY-NT and NE-NT: .07 (±.04) unit/month). After exclusion of the younger subjects (n=3), the analysis yielded similar results (Z=-1.8, p=.06). No Glu/Cr effects were found for the right hippocampus (Z=-.21, p>.05). A within-group comparisons of baseline and follow-up metabolite ratios revealed in the total non-treated group an increase with time (Z=-2.2, p<.05) for the Glu/Cr in the left hippocampus. In the total treated group, there was a trend towards lower Glu/Cr after memantine treatment (Z=-1.8, p=.06). At follow-up the left Glu/Cr was significantly lower in the treated than the non-treated group (Z=-2.4, p<.05), no difference was found for the right hippocampus (Z=-1.4, p>.05).
No changes over time were found for NAA/Cr ratios. This was true both for the rates of NAA/Cr change in the left (Z=-.76, p>.05) and right (Z=-.65, p>.05) hippocampus, as well as for within-group comparisons of baseline and follow-up metabolite ratios in the treated and non-treated group. Consequently, there were no differences between the two groups in the left (Z=-.54, p>.05) or right (Z=-1.3, p>.05) NAA/Cr at follow-up.
Discussion
This is the first clinical data, showing that treatment with an uncompetitive NMDAr antagonist (memantine) decreases the Glu/Cr in the hippocampus. Others have observed an increase in cingulate glutamatergic turnover (Rowland et al.2005) and an increase in frontal and cingulate blood flow, a surrogate for glutamate release (Holcomb et al.2005), after acute ketamine administration. Moreover, in animal hippocampal homogenates ketamine stimulated the phosphate-activated glutaminase (PAG) activity (an enzyme converting glutamine to Glu) (Dawson and Wallace1993).
Although both ketamine and memantine represent the same class of drugs, they differ in blocking kinetics and voltage dependence (Danysz and Parsons1998). In addition, the above experiments consisted of an acute challenge while we measured steady-state glutamate levels after 6 months of treatment. There is no information about brain glutamate changes in response to chronic treatment with NMDAr antagonists in humans and the information from animal models is limited. A recent paper reports on the decrease in CSF glutamate in rats treated with ketamine (Tomiya et al.2007), consistent with our finding. Moreover, repetitive administration of ketamine was shown to reduce PAG activity (Moran et al.1999). This suggests that chronic administration of uncompetitive NMDAr antagonist may result in glutamate decrease. An increased glutamate concentration in the CSF of AD patients was found (D’Aniello et al.2005;Jimenez-Jimenez et al.1998), and consequently, changes in the opposite direction, a reduction in glutamate, might be of beneficial effect.
NMDAr activation induced the endocytosis of EAAC1 (neuronal transporter excitatory amino acid carrier 1) in hippocampal cultures. An NMDAr antagonist inhibited this effect (Waxman et al.2007). It is hence possible that chronic treatment with NMDA antagonist can change glutamate uptake. As some glutamate uptake is also fueling de novo synthesis of GABA (Gonzalez et al.2007), a change in glutamate uptake might change its total concentration.
Excitotoxicity plays a major role in AD (Lipton2004). Atrophy-corrected NAA concentrations (Schuff et al.1997) and NAA/Cr hippocampal ratios (Block et al.2002) are lower in AD (reflecting cell death). Although cortical Glu/Cr reductions were also observed (Hattori et al.2002), no information on hippocampal levels is available. There is an evidence from MRS studies of other excitotoxicity related conditions like amyotrophic lateral sclerosis (Pioro et al.1999) and Huntington disease (Reynolds et al.2005) that glutamate is elevated in vulnerable regions. Thus, the decrease in hippocamal glutamate with memantine (without changes in NAA/Cr) is a potential explanation for the therapeutic effects.
The lack of Glu/Cr and NAA/Cr differences across diagnostic groups at baseline and the laterality effect also merit a discussion. With respect to this first finding, we believe it is most likely due to small sample sizes. With regard to the second: In AD left hemisphere seem to be more affected with more pronounced atrophy of the left MTL and other cortical regions (see in (Glodzik-Sobanska et al.2005)). Memantine is expected to work better for more advanced conditions, better blocking higher than lower receptor activity (Wenk et al.2006). Thus, the effects in the left hippocampus might be consistent with greater pathological involvement of the left hemisphere.
Another possible explanation for our findings cannot be dismissed: The decrease in Glu/Cr ratio may represent neuronal loss, not a drug effect. Few arguments speak against this possibility: First, no changes in NAA/Cr were found. Second, the IE-T group did not represent advanced stage of AD pathology: 4 MCI and 3 AD subjects, with the mean MMSE score above 24. Still, glutamate might be more sensitive marker of cellular dysfunction, and thus its decrease would be observed earlier. This possibility must be considered, as non-treated subjects with cognitive impairment were not studied.
The Glu/Cr CVs (coefficients of variation) were high in treated and non-treated groups. This is a possible limitation of our study. As additionally for all Glu/Cr measurements the means approach zero and SNR falls below 3, relying on the CVs to estimate the detection limits is somewhat problematic. Nevertheless, in the analysis of this data we did not rely on one experiment but rather on the pattern of changes amongst all subjects. Although there was no difference from baseline, at follow-up the left Glu/Cr was significantly lower in the treated than non-treated group, and a within-group comparisons of baseline and follow-up metabolite ratios revealed a decrease in Glu/Cr in the treated group. The decrease was not observed in any of the subjects in the non-treated group.
Finally, regarding our choice of the hippocampus: the action of memantine is not limited to this structure. However, because hippocampal intrinsic neurons are glutamatergic (Greenamyre and Porter1994) and hippocampal cells bear a great number of glutamatergic receptors (Kornhuber et al.1989), it seemed to be an anatomy of choice for our examination. The best spectra were obtained from the posterior parts of the hippocampus, underscoring shimming difficulties in the anterior part. Others also encountered this problem (Schuff et al.1997). The CSF surrounds posterior part of the hippocampus laterally and in most cases medially. Therefore, voxels which may have partial volume outside the hippocampus will have this volume full of CSF. Consequently, taking ratios would eliminate the CSF scaler. It is still possible, that, due to variable hippocampal thickness and shape, voxels might have contained parahippocampal white matter. However, small voxel volumes and uniform tissue sampling should reduce the contamination from CSF and other anatomical variations.
Conclusion
Overall, the results suggest that memantine decreases hippocampal Glu/Cr, possibly consistent with this drug’s anti-excitotoxic properties. As decline in neuronal function cannot be entirely rejected, these data require replication with larger samples sizes.
Acknowledgments
We thank Drs. Andrew A. Maudsley of the University of Miami and Brian J. Soher of Duke University for the use of the SITools-FITT spectral modeling software, Ms. Rachel Mistur and Schantel Williams for cognitive testing and Ms. Nissa Perry for coordination of the MR evaluations. This study was supported by Forest Laboratories, Inc., NIH grants: AG12101, AG 22374, AG0305, P30 AG08051, EB01015 and NS050520, and a grant from the Mentored Medical Student Clinical Research (MMSCR) Program NCRR M01RR00096 (KGK).
Abbreviations
- AD
Alzheimer’s disease
- BCRS
Brief Cognitive Rating Scale
- Cr
creatine
- CSF
cerebrospinal fluid
- FOV
field of view
- GDS
Global Deterioration Scale
- Glu
glutamate
- 1H-MRS
proton magnetic resonance spectroscopy
- IE-T
treated cognitively impaired elderly
- MCI
mild cognitive impairment
- NAA
N-acetylaspartate
- NE-NT
non-treated elderly controls
- NE-T
treated cognitively normal elderly
- NY-NT
non-treated young controls
- NMDAr
N-methyl-D-aspartic acid receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition. Washington, D.C.: American Psychiatric Association; 1994. [Google Scholar]
- Antuono PG, Jones J, Wang Y, Li SJ. Decreased glutamate 1 glutamine in Alzheimer’s disease detected in vivo with 1H-MRS at 0.5 T. Neurol. 2001;56:737–742. doi: 10.1212/wnl.56.6.737. [DOI] [PubMed] [Google Scholar]
- Bakchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results of a randomised, double-blind, placebo-controlled 6-month study. Journal of Alzheimer’s Disease. 2007:471–479. doi: 10.3233/jad-2007-11409. [DOI] [PubMed] [Google Scholar]
- Bernstein MA, Fain SB, Riederer SJ. Effect of windowing and zero-filled reconstruction of MRI data on spatial resolution and acquisition strategy. J Magn Res Imaging. 2001;14:270–280. doi: 10.1002/jmri.1183. [DOI] [PubMed] [Google Scholar]
- Block W, Jessen F, Traber F, Flacke S, Manka C, Lamerichs R, et al. Regional N-acetylaspartate reduction in the hippocampus detected with fast proton magnetic resonance spectroscopic imaging in patients with Alzheimer disease. Arch Neurol. 2002;59:828–834. doi: 10.1001/archneur.59.5.828. [DOI] [PubMed] [Google Scholar]
- Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Annals New York Academy of Sciences. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- Brooker HR, Mareci TH, Mao JT. Selective Fourier transform localization. Magn Res Med. 1987;5:417–433. doi: 10.1002/mrm.1910050503. [DOI] [PubMed] [Google Scholar]
- Chen JG, Charles HC, Barboriak DP, Doraiswamy PM. Magnetic resonance spectroscopy in Alzheimer’s disease: focus on N-acetylaspartate. Acta Neurol Scand Suppl. 2000;176:20–26. doi: 10.1034/j.1600-0404.2000.00303.x. [DOI] [PubMed] [Google Scholar]
- D’Aniello A, Fisher G, Migliaccio N, Cammisa G, D’Aniello E, Spinelli P. Amino acids and transaminases activity in ventricular CSF and in brain of normal and Alzheimer patients. Neuroscience Letters. 2005;388:49–53. doi: 10.1016/j.neulet.2005.06.030. [DOI] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. Glycine and N-Methyl-D-Aspartate Receptors: Physiological Significance and Possible Therapeutic Applications. Pharmacological Reviews. 1998;50:597–664. [PubMed] [Google Scholar]
- Dawson RJ, Wallace DR. Regulation of phosphate-activated glutaminase (PAG) by glutamate analogues. Neurochemical Research. 1993;18:125–132. doi: 10.1007/BF01474674. [DOI] [PubMed] [Google Scholar]
- Du YP, Parker DL, Davis WL, Cao G. Reduction of partial-volume artifacts with zero-filled interpolation in three-dimensional MR angiography. J Magn Res Imaging. 1994;4:733–741. doi: 10.1002/jmri.1880040517. [DOI] [PubMed] [Google Scholar]
- Ernst RR, Bodenhausen G, Wokaun A. The International Series of Monographs on Chemistry. Oxford: Claredon Press; 1987. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. [Google Scholar]
- Folstein M. The Mini-Mental State Examination. In: Crook T, Ferris SH, Bartus R, editors. Assessment in Geriatric Psychopharmacology. New Canaan: Mark Powley Associates; 1983. pp. 47–51. [Google Scholar]
- Glodzik-Sobanska L, Rusinek H, Mosconi L, Li Y, Zhan J, De Santi S, et al. The role of quantitative structural imaging in the early diagnosis of Alzheimer’s disease. Neuroimaging Clinics of North America. 2005;15:803–826. doi: 10.1016/j.nic.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Goelman G, Liu S, Gonen O. Reducing voxel bleed in Hadamard-encoded MRI and MRS. Magn Reson Med. 2006a;55:1460–1465. doi: 10.1002/mrm.20903. [DOI] [PubMed] [Google Scholar]
- Goelman G, Liu S, Hess D, Gonen O. Optimizing the efficiency of high-field multivoxel spectroscopic imaging by multiplexing in space and time. Magn Reson Med. 2006b;56:34–40. doi: 10.1002/mrm.20942. [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Susarla BTS, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04881.x. in press. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Porter RH. Anatomy and physiology of glutamate in the CNS. Neurol. 1994;44:S7–S13. [PubMed] [Google Scholar]
- Hattori N, Abe K, Sakoda S, Sawada T. Proton MR spectroscopic study at 3 Tesla on glutamate/glutamine in Alzheimer’s disease. NeuroReport. 2002;13:183–186. doi: 10.1097/00001756-200201210-00041. [DOI] [PubMed] [Google Scholar]
- Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA. Effects of Noncompetitive NMDA Receptor Blockade on Anterior Cingulate Cerebral Blood Flow in Volunteers with Schizophrenia. Neuropsychopharmacology. 2005;30:2275–2282. doi: 10.1038/sj.npp.1300824. [DOI] [PubMed] [Google Scholar]
- Hu J, Javaid T, Arias-Mendoza F, Liu Z, McNamara R, Brown TR. A fast, reliable, automatic shimming procedure using 1H chemical-shift-imaging spectroscopy. J Magn Reson. 1995;108:213–219. doi: 10.1006/jmrb.1995.1126. [DOI] [PubMed] [Google Scholar]
- Jimenez-Jimenez FJ, Molina JA, Gomez P, Vargas C, de Bustos F, Benito-Leon J, et al. Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. J Neural Transm. 1998;105:269–277. doi: 10.1007/s007020050056. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Mack-Bukhardt F, Riederer P, Hebenstreit GF, Reynolds GP. [3H]MK-801 binding sites in postmortem brain regions of schizophrenic patients. J Neural Transm. 1989;77:231–236. doi: 10.1007/BF01248936. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Failures and Successes of NMDA Receptor Antagonists: Molecular Basis for the Use of Open-Channel Blockers like Memantine in the Treatment of Acute and Chronic Neurologic Insults. NeuroRx. 2004;1:101–110. doi: 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareci TH, Brooker HR. Essential considerations for spectral localization using indirect gradient encoding of spatial information. J Magn Reson. 1991;92:229–246. [Google Scholar]
- Marion D, Ikura M, Bax A. Improved solvent suppression in one- and two-dimensional NMR spectra by convolution of time domain data. J Magn Reson. 1989;84:425–430. [Google Scholar]
- Moran J, Alavez S, Rivera-gaxiola M, Valencia A, Hurtado S. Effect of nmda antagonists on the activity ofglutaminase and aspartate aminotransferase in thedeveloping rat cerebellum. International Journal of Developmental Neuroscience. 1999;17:57–65. doi: 10.1016/s0736-5748(98)00063-x. [DOI] [PubMed] [Google Scholar]
- Ogg RJ, Kingsley PB, Taylor JS. WET, aT1-and B1-insensitive water suppresion method for in vivo localized 1H NMR spectroscopy. J Magn Reson. 1994;104:1–10. doi: 10.1006/jmrb.1994.1048. [DOI] [PubMed] [Google Scholar]
- Ottersen OP. Excitatory aminoacid neurotransmitters:anatomical systems. Excitatory Amino Acids Antagonists. 1991:14–38. [Google Scholar]
- Pioro EP, Majors A, Mitsumoto H, Nelson DR, Ng T. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurol. 1999;53:71–79. doi: 10.1212/wnl.53.1.71. [DOI] [PubMed] [Google Scholar]
- Reisberg B, London E, Ferris SH, Borenstein J, Scheier L, de Leon MJ. The Brief Cognitive Rating Scale: Language, motoric, and mood concomitants in primary degenerative dementia. Psychopharmacology Bulletin. 1983;19:702–708. [Google Scholar]
- Reisberg B, Sclan SG, Franssen EH, de Leon MJ, Kluger A, Torossian CL, et al. Clinical stages of normal aging and Alzheimer’s disease: The GDS staging system. Neurosci Res Communications. 1993;13(Suppl 1):551–554. [Google Scholar]
- Reynolds NC, Prost RW, Mark LP. Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington’s disease. Brain Res. 2005;1031:82–89. doi: 10.1016/j.brainres.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. American Journal of Psychiary. 2005;162:394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- Schuff N, Amend D, Ezekiel F, Steinman SK, Tanabe JL, Norman D, et al. Changes of hippocampal N-acetyl aspartate and volume in Alzheimer’s disease. A proton MR spectroscopic imaging and MRI study. Neurol. 1997;49:1513–1521. doi: 10.1212/wnl.49.6.1513. [DOI] [PubMed] [Google Scholar]
- Soher BJ, Young K, Govindaraju V, Maudsley AA. Automated spectral analysis III: application to in vivo proton MR spectroscopy and spectroscopic imaging. Magn Reson Med. 1998;40:822–831. doi: 10.1002/mrm.1910400607. [DOI] [PubMed] [Google Scholar]
- Tomiya M, Fukushima T, Kawai J, Aoyama C, Mitsuhashi S, Santa T, et al. Alterations of plasma and cerebrospinal fluid glutamate levels in rats treated with the N-methyl-D-aspartate receptor antagonist, ketamine. Biomedical Chromatrography. 2007;20:628–633. doi: 10.1002/bmc.677. [DOI] [PubMed] [Google Scholar]
- Vermathen P, Laxer KD, Matson GB, Weiner MW. Hippocampal structures: anteroposterior N-acetylaspartate differences in patients with epilepsy and control subjects as shown with proton MR spectroscopic imaging. Radiology. 2000;214:403–410. doi: 10.1148/radiology.214.2.r00fe43403. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Baconguis I, Lynch DR, Robinson MB. N-Methyl-D-aspartate Receptor-dependent Regulation of the Glutamate Transporter Excitatory Amino Acid Carrier 1. Journal of Biological Chemistry. 2007;282:17594–17607. doi: 10.1074/jbc.M702278200. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Parsons CG, Danysz W. Potential role of N-methyl-D-aspartate receptors as executors of neurodegeneration resulting from diverse insults: focus on memantine. Behavioural Pharmacology. 2006;17:411–424. doi: 10.1097/00008877-200609000-00007. [DOI] [PubMed] [Google Scholar]
- Zaaraoui W, Fleysher L, Fleysher R, Liu S, Soher BJ, Gonen O. Human brain-structure resolved T2 relaxation times of proton metabolites at 3 tesla. Magn Reson Med. 2007;57:983–989. doi: 10.1002/mrm.21250. [DOI] [PubMed] [Google Scholar]